
Multidimensional Gene Regulatory Landscape of Motor Organ Pulvinus in the Model Legume Medicago truncatula

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
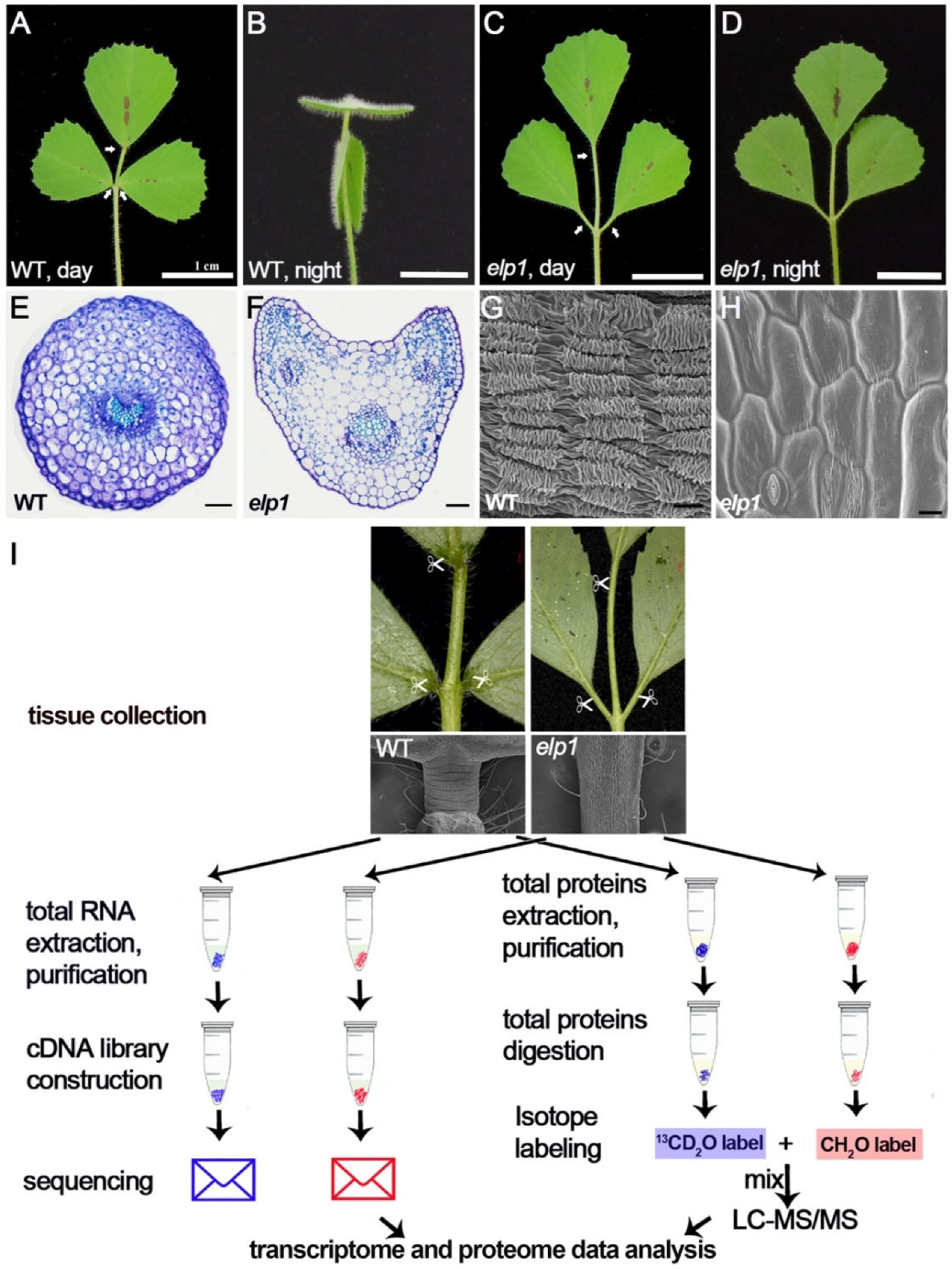
2.1. Collection of WT Pulvini and the Defecitve Pulvinus of elp1 Mutant for Transcriptome and Proteome Analyses
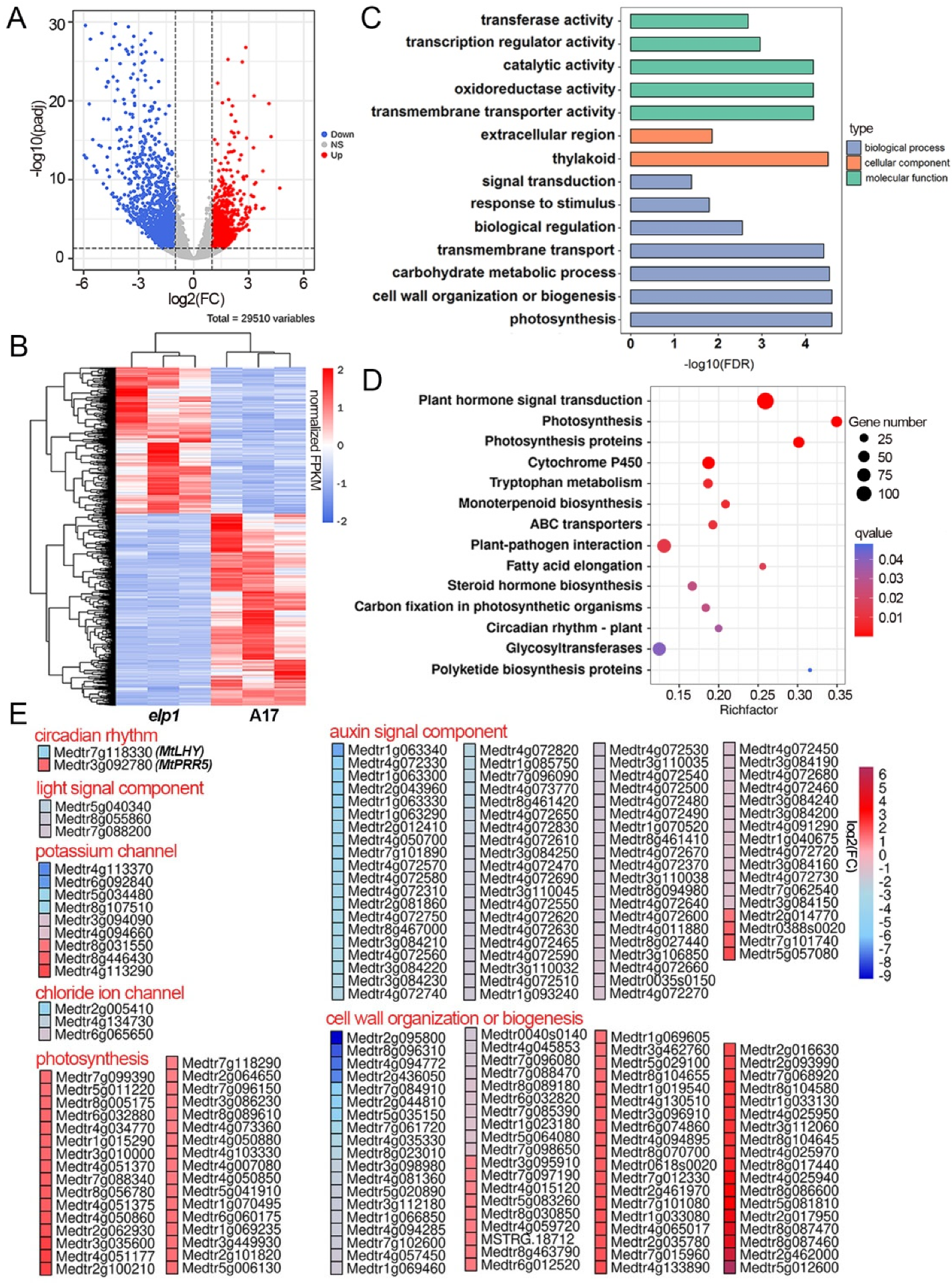
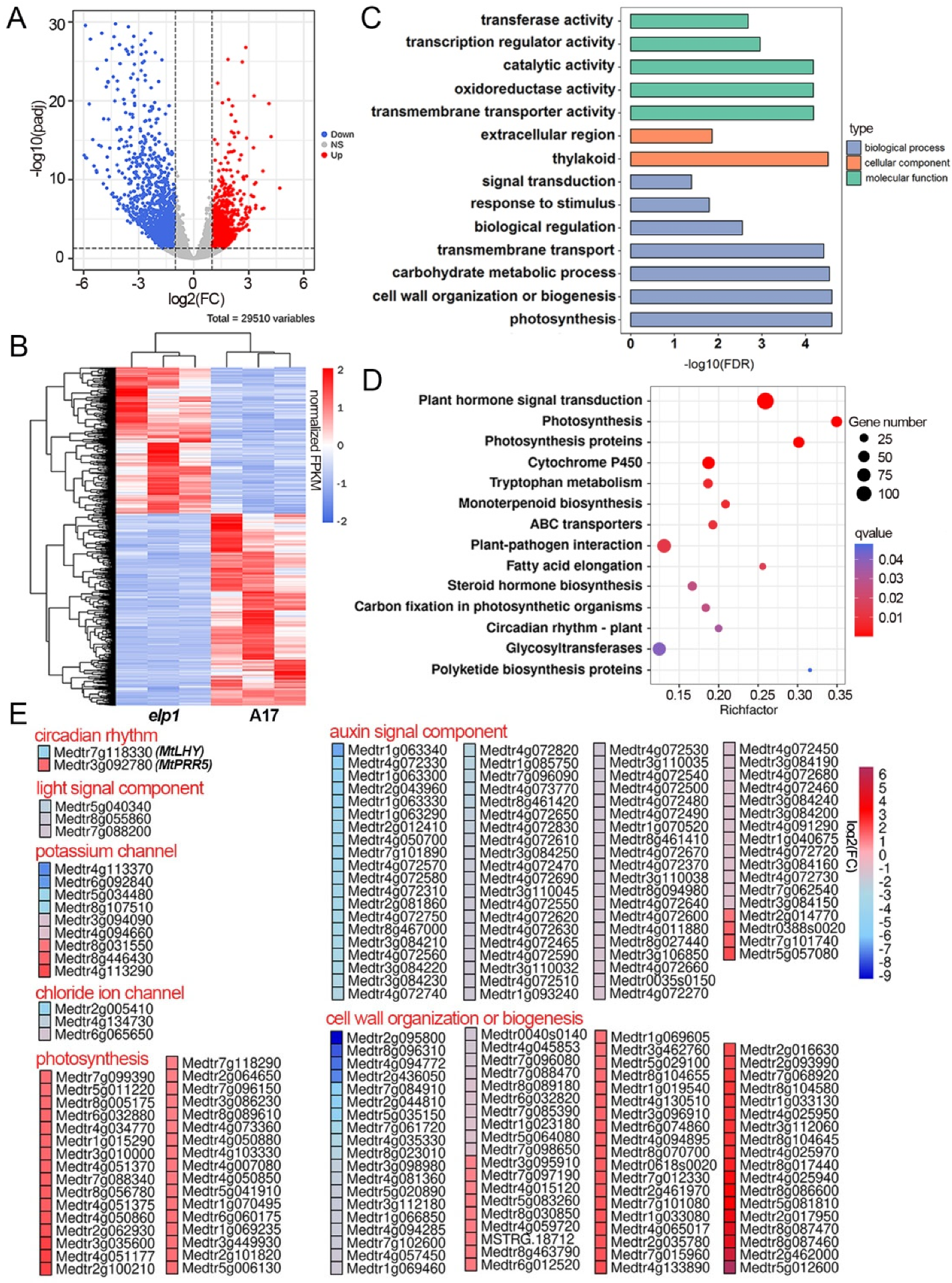
2.2. Multiple DEGs Are Involved in Pulvinus Development and Signal Transduction
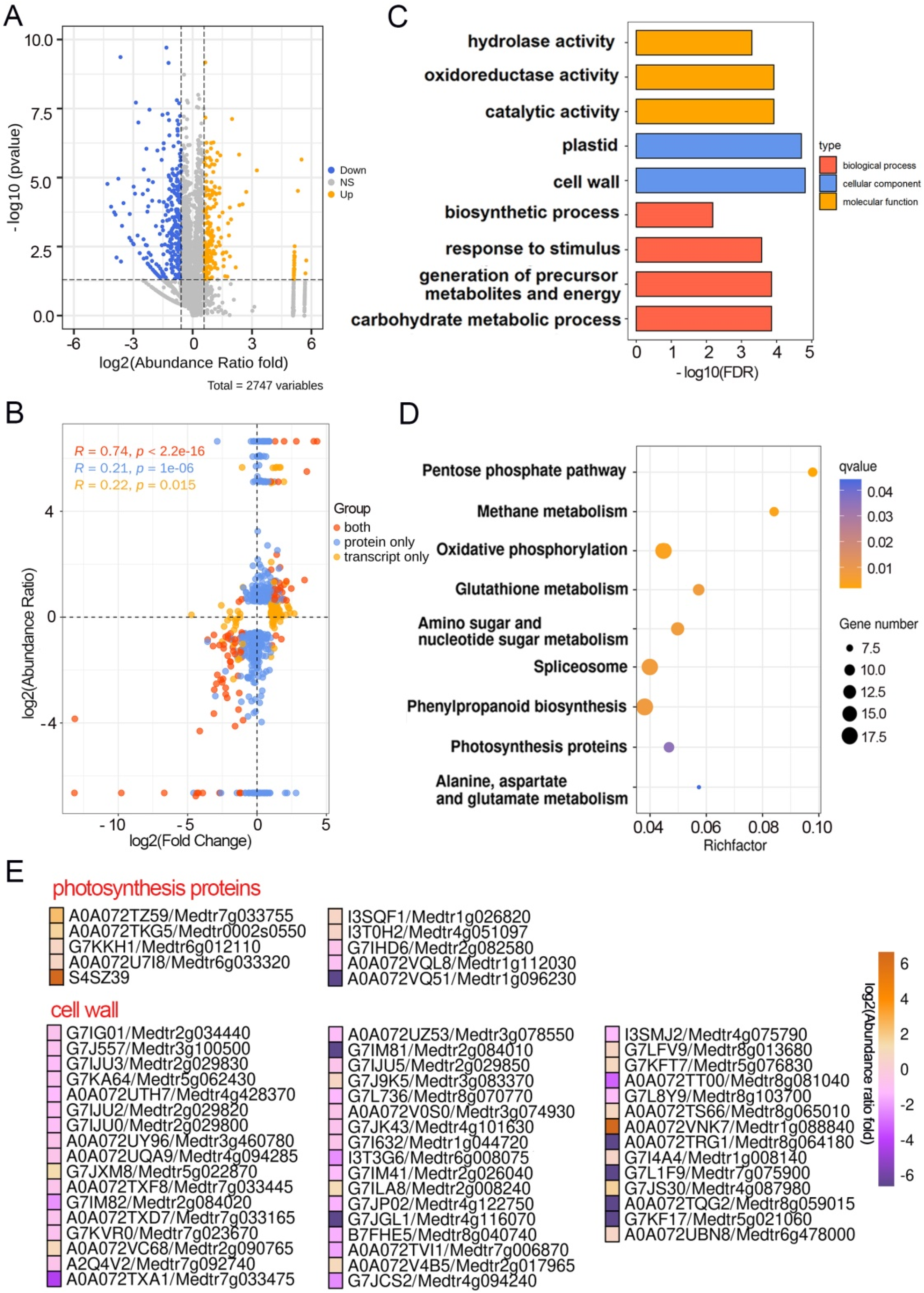
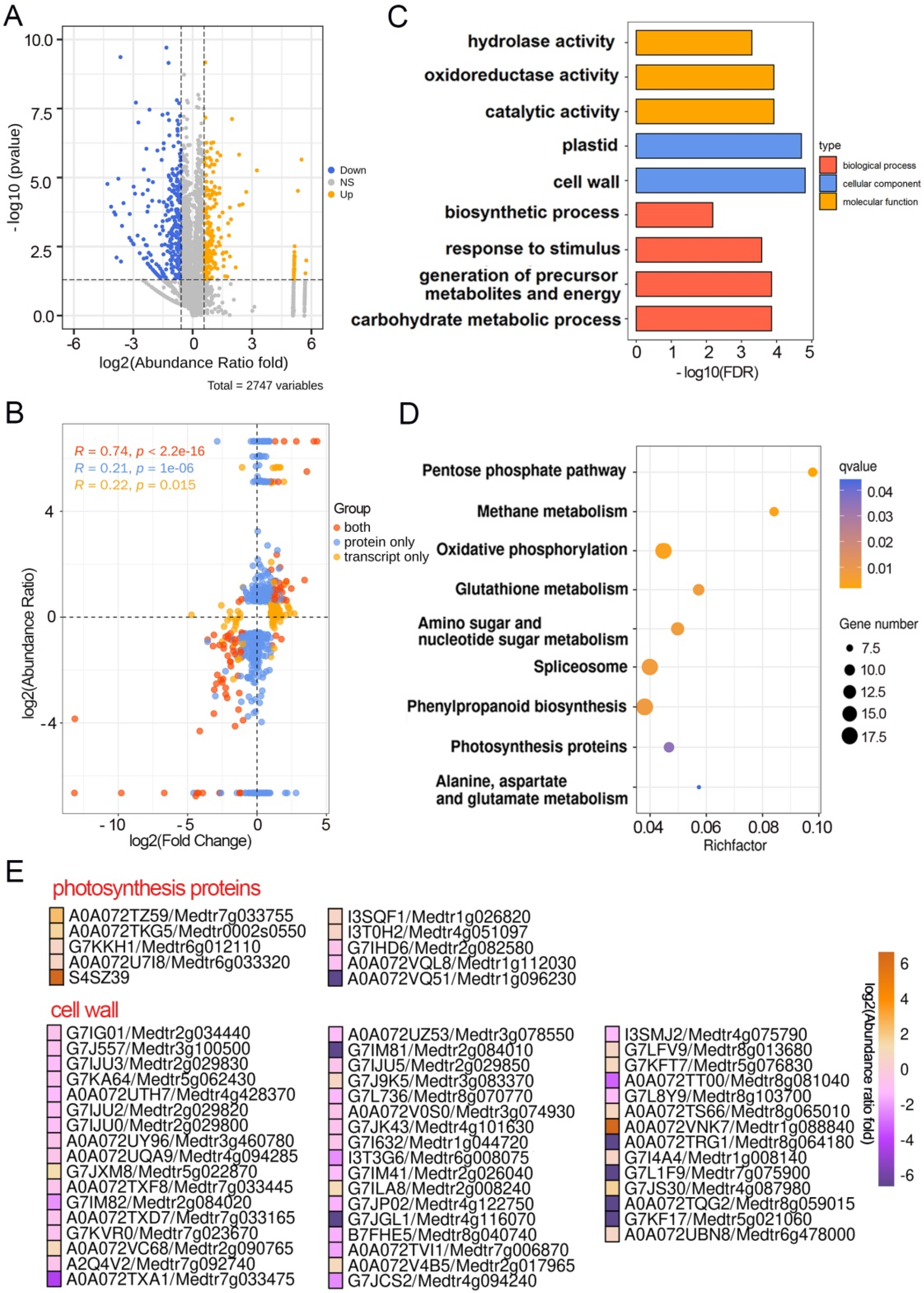
2.3. Multiple DEPs Are Involved in Pulvinus Development
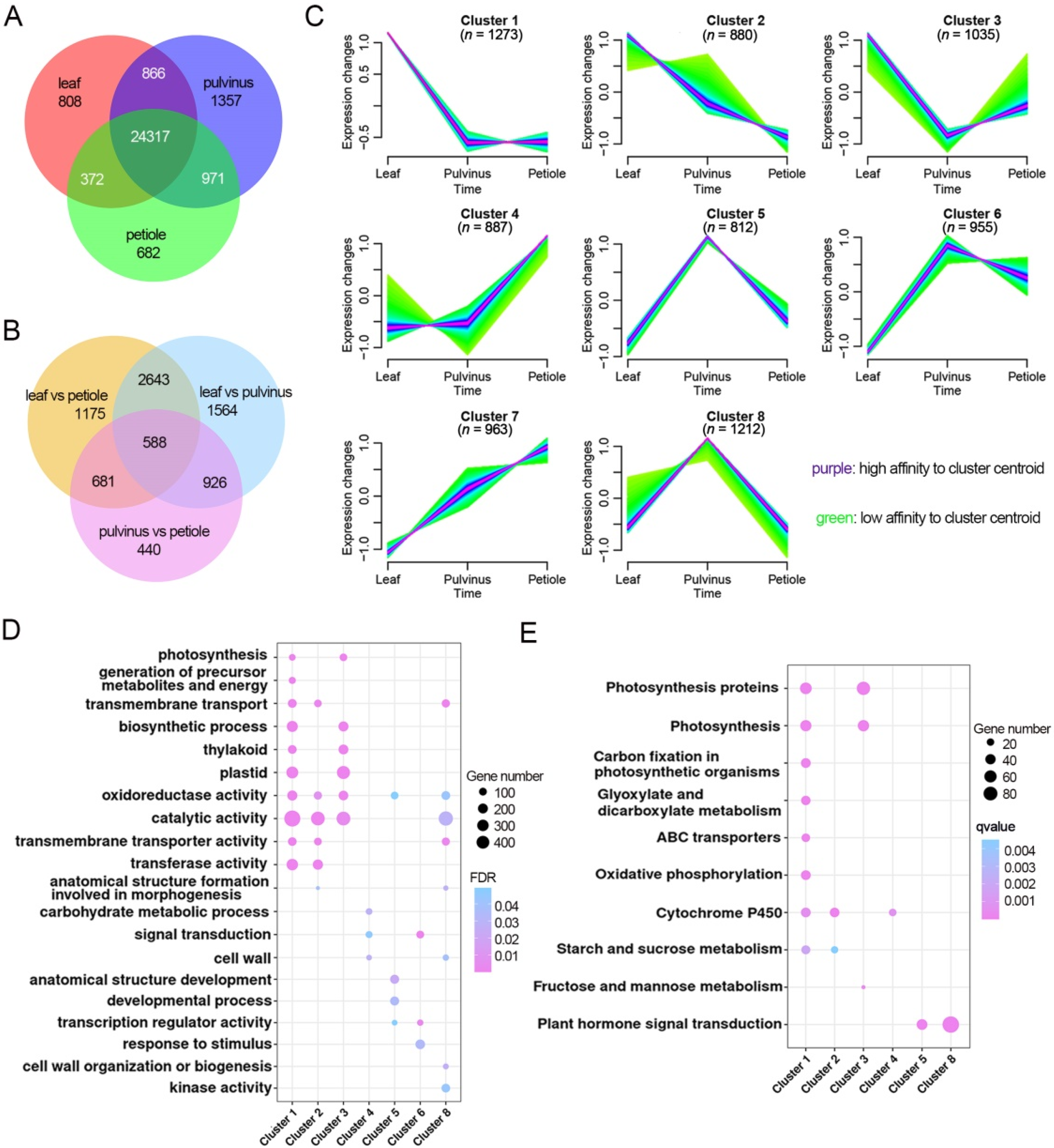
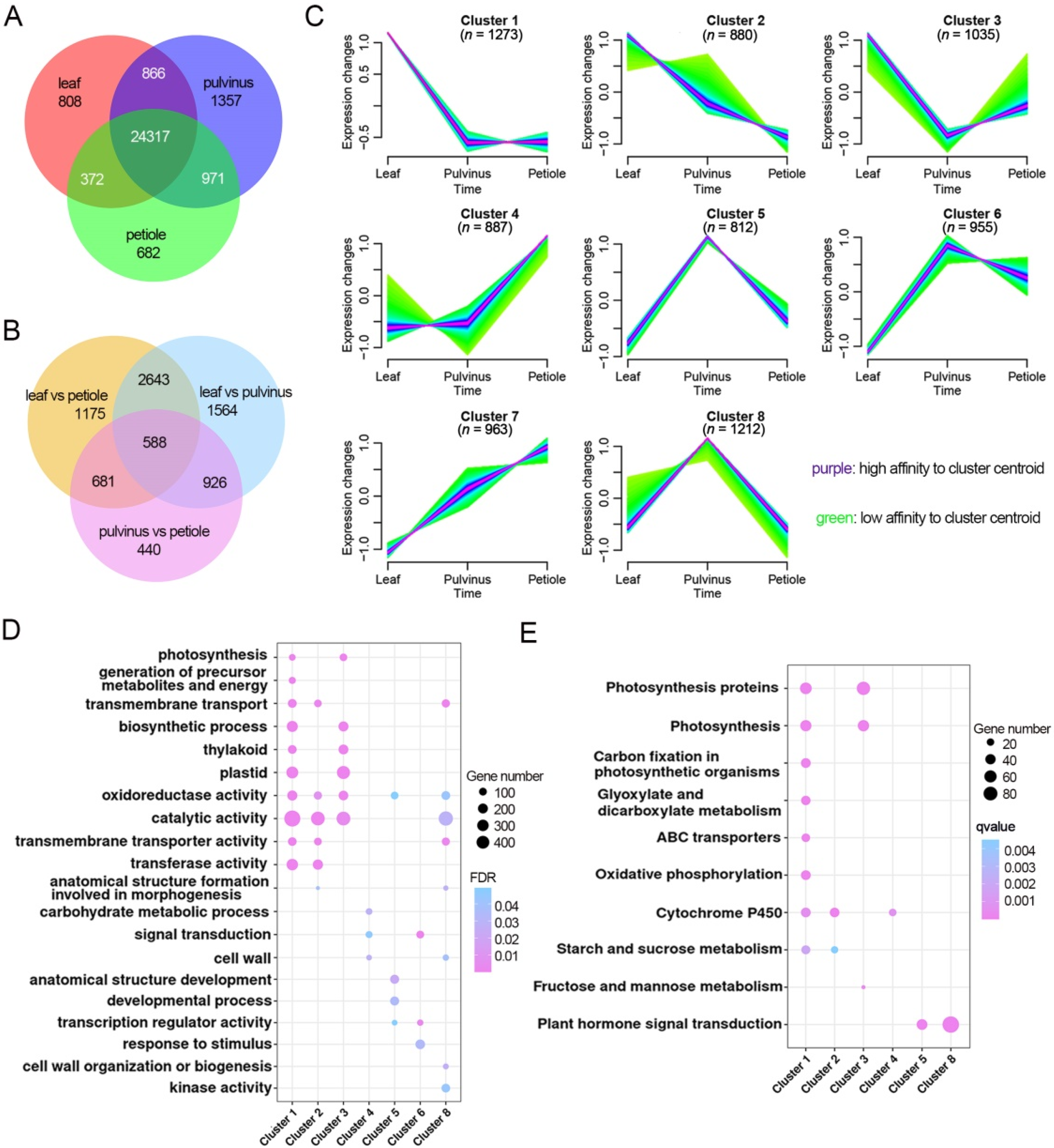
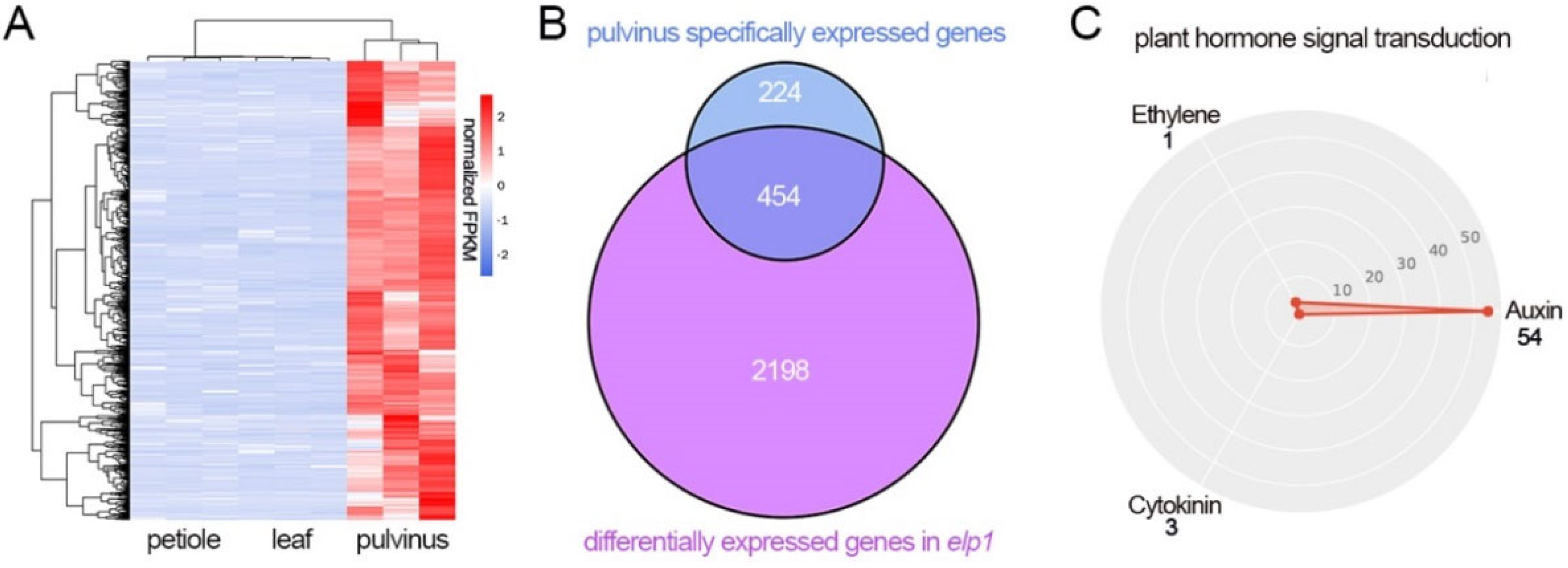
2.4. Leaflet, Pulvinus and Petiole Tissue Display Different Gene Regulatory Landscape
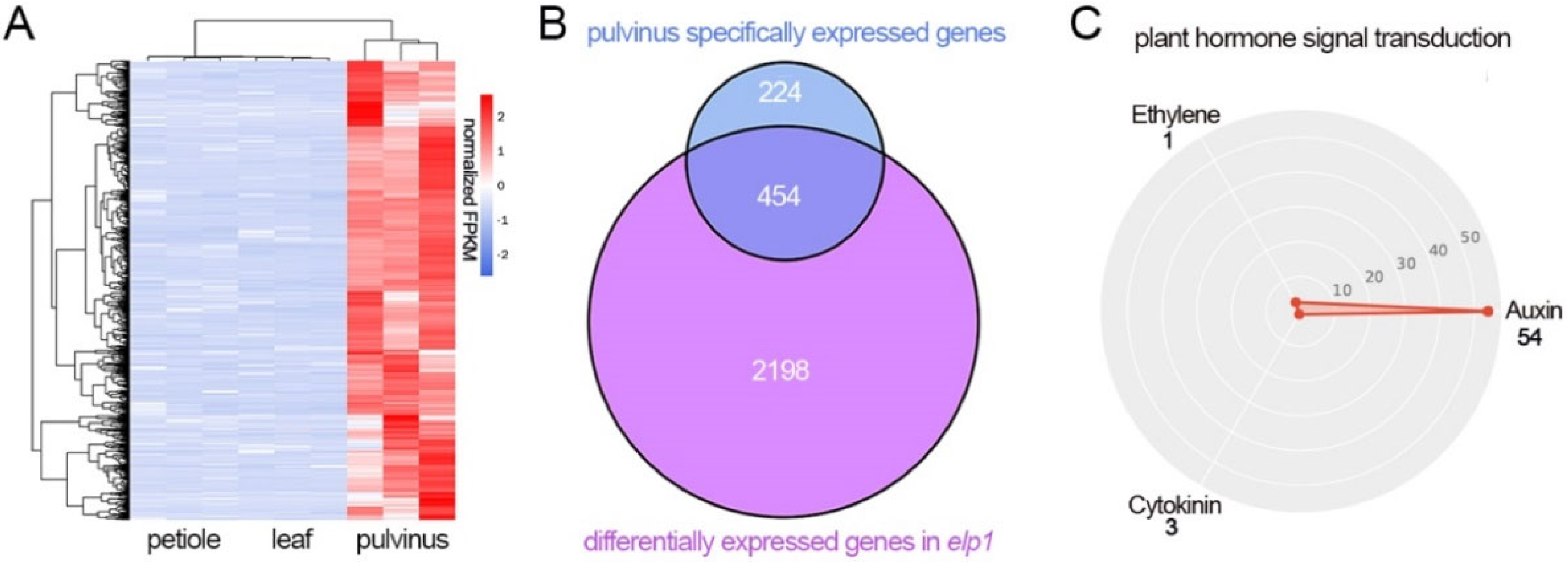
2.5. The Expression Level of Multiple Pulvinus Specific Expression Is Altered in elp1 Mutant
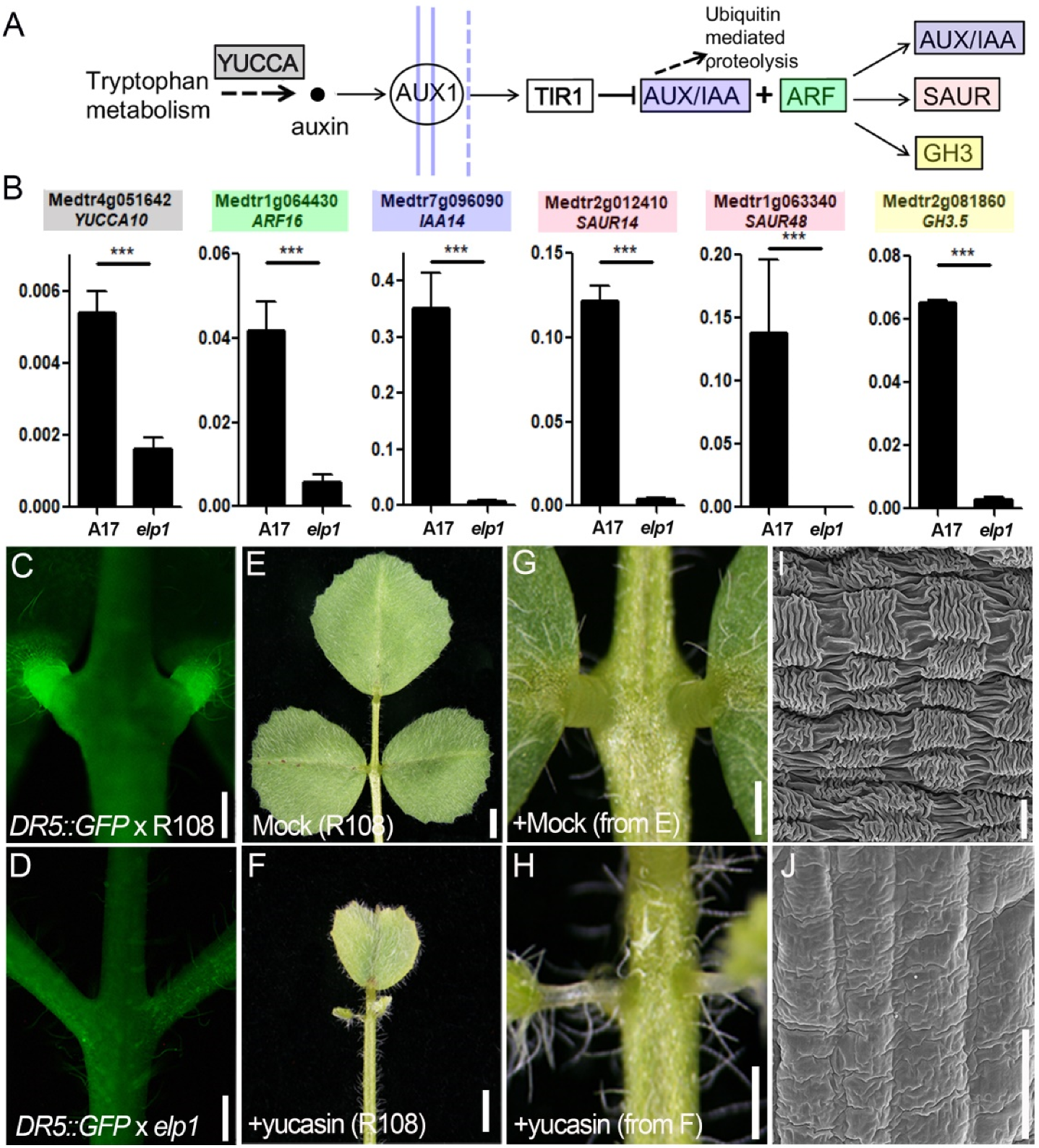
2.6. The Auxin Is Involved in the ELP1 Regulated Pulvinus Development
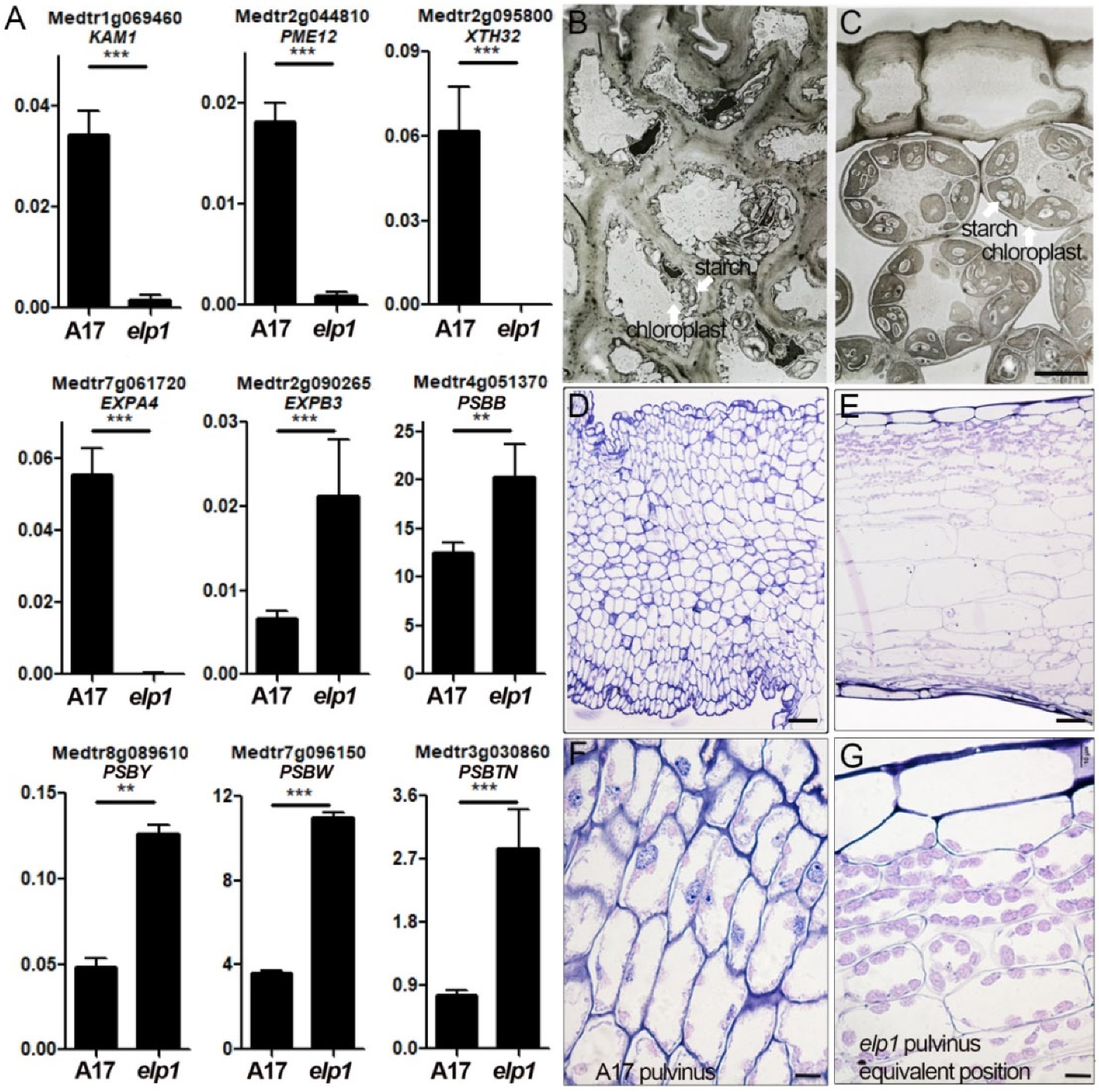
2.7. Both the Cell Wall Compositiona and Chloroplast Enrichment Are Altered in elp1 Mutants
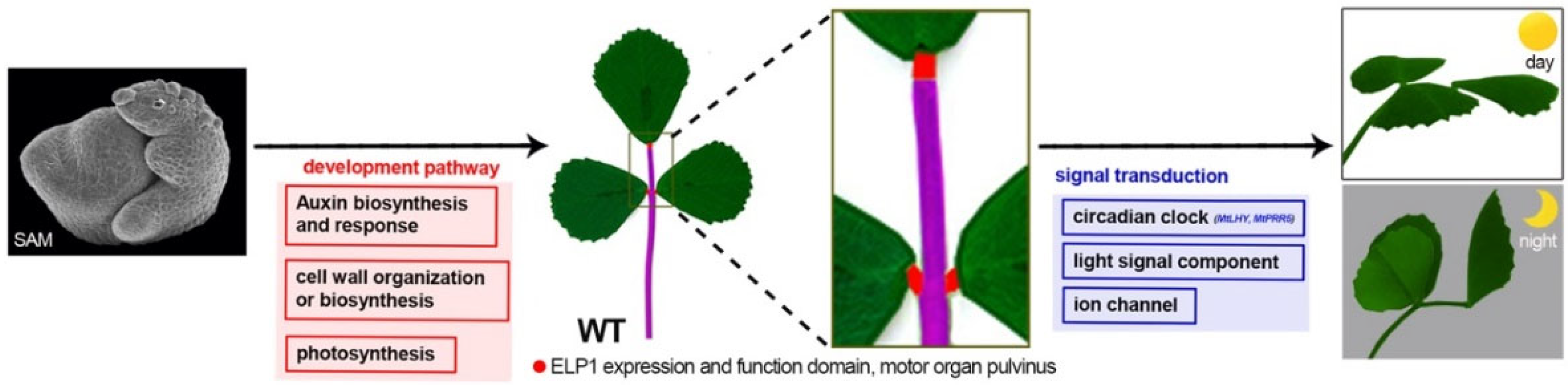
3. Discussion
3.1. Comparison Analysis of Multi-Omics Data
3.2. The Signal Transduction and Developmental Pathway Are Altered in elp1 Mutant
3.3. ELP1 Plays a Key Role in the Development of Pulvinus
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. RNA Extraction and Quantitative Real-Time PCR (RT-qPCR)
