
Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review

Abstract
:1. Introduction
1.1. Clinical Features
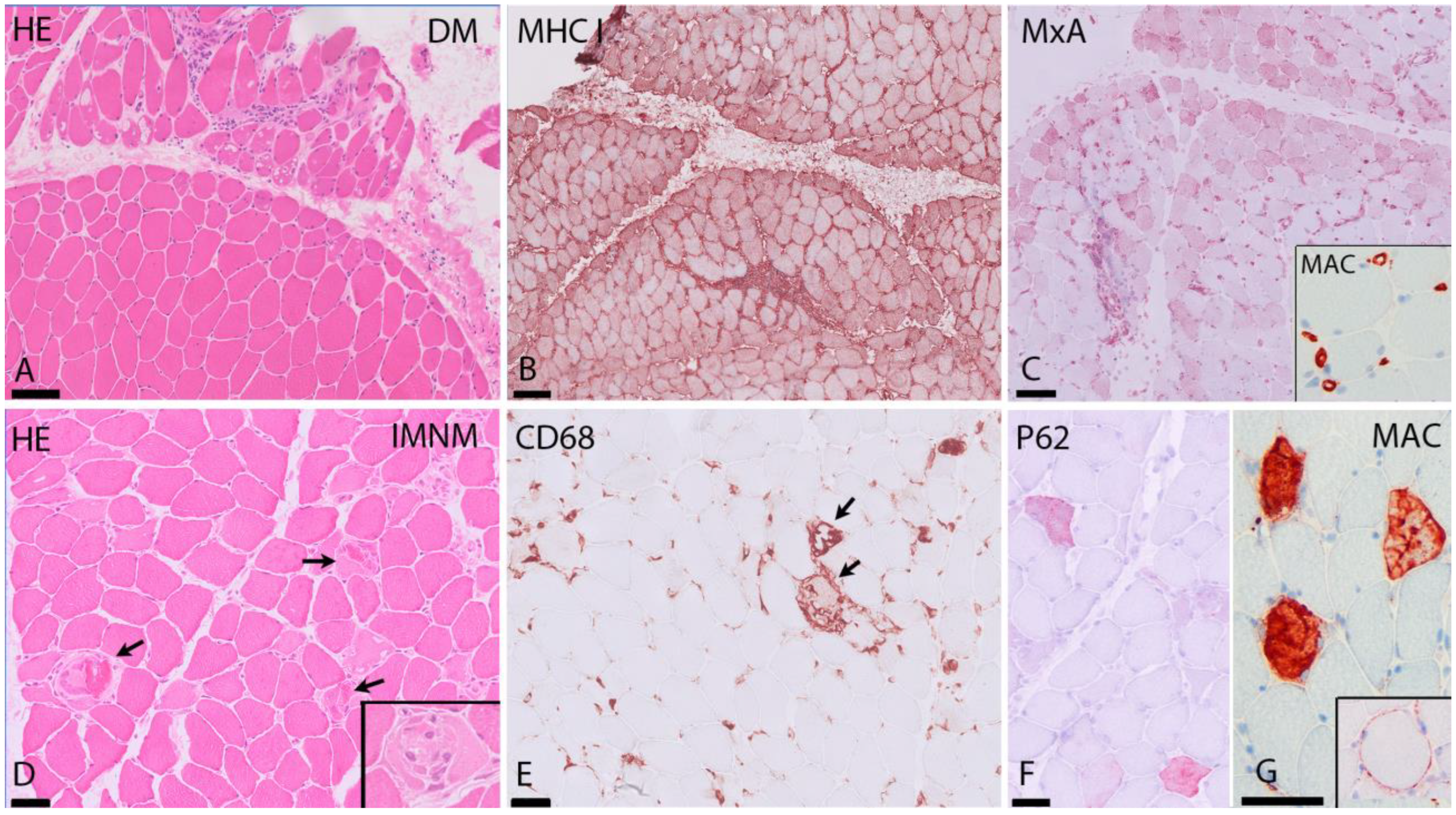
1.2. Muscle Biopsy
2. Pathophysiological Mechanisms
3. Humoral Immunity in DM and IMNM: The Role of B Cells and Antibodies
3.1. B Cells
3.1.1. B Cells in DM
3.1.2. B Cells in IMNM
3.2. Autoantibodies
3.2.1. Autoantibodies in DM
Anti-MDA-5 DM
Anti-TIF1-γ DM
3.2.2. Autoanntibodies in IMNM
Anti-SRP IMNM
Anti-HMGCR IMNM
4. Derangement of the Complement System in DM and IMNM
4.1. Complement in DM
4.2. Complement in IMNM
5. Interferon Pathway
5.1. Interferon Overexpression in DM
5.2. Interferon Overexpression in IMNM
6. Pharmacological Treatment
6.1. General Treatment Recommendations
6.2. Intensive Treatment in Rapidly Progressive or Refractory DM and IMNM
6.2.1. IVIG
6.2.2. Plasmapheresis
6.2.3. Cyclophosphamide
7. Pharmacological Compounds Targeting B Cells, Interferon Pathway and Complement
7.1. B Cell Depletion
7.2. Interferon Pathway
7.2.1. Anti-IFN-α Antibody
7.2.2. Janus Kinases (JAK) Inhibitors
7.3. Complement Inhibition
Anti-C5 Monoclonal Antibodies
8. New Pharmacological Compounds Targeting Other Pathways
8.1. Tumor Necrosis Factor (TNF)-α
8.2. Interleukine (IL)-Receptor Antagonists
8.3. Inhibition of T-Cell Costimulation
9. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mariampillai, K.; Granger, B.; Amelin, D.; Guiguet, M.; Hachulla, E.; Maurier, F.; Meyer, A.; Tohme, A.; Charuel, J.L. Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol. 2018, 75, 1528–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammen, A.L.; Allenbach, Y.; Stenzel, W.; Benveniste, O.; ENMC 239th Workshop Study Group. 239th ENMC International Workshop: Classification of dermatomyositis, Amsterdam, the Netherlands, 14–16 December 2018. Neuromuscul. Disord. 2020, 30, 70–92. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J. Current Classification and Management of Inflammatory Myopathies. J. Neuromuscul. Dis. 2018, 5, 109–129. [Google Scholar] [PubMed]
- Tanboon, J.; Uruha, A.; Stenzel, W.; Nishino, I. Where are we moving in the classification of idiopathic inflammatory myopathies? Curr. Opin. Neurol. 2020, 33, 590–603. [Google Scholar] [CrossRef]
- van der Meulen, M.F.; Bronner, I.M.; Hoogendijk, J.E.; Burger, H.; van Venrooij, W.J.; Voskuyl, A.E.; Dinant, H.J.; Linssen, W.H.; Wokke, J.H.; de Visser, M. Polymyositis: An overdiagnosed entity. Neurology 2003, 61, 316–321. [Google Scholar] [CrossRef]
- Loarce-Martos, J.; Lilleker, J.B.; Parker, M.; McHugh, N.; Chinoy, H. Polymyositis: Is there anything left? A retrospective diagnostic review from a tertiary myositis centre. Rheumatology 2021, 60, 3398–3403. [Google Scholar] [CrossRef]
- Hoogendijk, J.E.; Amato, A.A.; Lecky, B.R.; Choy, E.H.; Lundberg, I.E.; Rose, M.R.; Vencovsky, J.; de Visser, M.; Hughes, R.A. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, Naarden, The Netherlands, 10–12 October 2003. Neuromuscul. Disord. 2004, 14, 337–345. [Google Scholar] [CrossRef]
- Allenbach, Y.; Mammen, A.L.; Benveniste, O.; Stenzel, W.; Immune-Mediated Necrotizing Myopathies Working Group. 224th ENMC International Workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscul. Disord. 2018, 28, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Connors, G.R.; Christopher-Stine, L.; Oddis, C.V.; Danoff, S.K. Interstitial lung disease associated with the idiopathic inflammatory myopathies: What progress has been made in the past 35 years? Chest 2010, 138, 1464–1474. [Google Scholar] [CrossRef] [Green Version]
- van de Vlekkert, J.; Hoogendijk, J.E.; de Visser, M. Long-term follow-up of 62 patients with myositis. J. Neurol. 2014, 261, 992–998. [Google Scholar] [CrossRef]
- Nuno-Nuno, L.; Joven, B.E.; Carreira, P.E.; Maldonado-Romero, V.; Larena-Grijalba, C.; Llorente Cubas, I.; Tomero, E.; Barbadillo-Mateos, M.C.; Garcia de la Pena Lefebvre, P.; Ruiz-Gutierrez, L.; et al. Overlap myositis, a distinct entity beyond primary inflammatory myositis: A retrospective analysis of a large cohort from the REMICAM registry. Int. J. Rheum. Dis. 2019, 22, 1393–1401. [Google Scholar] [PubMed]
- Fredi, M.; Cavazzana, I.; Franceschini, F. The clinico-serological spectrum of overlap myositis. Curr. Opin. Rheumatol. 2018, 30, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Zanframundo, G.; Faghihi-Kashani, S.; Scire, C.A.; Bonella, F.; Corte, T.J.; Doyle, T.J.; Fiorentino, D.; Gonzalez-Gay, M.A.; Hudson, M.; Kuwana, M.; et al. Defining anti-synthetase syndrome: A systematic literature review. Clin. Exp. Rheumatol. 2022, 40, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Review: An update on inflammatory and autoimmune myopathies. Neuropathol. Appl. Neurobiol. 2011, 37, 226–242. [Google Scholar] [CrossRef]
- Lloyd, T.E.; Mammen, A.L.; Amato, A.A.; Weiss, M.D.; Needham, M.; Greenberg, S.A. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology 2014, 83, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Benveniste, O.; Stenzel, W.; Hilton-Jones, D.; Sandri, M.; Boyer, O.; van Engelen, B.G. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: The inflammatory egg comes before the degenerative chicken. Acta Neuropathol. 2015, 129, 611–624. [Google Scholar] [CrossRef]
- Keller, C.W.; Schmidt, J.; Lunemann, J.D. Immune and myodegenerative pathomechanisms in inclusion body myositis. Ann. Clin. Transl. Neurol. 2017, 4, 422–445. [Google Scholar] [CrossRef]
- Bellutti Enders, F.; Bader-Meunier, B.; Baildam, E.; Constantin, T.; Dolezalova, P.; Feldman, B.M.; Lahdenne, P.; Magnusson, B.; Nistala, K.; Ozen, S.; et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann. Rheum. Dis. 2017, 76, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Allenbach, Y.; Benveniste, O. Peculiar clinicopathological features of immune-mediated necrotizing myopathies. Curr. Opin. Rheumatol. 2018, 30, 655–663. [Google Scholar] [CrossRef]
- Kao, A.H.; Lacomis, D.; Lucas, M.; Fertig, N.; Oddis, C.V. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004, 50, 209–215. [Google Scholar] [CrossRef]