4.3. RNA Sequencing Assay and Data Analysis
4.4. New Gene Identification and Conservation Analysis
4.5. Differential Protein Expression Analysis
4.6. Functional Annotations and Enrichments
4.7. Paraffin Sections
4.8. Semi-Thin Sections
4.9. Scanning Electron Microscopy (SEM) Analysis
4.10. Transmission Electron Microscopy (TEM) Analysis
4.11. Yucasin Treatment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darwin, C. The Power of Movement in Plants; John Murray: London, UK, 1880. [Google Scholar]
- Satter, R.L.; Galston, A.W. Potassium Flux: A Common Feature of Albizzia Leaflet Movement Controlled by Phytochrome or Endogenous Rhythm. Science 1971, 174, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Coté, G.G.; Crain, R.C. Potassium Channels in Samanea saman Protoplasts Controlled by Phytochrome and the Biological Clock. Science 1993, 260, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.; Ehrenstein, G.; Iwasa, K.; Mischke, C.; Bare, C.; Satter, R.L. Potassium Channels in Motor Cells of Samanea saman. Plant Physiol. 1988, 88, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshelion, M.; Becker, D.; Czempinski, K.; Mueller-Roeber, B.; Attali, B.; Hedrich, R.; Moran, N. Diurnal and circadian regulation of putative potassium channels in a leaf moving organ. Plant Physiol. 2002, 128, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Moshelion, M.; Becker, D.; Biela, A.; Uehlein, N.; Hedrich, R.; Otto, B.; Levi, H.; Moran, N.; Kaldenhoff, R. Plasma membrane aquaporins in the motor cells of Samanea saman: Diurnal and circadian regulation. Plant Cell 