- Lilleker, J.B.; Vencovsky, J.; Wang, G.; Wedderburn, L.R.; Diederichsen, L.P.; Schmidt, J.; Oakley, P.; Benveniste, O.; Danieli, M.G.; Danko, K.; et al. The EuroMyositis registry: An international collaborative tool to facilitate myositis research. Ann. Rheum. Dis. 2018, 77, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.E.; Fujimoto, M.; Vencovsky, J.; Aggarwal, R.; Holmqvist, M.; Christopher-Stine, L.; Mammen, A.L.; Miller, F.W. Idiopathic inflammatory myopathies. Nat. Rev. Dis. Primers 2021, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wu, C.Y.; Huang, Y.L.; Wang, C.B.; Shen, J.L.; Chang, Y.T. Cancer risks of dermatomyositis and polymyositis: A nationwide cohort study in Taiwan. Arthritis Res. Ther. 2010, 12, R70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.L.; Zhang, Y.; Sigurgeirsson, B.; Pukkala, E.; Mellemkjaer, L.; Airio, A.; Evans, S.R.; Felson, D.T. Frequency of specific cancer types in dermatomyositis and polymyositis: A population-based study. Lancet 2001, 357, 96–100. [Google Scholar] [CrossRef]
- Musset, L.; Allenbach, Y.; Benveniste, O.; Boyer, O.; Bossuyt, X.; Bentow, C.; Phillips, J.; Mammen, A.; Van Damme, P.; Westhovens, R.; et al. Anti-HMGCR antibodies as a biomarker for immune-mediated necrotizing myopathies: A history of statins and experience from a large international multi-center study. Autoimmun. Rev. 2016, 15, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Mammen, A.L.; Chung, T.; Christopher-Stine, L.; Rosen, P.; Rosen, A.; Doering, K.R.; Casciola-Rosen, L.A. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011, 63, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allenbach, Y.; Drouot, L.; Rigolet, A.; Charuel, J.L.; Jouen, F.; Romero, N.B.; Maisonobe, T.; Dubourg, O.; Behin, A.; Laforet, P.; et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: Inconstant exposure to statin. Medicine 2014, 93, 150–157. [Google Scholar] [CrossRef]
- Suarez-Calvet, X.; Gallardo, E.; Pinal-Fernandez, I.; De Luna, N.; Lleixa, C.; Diaz-Manera, J.; Rojas-Garcia, R.; Castellvi, I.; Martinez, M.A.; Grau, J.M.; et al. RIG-I expression in perifascicular myofibers is a reliable biomarker of dermatomyositis. Arthritis Res. Ther. 2017, 19, 174. [Google Scholar] [CrossRef] [Green Version]
- Pinal-Fernandez, I.; Casciola-Rosen, L.A.; Christopher-Stine, L.; Corse, A.M.; Mammen, A.L. The Prevalence of Individual Histopathologic Features Varies according to Autoantibody Status in Muscle Biopsies from Patients with Dermatomyositis. J. Rheumatol. 2015, 42, 1448–1454. [Google Scholar] [CrossRef]
- Greenberg, S.A. Dermatomyositis and type 1 interferons. Curr. Rheumatol. Rep. 2010, 12, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Uruha, A.; Allenbach, Y.; Charuel, J.L.; Musset, L.; Aussy, A.; Boyer, O.; Mariampillai, K.; Landon-Cardinal, O.; Rasmussen, C.; Bolko, L.; et al. Diagnostic potential of sarcoplasmic myxovirus resistance protein A expression in subsets of dermatomyositis. Neuropathol. Appl. Neurobiol. 2019, 45, 513–522. [Google Scholar] [CrossRef]
- Tanboon, J.; Inoue, M.; Saito, Y.; Tachimori, H.; Hayashi, S.; Noguchi, S.; Okiyama, N.; Fujimoto, M.; Nishino, I. Dermatomyositis: Muscle Pathology According to Antibody Subtypes. Neurology 2022, 98, e739–e749. [Google Scholar] [CrossRef]
- Uruha, A.; Nishikawa, A.; Tsuburaya, R.S.; Hamanaka, K.; Kuwana, M.; Watanabe, Y.; Suzuki, S.; Suzuki, N.; Nishino, I. Sarcoplasmic MxA expression: A valuable marker of dermatomyositis. Neurology 2017, 88, 493–500. [Google Scholar] [CrossRef]
- Uruha, A.; Goebel, H.H.; Stenzel, W. Updates on the Immunopathology in Idiopathic Inflammatory Myopathies. Curr. Rheumatol. Rep. 2021, 23, 56. [Google Scholar] [CrossRef]
- Franzi, S.; Salajegheh, M.; Nazareno, R.; Greenberg, S.A. Type 1 interferons inhibit myotube formation independently of upregulation of interferon-stimulated gene 15. PLoS ONE 2013, 8, e65362. [Google Scholar] [CrossRef]
- Lahoria, R.; Selcen, D.; Engel, A.G. Microvascular alterations and the role of complement in dermatomyositis. Brain 2016, 139 Pt 7, 1891–1903. [Google Scholar] [CrossRef] [Green Version]
- De Paepe, B. Vascular changes and perifascicular muscle fiber damage in dermatomyositis: Another question of the chicken or the egg that is on our mind. Ann. Transl. Med. 2017, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- De Visser, M.; Emslie-Smith, A.M.; Engel, A.G. Early ultrastructural alterations in adult dermatomyositis. Capillary abnormalities precede other structural changes in muscle. J. Neurol. Sci. 1989, 94, 181–192. [Google Scholar] [CrossRef]
- Bronner, I.M.; Hoogendijk, J.E.; Veldman, H.; Ramkema, M.; van den Bergh Weerman, M.A.; Rozemuller, A.J.; de Visser, M. Tubuloreticular structures in different types of myositis: Implications for pathogenesis. Ultrastruct. Pathol. 2008, 32, 123–126. [Google Scholar] [CrossRef]
- Banker, B.Q. Dermatomyositis of childhood: Ultrastructural alterations of muscle and intramuscular blood vessels. J. Neuropathol. Exp. Neurol. 1975, 34, 46–75. [Google Scholar] [CrossRef]
- Jain, A.; Sharma, M.C.; Sarkar, C.; Bhatia, R.; Singh, S.; Gulati, S.; Handa, R. Detection of the membrane attack complex as a diagnostic tool in dermatomyositis. Acta Neurol. Scand. 2011, 123, 122–129. [Google Scholar] [CrossRef]
- Merlonghi, G.; Antonini, G.; Garibaldi, M. Immune-mediated necrotizing myopathy (IMNM): A myopathological challenge. Autoimmun. Rev. 2022, 21, 102993. [Google Scholar] [CrossRef]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [Green Version]
- Chung, T.; Christopher-Stine, L.; Paik, J.J.; Corse, A.; Mammen, A.L. The composition of cellular infiltrates in anti-HMG-CoA reductase-associated myopathy. Muscle Nerve 2015, 52, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Allenbach, Y.; Arouche-Delaperche, L.; Preusse, C.; Radbruch, H.; Butler-Browne, G.; Champtiaux, N.; Mariampillai, K.; Rigolet, A.; Hufnagl, P.; Zerbe, N.; et al. Necrosis in anti-SRP(+) and anti-HMGCR(+)myopathies: Role of autoantibodies and complement. Neurology 2018, 90, e507–e517. [Google Scholar] [CrossRef]
- Stockton, D.; Doherty, V.R.; Brewster, D.H. Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: A Scottish population-based cohort study. Br. J. Cancer 2001, 85, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Ramos, A.E.; Martin-Nares, E.; Hernandez-Molina, G. New Onset of Autoimmune Diseases Following COVID-19 Diagnosis. Cells 2021, 10, 3592. [Google Scholar] [CrossRef]
- Aschman, T.; Schneider, J.; Greuel, S.; Meinhardt, J.; Streit, S.; Goebel, H.H.; Buttnerova, I.; Elezkurtaj, S.; Scheibe, F.; Radke, J.; et al. Association Between SARS-CoV-2 Infection and Immune-Mediated Myopathy in Patients Who Have Died. JAMA Neurol. 2021, 78, 948–960. [Google Scholar] [CrossRef]
- Miller, F.W.; Chen, W.; O’Hanlon, T.P.; Cooper, R.G.; Vencovsky, J.; Rider, L.G.; Danko, K.; Wedderburn, L.R.; Lundberg, I.E.; Pachman, L.M.; et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. 2015, 16, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, S.; Chinoy, H.; Lamb, J.A. Genetics of idiopathic inflammatory myopathies: Insights into disease pathogenesis. Curr. Opin. Rheumatol. 2019, 31, 611–616. [Google Scholar] [CrossRef]