2002, 14, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Ishimaru, Y.; Munemasa, S.; Takeuchi, Y.; Washiyama, K.; Hamamoto, S.; Yoshikawa, N.; Mutara, Y.; Uozumi, N.; Ueda, M. Ion Channels Regulate Nyctinastic Leaf Opening in Samanea saman. Curr. Biol. 2018, 28, 2230–2238.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satter, R.L. Rhythmic and phytochrome-regulated changes in transmembrane potential in Samanea pulvini. Nature 1975, 255, 408–410. [Google Scholar] [CrossRef]
- Satter, R.L.; Guggino, S.E.; Lonergan, T.A.; Galston, A.W. The Effects of Blue and Far Red Light on Rhythmic Leaflet Movements in Samanea and Albizzia. Plant Physiol. 1981, 67, 965–968. [Google Scholar] [CrossRef] [Green Version]
- Hillman, W.S.; Koukkari, W.L. Phytochrome Effects in the Nyctinastic Leaf Movements of Albizzia julibrissin and Some Other Legumes. Plant Physiol. 1967, 42, 1413–1418. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Ishimaru, Y.; Takeuchi, Y.; Muraoka, Y. Plant nyctinasty—Who will decode the ‘Rosetta Stone’? New Phytol. 2019, 223, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Cote, G. Signal Transduction in Leaf Movement. Plant Physiol. 1995, 109, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Moreau, C.; Liu, Y.; Kawaguchi, M.; Hofer, J.; Ellis, N.; Chen, R. Conserved genetic determinant of motor organ identity in Medicago truncatula and related legumes. Proc. Natl. Acad. Sci. USA 2012, 109, 11723–11728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Han, L.; Fu, C.; Chai, M.; Zhang, W.; Li, G.; Tang, Y.; Wang, Z. Identification and characterization of petiolule- like pulvinus mutants with abolished nyctinastic leaf movement in the model legume Medicago truncatula. New Phytol. 