- Rothwell, S.; Chinoy, H.; Lamb, J.A.; Miller, F.W.; Rider, L.G.; Wedderburn, L.R.; McHugh, N.J.; Mammen, A.L.; Betteridge, Z.E.; Tansley, S.L.; et al. Focused HLA analysis in Caucasians with myositis identifies significant associations with autoantibody subgroups. Ann. Rheum. Dis. 2019, 78, 996–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.H.; Go, D.J.; Mimori, T.; Lee, S.J.; Kwon, H.M.; Park, J.W.; Park, M.H.; Song, E.Y.; Ha, Y.J.; Lee, E.Y.; et al. Novel susceptibility alleles in HLA region for myositis and myositis specific autoantibodies in Korean patients. Semin. Arthritis Rheum. 2019, 49, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.W.; Lamb, J.A.; Schmidt, J.; Nagaraju, K. Risk factors and disease mechanisms in myositis. Nat. Rev. Rheumatol. 2018, 14, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Goebels, N.; Michaelis, D.; Engelhardt, M.; Huber, S.; Bender, A.; Pongratz, D.; Johnson, M.A.; Wekerle, H.; Tschopp, J.; Jenne, D.; et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J. Clin. Investig. 1996, 97, 2905–2910. [Google Scholar] [CrossRef] [Green Version]
- Arouche-Delaperche, L.; Allenbach, Y.; Amelin, D.; Preusse, C.; Mouly, V.; Mauhin, W.; Tchoupou, G.D.; Drouot, L.; Boyer, O.; Stenzel, W.; et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann. Neurol. 2017, 81, 538–548. [Google Scholar] [CrossRef]
- Muro, Y.; Sugiura, K.; Hoshino, K.; Akiyama, M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology 2012, 51, 800–804. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Matsushita, M.; Tada, K.; Yamaji, K.; Takasaki, Y.; Tamura, N. Clinical characteristics and change in the antibody titres of patients with anti-MDA5 antibody-positive inflammatory myositis. Rheumatology 2017, 56, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Mizumaki, K.; Kano, M.; Yagi, N.; Tennichi, M.; Takeuchi, A.; Okamoto, Y.; Hamaguchi, Y.; Murakami, A.; Hasegawa, M.; et al. Antimelanoma differentiation-associated protein 5 antibody level is a novel tool for monitoring disease activity in rapidly progressive interstitial lung disease with dermatomyositis. Br. J. Dermatol. 2017, 176, 395–402. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Pinkus, J.L.; Pinkus, G.S.; Burleson, T.; Sanoudou, D.; Tawil, R.; Barohn, R.J.; Saperstein, D.S.; Briemberg, H.R.; Ericsson, M.; et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann. Neurol. 2005, 57, 664–678. [Google Scholar] [CrossRef]
- Fischer, N.; Preusse, C.; Radke, J.; Pehl, D.; Allenbach, Y.; Schneider, U.; Feist, E.; von Casteleyn, V.; Hahn, K.; Ruck, T.; et al. Sequestosome-1 (p62) expression reveals chaperone-assisted selective autophagy in immune-mediated necrotizing myopathies. Brain Pathol. 2020, 30, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Ceribelli, A.; De Santis, M.; Isailovic, N.; Gershwin, M.E.; Selmi, C. The Immune Response and the Pathogenesis of Idiopathic Inflammatory Myositis: A Critical Review. Clin. Rev. Allergy Immunol. 2017, 52, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Inflammatory Muscle Diseases. N. Engl. J. Med. 2015, 373, 393–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, N.J.; Tansley, S.L. Autoantibodies in myositis. Nat. Rev. Rheumatol. 2018, 14, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, Z.; McHugh, N. Myositis-specific autoantibodies: An important tool to support diagnosis of myositis. J. Intern. Med. 2016, 280, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Oddis, C.V.; Goudeau, D.; Koontz, D.; Qi, Z.; Reed, A.M.; Ascherman, D.P.; Levesque, M.C. Autoantibody levels in myositis patients correlate with clinical response during B cell depletion with rituximab. Rheumatology 2016, 55, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, F.H.C.; Miossi, R.; de Moraes, J.C.B.; Bonfa, E.; Shinjo, S.K. Favorable rituximab response in patients with refractory idiopathic inflammatory myopathies. Adv. Rheumatol. 2018, 58, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclair, V.; Galindo-Feria, A.S.; Dastmalchi, M.; Holmqvist, M.; Lundberg, I.E. Efficacy and safety of rituximab in anti-synthetase antibody positive and negative subjects with idiopathic inflammatory myopathy: A registry-based study. Rheumatology 2019, 58, 1214–1220. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Bradshaw, E.M.; Pinkus, J.L.; Pinkus, G.S.; Burleson, T.; Due, B.; Bregoli, L.; O’Connor, K.C.; Amato, A.A. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 2005, 65, 1782–1787. [Google Scholar] [CrossRef]
- Radke, J.; Koll, R.; Preusse, C.; Pehl, D.; Todorova, K.; Schonemann, C.; Allenbach, Y.; Aronica, E.; de Visser, M.; Heppner, F.L.; et al. Architectural B-cell organization in skeletal muscle identifies subtypes of dermatomyositis. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e451. [Google Scholar] [CrossRef] [Green Version]
- Brunn, A.; Hans, V.J.; Vogelgesang, S.; Deckert, M. Inflammatory myopathy with abundant macrophages and dermatomyositis: Two stages of one disorder or two distinct entities? Acta Neuropathol. 2009, 118, 793–801. [Google Scholar] [CrossRef]
- De Bleecker, J.L.; Engel, A.G.; Butcher, E.C. Peripheral lymphoid tissue-like adhesion molecule expression in nodular infiltrates in inflammatory myopathies. Neuromuscul. Disord. 1996, 6, 255–260. [Google Scholar] [CrossRef]
- McIntyre, D.; Zuckerman, N.S.; Field, M.; Mehr, R.; Stott, D.I. The VH repertoire and clonal diversification of B cells in inflammatory myopathies. Eur. J. Immunol. 2014, 44, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, W.; Liu, Y.; Wang, Z.; Yuan, Y. Factors associated with refractory autoimmune necrotizing myopathy with anti-signal recognition particle antibodies. Orphanet J. Rare Dis. 2020, 15, 181. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Nishikawa, A.; Kuwana, M.; Nishimura, H.; Watanabe, Y.; Nakahara, J.; Hayashi, Y.K.; Suzuki, N.; Nishino, I. Inflammatory myopathy with anti-signal recognition particle antibodies: Case series of 100 patients. Orphanet J. Rare Dis. 2015, 10, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rietveld, A.; Lim, J.; de Visser, M.; van Engelen, B.; Pruijn, G.; Benveniste, O.; van der Kooi, A.; Saris, C. Autoantibody testing in idiopathic inflammatory myopathies. Pract. Neurol. 2019, 19, 284–294. [Google Scholar] [CrossRef]
- Aussy, A.; Boyer, O.; Cordel, N. Dermatomyositis and Immune-Mediated Necrotizing Myopathies: A Window on Autoimmunity and Cancer. Front. Immunol. 2017, 8, 992. [Google Scholar] [CrossRef]