2012, 196, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, G. Linkage relations of tendrilled acacia (tac) and apulvinic. Pisum Newsl. 1984, 16, 46–48. [Google Scholar]
- Kawaguchi, M. SLEEPLESS, a gene conferring nyctinastic movement in legume. J. Plant Res. 2003, 116, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yang, S.; Cheng, W.; Fu, Y.; Leng, J.; Yuan, X.; Jiang, N.; Ma, J.; Feng, X. GmILPA1, Encoding an APC8-like Protein, Controls Leaf Petiole Angle in Soybean. Plant Physiol. 2017, 174, 1167–1176. [Google Scholar] [PubMed] [Green Version]
- Zhou, S.; Yang, T.; Mao, Y.; Liu, Y.; Guo, S.; Wang, R.; Fangyue, G.; He, L.; Zhao, B.; Bai, Q.; et al. The F-box protein MIO1/SLB1 regulates organ size and leaf movement in Medicago truncatula. J. Exp. Bot. 2021, 72, 2995–3011. [Google Scholar] [CrossRef]
- Kong, Y.; Meng, Z.; Wang, H.; Wang, Y.; Zhang, Y.; Hong, L.; Liu, R.; Wang, M.; Zhang, J.; Han, L.; et al. Brassinosteroid homeostasis is critical for the functionality of the Medicago truncatula pulvinus. Plant Physiol. 2021, 185, 1745–1763. [Google Scholar] [CrossRef]
- Zhao, W.; Bai, Q.; Zhao, B.; Wu, Q.; Wang, C.; Liu, Y.; Yang, T.; Liu, Y.; He, H.; Du, S.; et al. The geometry of the compound leaf plays a significant role in the leaf movement of Medicago truncatula modulated by mtdwarf4a. New Phytol. 2021, 230, 475–484. [Google Scholar] [CrossRef]
- Tang, H.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.; Zhou, S.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genom. 2014, 15, 312. [Google Scholar] [CrossRef] [Green Version]
- Carrere, S.; Verdier, J.; Gamas, P. MtExpress, a Comprehensive and Curated RNAseq-based Gene Expression Atlas for the Model Legume Medicago truncatula. Plant Cell Physiol. 2021, 62, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Han, L.; Liu, X.; Wang, H.; Wen, L.; Yu, X.; Xu, X.; Kong, F.; Fu, C.; Mysore, K.S.; et al. The nodulation and nyctinastic leaf movement is orchestrated by clock gene LHY in Medicago truncatula. J. Integr. Plant Biol. 2020, 62, 1880–1895. [Google Scholar] [CrossRef] [PubMed]
- Husbands, A.; Bell, E.M.; Shuai, B.; Smith, H.M.; Springer, P.S. LATERAL ORGAN BOUNDARIES defines a new family of DNA-binding transcription factors and can interact with specific bHLH proteins. Nucleic Acids Res. 2007, 35, 6663–6671. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.M.; Lin, W.-C.; Husbands, A.Y.; Yu, L.; Jaganatha, V.; Jablonska, B.; Mangeon, A.; Neff, M.M.; Girke, T.; Springer, P.S. Arabidopsis LATERAL ORGAN BOUNDARIES negatively regulates brassinosteroid accumulation to limit growth in organ boundaries. Proc. Natl. Acad. Sci. USA 2012, 109, 21146–21151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wang, X.; He, Y.; Xu, H.; Wang, L. Clock component OsPRR73 positively regulates rice salt tolerance by modulating OsHKT2;1 -mediated sodium homeostasis. EMBO J. 2020, 40, e105086. [Google Scholar] [CrossRef]