- Allenbach, Y.; Keraen, J.; Bouvier, A.M.; Jooste, V.; Champtiaux, N.; Hervier, B.; Schoindre, Y.; Rigolet, A.; Gilardin, L.; Musset, L.; et al. High risk of cancer in autoimmune necrotizing myopathies: Usefulness of myositis specific antibody. Brain 2016, 139, 2131–2135. [Google Scholar] [CrossRef] [Green Version]
- Selva-O’Callaghan, A.; Ros, J.; Gil-Vila, A.; Vila-Pijoan, G.; Trallero-Araguas, E.; Pinal-Fernandez, I. Malignancy and myositis, from molecular mimicry to tumor infiltrating lymphocytes. Neuromuscul. Disord. 2019, 29, 819–825. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.; Nagaraju, K.; Plotz, P.; Wang, K.; Levine, S.; Gabrielson, E.; Corse, A.; Rosen, A. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J. Exp. Med. 2005, 201, 591–601. [Google Scholar] [CrossRef]
- Mammen, A.L. Statin-Associated Autoimmune Myopathy. N. Engl. J. Med. 2016, 374, 664–669. [Google Scholar] [CrossRef]
- Platteel, A.C.M.; Wevers, B.A.; Lim, J.; Bakker, J.A.; Bontkes, H.J.; Curvers, J.; Damoiseaux, J.; Heron, M.; de Kort, G.; Limper, M.; et al. Frequencies and clinical associations of myositis-related antibodies in The Netherlands: A one-year survey of all Dutch patients. J. Transl. Autoimmun. 2019, 2, 100013. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, Z.; Tansley, S.; Shaddick, G.; Chinoy, H.; Cooper, R.G.; New, R.P.; Lilleker, J.B.; Vencovsky, J.; Chazarain, L.; Danko, K.; et al. Frequency, mutual exclusivity and clinical associations of myositis autoantibodies in a combined European cohort of idiopathic inflammatory myopathy patients. J. Autoimmun. 2019, 101, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Hida, A.; Yamashita, T.; Hosono, Y.; Inoue, M.; Kaida, K.; Kadoya, M.; Miwa, Y.; Yajima, N.; Maezawa, R.; Arai, S.; et al. Anti-TIF1-gamma antibody and cancer-associated myositis: A clinicohistopathologic study. Neurology 2016, 87, 299–308. [Google Scholar] [CrossRef]
- Mugii, N.; Hasegawa, M.; Matsushita, T.; Hamaguchi, Y.; Oohata, S.; Okita, H.; Yahata, T.; Someya, F.; Inoue, K.; Murono, S.; et al. Oropharyngeal Dysphagia in Dermatomyositis: Associations with Clinical and Laboratory Features Including Autoantibodies. PLoS ONE 2016, 11, e0154746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allenbach, Y.; Uzunhan, Y.; Toquet, S.; Leroux, G.; Gallay, L.; Marquet, A.; Meyer, A.; Guillaud, C.; Limal, N.; Gagnadoux, F.; et al. Different phenotypes in dermatomyositis associated with anti-MDA5 antibody: Study of 121 cases. Neurology 2020, 95, e70–e78. [Google Scholar] [CrossRef]
- Mamyrova, G.; Kishi, T.; Shi, M.; Targoff, I.N.; Huber, A.M.; Curiel, R.V.; Miller, F.W.; Rider, L.G.; Childhood Myositis Heterogeneity Collaborative Study Group. Anti-MDA5 autoantibodies associated with juvenile dermatomyositis constitute a distinct phenotype in North America. Rheumatology 2021, 60, 1839–1849. [Google Scholar] [CrossRef]
- Nombel, A.; Fabien, N.; Coutant, F. Dermatomyositis With Anti-MDA5 Antibodies: Bioclinical Features, Pathogenesis and Emerging Therapies. Front. Immunol. 2021, 12, 773352. [Google Scholar] [CrossRef]
- Fiorentino, D.; Chung, L.; Zwerner, J.; Rosen, A.; Casciola-Rosen, L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): A retrospective study. J. Am. Acad. Dermatol. 2011, 65, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.C.; Casciola-Rosen, L.; Samedy, L.A.; Werner, J.; Owoyemi, K.; Danoff, S.K.; Christopher-Stine, L. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: Expanding the clinical spectrum. Arthritis Care Res. 2013, 65, 1307–1315. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, H.; Nakashima, R.; Hosono, Y.; Imura, Y.; Yagita, M.; Yoshifuji, H.; Hirata, S.; Nojima, T.; Sugiyama, E.; Hatta, K.; et al. Multicenter Prospective Study of the Efficacy and Safety of Combined Immunosuppressive Therapy with High-Dose Glucocorticoid, Tacrolimus, and Cyclophosphamide in Interstitial Lung Diseases Accompanied by Anti-Melanoma Differentiation-Associated Gene 5-Positive Dermatomyositis. Arthritis Rheumatol. 2020, 72, 488–498. [Google Scholar]
- Wu, W.; Guo, L.; Fu, Y.; Wang, K.; Zhang, D.; Xu, W.; Chen, Z.; Ye, S. Interstitial Lung Disease in Anti-MDA5 Positive Dermatomyositis. Clin. Rev. Allergy Immunol. 2021, 60, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, Y.; Kuwana, M.; Hoshino, K.; Hasegawa, M.; Kaji, K.; Matsushita, T.; Komura, K.; Nakamura, M.; Kodera, M.; Suga, N.; et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: A multicenter cross-sectional study. Arch. Dermatol. 2011, 147, 391–398. [Google Scholar] [CrossRef] [PubMed]
- McPherson, M.; Economidou, S.; Liampas, A.; Zis, P.; Parperis, K. Management of MDA-5 antibody positive clinically amyopathic dermatomyositis associated interstitial lung disease: A systematic review. Semin. Arthritis Rheum. 2022, 53, 151959. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, W.; Wang, Y.; Guo, Z.; Sun, L.; Kuwana, M. Distinct profiles of myositis-specific autoantibodies in Chinese and Japanese patients with polymyositis/dermatomyositis. Clin. Rheumatol. 2015, 34, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, I.; Okura, Y.; Yamada, M.; Kawamura, N.; Kuwana, M.; Ariga, T. Anti-melanoma differentiation-associated gene 5 antibody is a diagnostic and predictive marker for interstitial lung diseases associated with juvenile dermatomyositis. J. Pediatr. 2011, 158, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Ueki, M.; Kobayashi, I.; Takezaki, S.; Tozawa, Y.; Okura, Y.; Yamada, M.; Kuwana, M.; Ariga, T. Myositis-specific autoantibodies in Japanese patients with juvenile idiopathic inflammatory myopathies. Mod. Rheumatol. 2019, 29, 351–356. [Google Scholar] [CrossRef]
- Zhang, S.H.; Zhao, Y.; Xie, Q.B.; Jiang, Y.; Wu, Y.K.; Yan, B. Aberrant activation of the type I interferon system may contribute to the pathogenesis of anti-melanoma differentiation-associated gene 5 dermatomyositis. Br. J. Dermatol. 2019, 180, 1090–1098. [Google Scholar] [CrossRef]
- Horai, Y.; Koga, T.; Fujikawa, K.; Takatani, A.; Nishino, A.; Nakashima, Y.; Suzuki, T.; Kawashiri, S.Y.; Iwamoto, N.; Ichinose, K.; et al. Serum interferon-alpha is a useful biomarker in patients with anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive dermatomyositis. Mod. Rheumatol. 2015, 25, 85–89. [Google Scholar] [CrossRef]
- Cassius, C.; Amode, R.; Delord, M.; Battistella, M.; Poirot, J.; How-Kit, A.; Lepelletier, C.; Jachiet, M.; de Masson, A.; Frumholtz, L.; et al. MDA5(+) Dermatomyositis Is Associated with Stronger Skin Type I Interferon Transcriptomic Signature with Upregulation of IFN-kappa Transcript. J. Investig. Dermatol. 2020, 140, 1276–1279.e7. [Google Scholar] [CrossRef]
- Deng, S.X.; Hanson, E.; Sanz, I. In vivo cell penetration and intracellular transport of anti-Sm and anti-La autoantibodies. Int. Immunol. 2000, 12, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Vlahakos, D.; Foster, M.H.; Ucci, A.A.; Barrett, K.J.; Datta, S.K.; Madaio, M.P. Murine monoclonal anti-DNA antibodies penetrate cells, bind to nuclei, and induce glomerular proliferation and proteinuria in vivo. J. Am. Soc. Nephrol. 1992, 2, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.N.; Gardner, L.; Levin, M. Antibodies to an intracellular antigen penetrate neuronal cells and cause deleterious effects. J. Clin. Cell Immunol. 2013, 4, 134. [Google Scholar] [CrossRef] [Green Version]
- Okiyama, N.; Ichimura, Y.; Shobo, M.; Tanaka, R.; Kubota, N.; Saito, A.; Ishitsuka, Y.; Watanabe, R.; Fujisawa, Y.; Nakamura, Y.; et al. Immune response to dermatomyositis-specific autoantigen, transcriptional intermediary factor 1gamma can result in experimental myositis. Ann. Rheum. Dis. 2021, 80, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, A.; Hamaguchi, Y.; Fujimoto, M.; Tokura, Y. TIF1gamma-overexpressing, highly progressive endometrial carcinoma in a patient with dermato-myositis positive for malignancy-associated anti-p155/140 autoantibody. Acta Derm. Venereol. 2013, 93, 715–716. [Google Scholar] [CrossRef]
- Fujimoto, M.; Murakami, A.; Kurei, S.; Okiyama, N.; Kawakami, A.; Mishima, M.; Sato, S.; Seishima, M.; Suda, T.; Mimori, T.; et al. Enzyme-linked immunosorbent assays for detection of anti-transcriptional intermediary factor-1 gamma and anti-Mi-2 autoantibodies in dermatomyositis. J. Dermatol. Sci. 2016, 84, 272–281. [Google Scholar] [CrossRef]
- Targoff, I.N.; Johnson, A.E.; Miller, F.W. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. 1990, 33, 1361–1370. [Google Scholar] [CrossRef]
- Christopher-Stine, L.; Casciola-Rosen, L.A.; Hong, G.; Chung, T.; Corse, A.M.; Mammen, A.L. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010, 62, 2757–2766. [Google Scholar] [CrossRef] [Green Version]
- Allenbach, Y.; Benveniste, O.; Stenzel, W.; Boyer, O. Immune-mediated necrotizing myopathy: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2020, 16, 689–701. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Immune-Mediated Necrotizing Myopathy. Curr. Rheumatol. Rep. 2018, 20, 21. [Google Scholar] [CrossRef]
- Watanabe, Y.; Uruha, A.; Suzuki, S.; Nakahara, J.; Hamanaka, K.; Takayama, K.; Suzuki, N.; Nishino, I. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1038–1044. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Parks, C.; Werner, J.L.; Albayda, J.; Paik, J.; Danoff, S.K.; Casciola-Rosen, L.; Christopher-Stine, L.; Mammen, A.L. Longitudinal Course of Disease in a Large Cohort of Myositis Patients With Autoantibodies Recognizing the Signal Recognition Particle. Arthritis Care Res. 2017, 69, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Carrino, J.A.; Lahouti, A.H.; Basharat, P.; Albayda, J.; Paik, J.J.; Ahlawat, S.; Danoff, S.K.; Lloyd, T.E.; et al. Thigh muscle MRI in immune-mediated necrotising myopathy: Extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Ann. Rheum. Dis. 2017, 76, 681–687. [Google Scholar] [CrossRef]
- Hengstman, G.J.; ter Laak, H.J.; Vree Egberts, W.T.; Lundberg, I.E.; Moutsopoulos, H.M.; Vencovsky, J.; Doria, A.; Mosca, M.; van Venrooij, W.J.; van Engelen, B.G. Anti-signal recognition particle autoantibodies: Marker of a necrotising myopathy. Ann. Rheum. Dis. 2006, 65, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, O.; Drouot, L.; Jouen, F.; Charuel, J.L.; Bloch-Queyrat, C.; Behin, A.; Amoura, Z.; Marie, I.; Guiguet, M.; Eymard, B.; et al. Correlation of anti-signal recognition particle autoantibody levels with creatine kinase activity in patients with necrotizing myopathy. Arthritis Rheum. 2011, 63, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Mimori, T.; Mukai, R.; Kashiwagi, H.; Hardin, J.A. Characterization of human autoantibodies that selectively precipitate the 7SL RNA component of the signal recognition particle. J. Immunol. 1987, 138, 3219–3223. [Google Scholar]
- Reeves, W.H.; Nigam, S.K.; Blobel, G. Human autoantibodies reactive with the signal-recognition particle. Proc. Natl. Acad. Sci. USA 1986, 83, 9507–9511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romisch, K.; Miller, F.W.; Dobberstein, B.; High, S. Human autoantibodies against the 54 kDa protein of the signal recognition particle block function at multiple stages. Arthritis Res. Ther. 2006, 8, R39. [Google Scholar] [CrossRef] [Green Version]
- Rojana-udomsart, A.; Mitrpant, C.; Bundell, C.; Price, L.; Luo, Y.B.; Fabian, V.; Wilton, S.D.; Hollingsworth, P.; Mastaglia, F.L. Complement-mediated muscle cell lysis: A possible mechanism of myonecrosis in anti-SRP associated necrotizing myopathy (ASANM). J. Neuroimmunol. 2013, 264, 65–70. [Google Scholar] [CrossRef]
- Bergua, C.; Chiavelli, H.; Allenbach, Y.; Arouche-Delaperche, L.; Arnoult, C.; Bourdenet, G.; Jean, L.; Zoubairi, R.; Guerout, N.; Mahler, M.; et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann. Rheum. Dis. 2019, 78, 131–139. [Google Scholar] [CrossRef]
- Yanase, K.; Madaio, M.P. Nuclear localizing anti-DNA antibodies enter cells via caveoli and modulate expression of caveolin and p53. J. Autoimmun. 2005, 24, 145–151. [Google Scholar] [CrossRef]
- Yanase, K.; Smith, R.M.; Puccetti, A.; Jarett, L.; Madaio, M.P. Receptor-mediated cellular entry of nuclear localizing anti-DNA antibodies via myosin 1. J. Clin. Investig. 1997, 100, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.J.; Nahm, D.H.; Jang, Y.J. Mouse monoclonal autoantibodies penetrate mouse macrophage cells and stimulate NF-kappaB activation and TNF-alpha release. Immunol. Lett. 2009, 124, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.C.; Sun, G.H.; Lee, T.P.; Huang, J.C.; Yu, C.L.; Chen, C.H.; Tang, S.J.; Sun, K.H. Arginines in the CDR of anti-dsDNA autoantibodies facilitate cell internalization via electrostatic interactions. Eur. J. Immunol. 2008, 38, 3178–3190. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Segovia, D.; Ruiz-Arguelles, A.; Fishbein, E. Antibody to nuclear ribonucleoprotein penetrates live human mononuclear cells through Fc receptors. Nature 1978, 271, 67–69. [Google Scholar] [CrossRef]