- Fankhauser, C.; Yeh, K.-C.; Clark, J.; Lagarias, J.C.; Zhang, H.; Elich, T.D.; Chory, J. PKS1, a Substrate Phosphorylated by Phytochrome That Modulates Light Signaling in Arabidopsis. Science 1999, 284, 1539–1541. [Google Scholar] [CrossRef]
- Vanneste, S.; Friml, J. Auxin: A Trigger for Change in Plant Development. Cell 2009, 136, 1005–1016. [Google Scholar] [CrossRef]
- Weijers, D.; Wagner, D. Transcriptional Responses to the Auxin Hormone. Annu. Rev. Plant Biol. 2016, 67, 539–574. [Google Scholar] [CrossRef]
- Zhou, C.; Han, L.; Hou, C.; Metelli, A.; Qi, L.; Tadege, M.; Mysore, K.S.; Wang, Z.-Y. Developmental Analysis of aMedicago truncatula smooth leaf margin1Mutant Reveals Context-Dependent Effects on Compound Leaf Development. Plant Cell 2011, 23, 2106–2124. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Han, L.; Fu, C.; Wen, J.; Cheng, X.; Nakashima, J.; Ma, J.; Tang, Y.; Tan, Y.; Tadege, M.; et al. The trans-acting short interfering RNA3 pathway and no apical meristem antagonistically regulate leaf margin development and lateral organ separation, as revealed by analysis of an argonaute7/lobed leaflet1 mutant in Medicago truncatula. Plant Cell 2013, 25, 48454862. [Google Scholar]
- Peng, J.; Berbel, A.; Madueño, F.; Chen, R. AUXIN RESPONSE FACTOR3 Regulates Compound Leaf Patterning by Directly Repressing PALMATE-LIKE PENTAFOLIATA1 Expression in Medicago truncatula. Front. Plant Sci. 2017, 8, 1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Han, L.; Zhao, Y.; Wang, H.; Nakashima, J.; Tong, J.; Xiao, L.; Wang, Z. Transforming compound leaf patterning by manipulating REVOLUTA in Medicago