- Okazaki, Y.; Ohno, H.; Takase, K.; Ochiai, T.; Saito, T. Cell surface expression of calnexin, a molecular chaperone in the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 35751–35758. [Google Scholar] [CrossRef] [Green Version]
- McNally, A.K.; Anderson, J.M. Multinucleated giant cell formation exhibits features of phagocytosis with participation of the endoplasmic reticulum. Exp. Mol. Pathol. 2005, 79, 126–135. [Google Scholar] [CrossRef]
- Alshehri, A.; Choksi, R.; Bucelli, R.; Pestronk, A. Myopathy with anti-HMGCR antibodies: Perimysium and myofiber pathology. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e124. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.L.; Christopher-Stine, L.; Ghazarian, S.R.; Pak, K.S.; Kus, J.E.; Daya, N.R.; Lloyd, T.E.; Mammen, A.L. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. 2012, 64, 4087–4093. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Suzuki, S.; Nishimura, H.; Murata, K.Y.; Kurashige, T.; Ikawa, M.; Asahi, M.; Konishi, H.; Mitsuma, S.; Kawabata, S.; et al. Statins and myotoxic effects associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies: An observational study in Japan. Medicine 2015, 94, e416. [Google Scholar] [CrossRef]
- Trapani, L.; Segatto, M.; La Rosa, P.; Fanelli, F.; Moreno, S.; Marino, M.; Pallottini, V. 3-hydroxy 3-methylglutaryl coenzyme A reductase inhibition impairs muscle regeneration. J. Cell Biochem. 2012, 113, 2057–2063. [Google Scholar] [CrossRef]
- Martini, C.; Trapani, L.; Narciso, L.; Marino, M.; Trentalance, A.; Pallottini, V. 3-hydroxy 3-methylglutaryl coenzyme A reductase increase is essential for rat muscle differentiation. J. Cell Physiol. 2009, 220, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Group, S.C.; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef]
- Zwarthoff, S.A.; Berends, E.T.M.; Mol, S.; Ruyken, M.; Aerts, P.C.; Jozsi, M.; de Haas, C.J.C.; Rooijakkers, S.H.M.; Gorham, R.D., Jr. Functional Characterization of Alternative and Classical Pathway C3/C5 Convertase Activity and Inhibition Using Purified Models. Front. Immunol. 2018, 9, 1691. [Google Scholar] [CrossRef] [Green Version]
- Bayly-Jones, C.; Bubeck, D.; Dunstone, M.A. The mystery behind membrane insertion: A review of the complement membrane attack complex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160221. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, S.A. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology 2007, 69, 2008–2019. [Google Scholar] [CrossRef]
- Leddy, J.P.; Griggs, R.C.; Klemperer, M.R.; Frank, M.M. Hereditary complement (C2) deficiency with dermatomyositis. Am. J. Med. 1975, 58, 83–91. [Google Scholar] [CrossRef]
- Campo, A.; Hausmann, G.; Martí, R.M.; Estrach, T.; Grau, J.M.; Porcel, J.M.; Herrero, C. Complement activation products (C3a and C5b-9) as markers of activity of dermatomyositis. Comparison with usual biochemical parameters. Actas Dermosifiliogr. 2007, 98, 403–414. [Google Scholar] [CrossRef]
- Basta, M.; Dalakas, M.C. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J. Clin. Investig. 1994, 94, 1729–1735. [Google Scholar] [CrossRef] [Green Version]
- Pytel, P. C4d staining as immunohistochemical marker in inflammatory myopathies. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 696–704. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Amato, A.A. Uncertainties in the pathogenesis of adult dermatomyositis. Curr. Opin. Neurol. 2004, 17, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Emslie-Smith, A.M.; Engel, A.G. Microvascular changes in early and advanced dermatomyositis: A quantitative study. Ann. Neurol. 1990, 27, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Kissel, J.T.; Mendell, J.R.; Rammohan, K.W. Microvascular deposition of complement membrane attack complex in dermatomyositis. N. Engl. J. Med. 1986, 314, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, J.N.; Engel, W.K. Vascular deposits of immunoglobulin and complement in idiopathic inflammatory myopathy. N. Engl. J. Med. 1972, 286, 333–338. [Google Scholar] [CrossRef]
- Braczynski, A.K.; Harter, P.N.; Zeiner, P.S.; Drott, U.; Tews, D.S.; Preusse, C.; Penski, C.; Dunst, M.; Weis, J.; Stenzel, W.; et al. C5b-9 deposits on endomysial capillaries in non-dermatomyositis cases. Neuromuscul. Disord. 2016, 26, 283–291. [Google Scholar] [CrossRef]
- Ichikawa, E.; Furuta, J.; Kawachi, Y.; Imakado, S.; Otsuka, F. Hereditary complement (C9) deficiency associated with dermatomyositis. Br. J. Dermatol. 2001, 144, 1080–1083. [Google Scholar] [CrossRef]
- Knauss, S.; Preusse, C.; Allenbach, Y.; Leonard-Louis, S.; Touat, M.; Fischer, N.; Radbruch, H.; Mothes, R.; Matyash, V.; Bohmerle, W.; et al. PD1 pathway in immune-mediated myopathies: Pathogenesis of dysfunctional T cells revisited. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e558. [Google Scholar] [CrossRef] [Green Version]
- Albazli, K.; Kaminski, H.J.; Howard, J.F., Jr. Complement Inhibitor Therapy for Myasthenia Gravis. Front. Immunol. 2020, 11, 917. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, R.J.; Kong, S.W.; Yao, Y.; Jallal, B.; Kiener, P.A.; Pinkus, J.L.; Beggs, A.H.; Amato, A.A.; Greenberg, S.A. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007, 56, 3784–3792. [Google Scholar] [CrossRef]
- Baechler, E.C.; Bauer, J.W.; Slattery, C.A.; Ortmann, W.A.; Espe, K.J.; Novitzke, J.; Ytterberg, S.R.; Gregersen, P.K.; Behrens, T.W.; Reed, A.M. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol. Med. 2007, 13, 59–68. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Role of type I interferons in the activation of autoreactive B cells. Immunol. Cell Biol. 2012, 90, 498–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Luo, H.; Zhang, H.; Zuo, X.; Wang, L.; Zhu, H. Using multi-omics methods to understand dermatomyositis/polymyositis. Autoimmun Rev. 2017, 16, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, M.; Kong, S.W.; Pinkus, J.L.; Walsh, R.J.; Liao, A.; Nazareno, R.; Amato, A.A.; Krastins, B.; Morehouse, C.; Higgs, B.W.; et al. Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann. Neurol 2010, 67, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Derfoul, A.; Pak, K.; Plotz, P.; Miller, F.W.; Milisenda, J.C.; Grau-Junyent, J.M.; Selva-O’Callaghan, A.; Paik, J.; et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology 2019, 93, e1193–e1204. [Google Scholar] [CrossRef]
- Amici, D.R.; Pinal-Fernandez, I.; Christopher-Stine, L.; Mammen, A.L.; Mendillo, M.L. A network of core and subtype-specific gene expression programs in myositis. Acta Neuropathol. 2021, 142, 887–898. [Google Scholar] [CrossRef]
- Liao, A.P.; Salajegheh, M.; Nazareno, R.; Kagan, J.C.; Jubin, R.G.; Greenberg, S.A. Interferon beta is associated with type 1 interferon-inducible gene expression in dermatomyositis. Ann. Rheum. Dis. 2011, 70, 831–836. [Google Scholar] [CrossRef]
- Huard, C.; Gulla, S.V.; Bennett, D.V.; Coyle, A.J.; Vleugels, R.A.; Greenberg, S.A. Correlation of cutaneous disease activity with type 1 interferon gene signature and interferon beta in dermatomyositis. Br. J. Dermatol. 2017, 176, 1224–1230. [Google Scholar] [CrossRef] [PubMed]
- Oddis, C.V.; Aggarwal, R. Treatment in myositis. Nat. Rev. Rheumatol. 2018, 14, 279–289. [Google Scholar] [CrossRef]
- Gordon, P.A.; Winer, J.B.; Hoogendijk, J.E.; Choy, E.H. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst. Rev. 2012, 2012, CD003643. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Illa, I.; Dambrosia, J.M.; Soueidan, S.A.; Stein, D.P.; Otero, C.; Dinsmore, S.T.; McCrosky, S. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N. Engl. J. Med. 1993, 329, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Charles-Schoeman, C.; Schessl, J.; Dimachkie, M.M.; Beckmann, I.; Levine, T. Prospective, double-blind, randomized, placebo-controlled phase III study evaluating efficacy and safety of octagam 10% in patients with dermatomyositis (“ProDERM Study”). Medicine 2021, 100, e23677. [Google Scholar] [CrossRef]
- Vermaak, E.; Tansley, S.L.; McHugh, N.J. The evidence for immunotherapy in dermatomyositis and polymyositis: A systematic review. Clin. Rheumatol. 2015, 34, 2089–2095. [Google Scholar] [CrossRef] [Green Version]
- van de Vlekkert, J.; Hoogendijk, J.E.; de Haan, R.J.; Algra, A.; van der Tweel, I.; van der Pol, W.L.; Uijtendaal, E.V.; de Visser, M.; Dexa Myositis, T. Oral dexamethasone pulse therapy versus daily prednisolone in sub-acute onset myositis, a randomised clinical trial. Neuromuscul. Disord. 2010, 20, 382–389. [Google Scholar] [CrossRef]
- Newman, E.D.; Scott, D.W. The Use of Low-dose Oral Methotrexate in the Treatment of Polymyositis and Dermatomyositis. J. Clin. Rheumatol. 1995, 1, 99–102. [Google Scholar] [CrossRef]
- Majithia, V.; Harisdangkul, V. Mycophenolate mofetil (CellCept): An alternative therapy for autoimmune inflammatory myopathy. Rheumatology 2005, 44, 386–389. [Google Scholar] [CrossRef] [Green Version]
- Pisoni, C.N.; Cuadrado, M.J.; Khamashta, M.A.; Hughes, G.R.; D’Cruz, D.P. Mycophenolate mofetil treatment in resistant myositis. Rheumatology 2007, 46, 516–518. [Google Scholar] [CrossRef] [Green Version]
- Danieli, M.G.; Calcabrini, L.; Calabrese, V.; Marchetti, A.; Logullo, F.; Gabrielli, A. Intravenous immunoglobulin as add on treatment with mycophenolate mofetil in severe myositis. Autoimmun. Rev. 2009, 9, 124–127. [Google Scholar] [CrossRef]
- Goswami, R.P.; Haldar, S.N.; Chatterjee, M.; Vij, P.; van der Kooi, A.J.; Lim, J.; Raaphorst, J.; Bhadu, D.; Gelardi, C.; Danieli, M.G.; et al. Efficacy and safety of intravenous and subcutaneous immunoglobulin therapy in idiopathic inflammatory myopathy: A systematic review and meta-analysis. Autoimmun. Rev. 2022, 21, 102997. [Google Scholar] [CrossRef] [PubMed]
- Seite, J.F.; Hillion, S.; Harbonnier, T.; Pers, J.O. Review: Intravenous immunoglobulin and B cells: When the product regulates the producer. Arthritis Rheumatol. 2015, 67, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Bounfour, T.; Bouaziz, J.D.; Bezier, M.; Cordoliani, F.; Saussine, A.; Petit, A.; Juillard, C.; Bagot, M.; Rybojad, M. Clinical efficacy of intravenous immunoglobulins for the treatment of dermatomyositis skin lesions without muscle disease. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1150–1157. [Google Scholar] [CrossRef]
- Femia, A.N.; Eastham, A.B.; Lam, C.; Merola, J.F.; Qureshi, A.A.; Vleugels, R.A. Intravenous immunoglobulin for refractory cutaneous dermatomyositis: A retrospective analysis from an academic medical center. J. Am. Acad. Dermatol. 2013, 69, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Mammen, A.L.; Tiniakou, E. Intravenous Immune Globulin for Statin-Triggered Autoimmune Myopathy. N. Engl. J. Med. 2015, 373, 1680–1682. [Google Scholar] [CrossRef] [Green Version]
- Kassardjian, C.D.; Lennon, V.A.; Alfugham, N.B.; Mahler, M.; Milone, M. Clinical Features and Treatment Outcomes of Necrotizing Autoimmune Myopathy. JAMA Neurol. 2015, 72, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Eftimov, F.; Verhamme, C.; Brusse, E.; Hoogendijk, J.E.; Saris, C.G.J.; Raaphorst, J.; De Haan, R.J.; van Schaik, I.N.; Aronica, E.; et al. Intravenous immunoglobulins as first-line treatment in idiopathic inflammatory myopathies: A pilot study. Rheumatology 2021, 60, 1784–1792. [Google Scholar] [CrossRef]
- Detert, J.; Bastian, H.; Listing, J.; Weiss, A.; Wassenberg, S.; Liebhaber, A.; Rockwitz, K.; Alten, R.; Kruger, K.; Rau, R.; et al. Induction therapy with adalimumab plus methotrexate for 24 weeks followed by methotrexate monotherapy up to week 48 versus methotrexate therapy alone for DMARD-naive patients with early rheumatoid arthritis: HIT HARD, an investigator-initiated study. Ann. Rheum. Dis. 2013, 72, 844–850. [Google Scholar] [CrossRef]
- Reeves, H.M.; Winters, J.L. The mechanisms of action of plasma exchange. Br. J. Haematol. 2014, 164, 342–351. [Google Scholar] [CrossRef]
- Miller, F.W.; Leitman, S.F.; Cronin, M.E.; Hicks, J.E.; Leff, R.L.; Wesley, R.; Fraser, D.D.; Dalakas, M.; Plotz, P.H. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N. Engl. J. Med. 1992, 326, 1380–1384. [Google Scholar] [CrossRef] [PubMed]
- Shirakashi, M.; Nakashima, R.; Tsuji, H.; Tanizawa, K.; Handa, T.; Hosono, Y.; Akizuki, S.; Murakami, K.; Hashimoto, M.; Yoshifuji, H.; et al. Efficacy of plasma exchange in anti-MDA5-positive dermatomyositis with interstitial lung disease under combined immunosuppressive treatment. Rheumatology 2020, 59, 3284–3292. [Google Scholar] [CrossRef]
- Hervier, B.; Uzunhan, Y. Inflammatory Myopathy-Related Interstitial Lung Disease: From Pathophysiology to Treatment. Front. Med. 2019, 6, 326. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Charles-Schoeman, C. Oral cyclophosphamide in treatment of patients with refractory idiopathic inflammatory myopathies: A retrospective observational study. Clin. Rheumatol. 2018, 37, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.E.; Vencovsky, J.; Alexanderson, H. Therapy of myositis: Biological and physical. Curr. Opin. Rheumatol. 2014, 26, 704–711. [Google Scholar] [CrossRef]