truncatula. Plant J. 2019, 100, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; He, L.; Jiang, C.; Liu, Y.; He, H.; Bai, Q.; Zhou, S.; Zheng, X.; Wen, J.; Mysore, K.S.; et al. Lateral Leaflet Suppression 1 (LLS1), encoding the MtYUCCA1 protein, regulates lateral leaflet development in Medicago truncatula. New Phytol. 2020, 227, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Lu, S.; Chai, M.; Zhou, C.; Sun, L.; Tang, Y.; Nakashima, J.; Kolape, J.; Wen, Z.; Behzadirad, M.; et al. Functional characterization of PETIOLULE-LIKE PULVINUS (PLP) gene in abscission zone development in Medicago truncatula and its application to genetic improvement of alfalfa. Plant Biotechnol. J. 2020, 19, 351–364. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Jain, A.; Tuteja, G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 2019, 35, 1966–1967. [Google Scholar] [CrossRef]
- Lomsadze, A.; Burns, P.D.; Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014, 42, e119. [Google Scholar] [CrossRef]
- Hoff, K.J.; Stanke, M. WebAUGUSTUS—A web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 2013, 41, W123–W128. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Vesztrocy, A.W.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Wen, J.; Tadege, M.; Li, G.; Liu, Y.; Mysore, K.S.; Ratet, P.; Chen, R. Control of Compound Leaf Development by FLORICAULA/LEAFY Ortholog SINGLE LEAFLET1 in Medicago truncatula. Plant Physiol. 2008, 146, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; He, L.; Liu, Y.; Mao, Y.; Wang, C.; Zhao, B.; Li, Y.; He, H.; Guo, S.; Zhang, L.; et al. A study of male fertility control in Medicago truncatula uncovers an evolutionarily conserved recruitment of two tapetal bHLH subfamilies in plant sexual reproduction. New Phytol. 2020, 228, 1115–1133. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Q.; Yang, W.; Qin, G.; Zhao, B.; He, L.; Zhang, X.; Zhao, W.; Zhou, D.; Liu, Y.; Liu, Y.; et al. Multidimensional Gene Regulatory Landscape of Motor Organ Pulvinus in the Model Legume Medicago truncatula. Int. J. Mol. Sci. 2022, 23, 4439. https://doi.org/10.3390/ijms23084439
Bai Q, Yang W, Qin G, Zhao B, He L, Zhang X, Zhao W, Zhou D, Liu Y, Liu Y, et al. Multidimensional Gene Regulatory Landscape of Motor Organ Pulvinus in the Model Legume Medicago truncatula. International Journal of Molecular Sciences. 2022; 23(8):4439. https://doi.org/10.3390/ijms23084439
Chicago/Turabian StyleBai, Quanzi, Wenjing Yang, Guochen Qin, Baolin Zhao, Liangliang He, Xuan Zhang, Weiyue Zhao, Dian Zhou, Ye Liu, Yu Liu, and et al. 2022. "Multidimensional Gene Regulatory Landscape of Motor Organ Pulvinus in the Model Legume Medicago truncatula" International Journal of Molecular Sciences 23, no. 8: 4439. https://doi.org/10.3390/ijms23084439
APA StyleBai, Q., Yang, W., Qin, G., Zhao, B., He, L., Zhang, X., Zhao, W., Zhou, D., Liu, Y., Liu, Y., He, H., Tadege, M., Xiong, Y., Liu, C., & Chen, J. (2022). Multidimensional Gene Regulatory Landscape of Motor Organ Pulvinus in the Model Legume Medicago truncatula. International Journal of Molecular Sciences, 23(8), 4439. https://doi.org/10.3390/ijms23084439