- Moghadam-Kia, S.; Aggarwal, R.; Oddis, C.V. Biologics for idiopathic inflammatory myopathies. Curr. Opin. Rheumatol. 2017, 29, 645–651. [Google Scholar] [CrossRef]
- Oddis, C.V.; Reed, A.M.; Aggarwal, R.; Rider, L.G.; Ascherman, D.P.; Levesque, M.C.; Barohn, R.J.; Feldman, B.M.; Harris-Love, M.O.; Koontz, D.C.; et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheum. 2013, 65, 314–324. [Google Scholar] [CrossRef]
- Aggarwal, R.; Bandos, A.; Reed, A.M.; Ascherman, D.P.; Barohn, R.J.; Feldman, B.M.; Miller, F.W.; Rider, L.G.; Harris-Love, M.O.; Levesque, M.C.; et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 2014, 66, 740–749. [Google Scholar] [CrossRef]
- He, C.; Li, W.; Xie, Q.; Yin, G. Rituximab in the Treatment of Interstitial Lung Diseases Related to Anti-Melanoma Differentiation-Associated Gene 5 Dermatomyositis: A Systematic Review. Front. Immunol. 2021, 12, 820163. [Google Scholar] [CrossRef]
- Kaegi, C.; Steiner, U.C.; Wuest, B.; Crowley, C.; Boyman, O. Systematic review of safety and efficacy of belimumab in treating immune-mediated disorders. Allergy 2021, 76, 2673–2683. [Google Scholar] [CrossRef]
- Higgs, B.W.; Zhu, W.; Morehouse, C.; White, W.I.; Brohawn, P.; Guo, X.; Rebelatto, M.; Le, C.; Amato, A.; Fiorentino, D.; et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-alpha monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann. Rheum. Dis. 2014, 73, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Ladislau, L.; Suarez-Calvet, X.; Toquet, S.; Landon-Cardinal, O.; Amelin, D.; Depp, M.; Rodero, M.P.; Hathazi, D.; Duffy, D.; Bondet, V.; et al. JAK inhibitor improves type I interferon induced damage: Proof of concept in dermatomyositis. Brain 2018, 141, 1609–1621. [Google Scholar] [CrossRef]
- Paik, J.J.; Casciola-Rosen, L.; Shin, J.Y.; Albayda, J.; Tiniakou, E.; Leung, D.G.; Gutierrez-Alamillo, L.; Perin, J.; Florea, L.; Antonescu, C.; et al. Study of Tofacitinib in Refractory Dermatomyositis: An Open-Label Pilot Study of Ten Patients. Arthritis Rheumatol. 2021, 73, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Faguer, S.; Belliere, J.; Ribes, D. Complement C5-blocking Agent in Refractory Dermatomyositis. J. Rheumatol. 2018, 45, 1710–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, K.; Bookbinder, S.; Furie, R.; Oddis, C.; Mojcik, C.; Bombara, M.; Plotz, P.; Kissel, J. A pilot study of eculizumab in patients with dermatomyositis. Arthritis Rheum. 2002, 46, S489. [Google Scholar]
- Lundberg, I.E.; Dastmalchi, M. Possible pathogenic mechanisms in inflammatory myopathies. Rheum. Dis. Clin. N. Am. 2002, 28, 799–822. [Google Scholar] [CrossRef]
- Efthimiou, P. Tumor necrosis factor-alpha in inflammatory myopathies: Pathophysiology and therapeutic implications. Semin. Arthritis Rheum. 2006, 36, 168–172. [Google Scholar] [CrossRef]
- Efthimiou, P.; Schwartzman, S.; Kagen, L.J. Possible role for tumour necrosis factor inhibitors in the treatment of resistant dermatomyositis and polymyositis: A retrospective study of eight patients. Ann. Rheum. Dis. 2006, 65, 1233–1236. [Google Scholar] [CrossRef] [Green Version]
- Dastmalchi, M.; Grundtman, C.; Alexanderson, H.; Mavragani, C.P.; Einarsdottir, H.; Helmers, S.B.; Elvin, K.; Crow, M.K.; Nennesmo, I.; Lundberg, I.E. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann. Rheum. Dis. 2008, 67, 1670–1677. [Google Scholar] [CrossRef]
- Schiffenbauer, A.; Garg, M.; Castro, C.; Pokrovnichka, A.; Joe, G.; Shrader, J.; Cabalar, I.V.; Faghihi-Kashani, S.; Harris-Love, M.O.; Plotz, P.H.; et al. A randomized, double-blind, placebo-controlled trial of infliximab in refractory polymyositis and dermatomyositis. Semin. Arthritis Rheum. 2018, 47, 858–864. [Google Scholar] [CrossRef]
- Muscle Study, G. A randomized, pilot trial of etanercept in dermatomyositis. Ann. Neurol. 2011, 70, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Rosenbach, M.; Kim, E.J.; Kim, B.; Werth, V.P.; Dunham, J. Tumor necrosis factor inhibitor-associated dermatomyositis. Arch. Dermatol. 2010, 146, 780–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riolo, G.; Towheed, T.E. Anti-tumor necrosis factor inhibitor therapy-induced dermatomyositis and fasciitis. J. Rheumatol. 2012, 39, 192–194. [Google Scholar]
- Lepidi, H.; Frances, V.; Figarella-Branger, D.; Bartoli, C.; Machado-Baeta, A.; Pellissier, J.F. Local expression of cytokines in idiopathic inflammatory myopathies. Neuropathol. Appl. Neurobiol. 1998, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Rockette, H.; Venturupalli, S.; Marder, G.; Dimachkie, M.; Gazeley, D.; Ernste, F.C.; Crofford, L.; Moghadam-Kia, S.; Koontz, D.; et al. Tocilizumab in Myositis: Results of a Phase IIb Double-Blind Randomized Controlled Trial. Arthritis Rheumatol. 2020, 72, 958. [Google Scholar]
- Grundtman, C.; Salomonsson, S.; Dorph, C.; Bruton, J.; Andersson, U.; Lundberg, I.E. Immunolocalization of interleukin-1 receptors in the sarcolemma and nuclei of skeletal muscle in patients with idiopathic inflammatory myopathies. Arthritis Rheum. 2007, 56, 674–687. [Google Scholar] [CrossRef]
- Zong, M.; Dorph, C.; Dastmalchi, M.; Alexanderson, H.; Pieper, J.; Amoudruz, P.; Barbasso Helmers, S.; Nennesmo, I.; Malmstrom, V.; Lundberg, I.E. Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: A mechanistic study with 12 months follow-up. Ann. Rheum. Dis. 2014, 73, 913–920. [Google Scholar] [CrossRef]
- Lorenzetti, R.; Janowska, I.; Smulski, C.R.; Frede, N.; Henneberger, N.; Walter, L.; Schleyer, M.T.; Huppe, J.M.; Staniek, J.; Salzer, U.; et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J. Autoimmun. 2019, 101, 145–152. [Google Scholar] [CrossRef]
- Tjarnlund, A.; Tang, Q.; Wick, C.; Dastmalchi, M.; Mann, H.; Tomasova Studynkova, J.; Chura, R.; Gullick, N.J.; Salerno, R.; Ronnelid, J.; et al. Abatacept in the treatment of adult dermatomyositis and polymyositis: A randomised, phase IIb treatment delayed-start trial. Ann. Rheum. Dis. 2018, 77, 55–62. [Google Scholar] [CrossRef]

{kind=link}
| Characteristic | DM | IMNM | ASyS | OM |
|---|---|---|---|---|
| Muscle weakness | ++ | +++ | ++ | ++ |
| Malignancies | ++ | + | − | − |
| Rapid progression * | ++ | +++ | − | − |
| Raynaud’s | − | − | ++ | ++ |
| Skin involvement | +++ | + | ++ | + |
| Interstitial lung disease | + | − | +++ | ++ |
| Peri/myocarditis ** | + | + | + | + |
| Diagnosis | Demographic Characteristics | Clinical Features | Autoantibodies | Other Features |
|---|---|---|---|---|
| Dermatomyositis (DM) | All ages Peak incidence 30–50 years 70% female | Symmetrical weakness Proximal > distal weakness Neck flexion > extension weakness Dysphagia Subacute onset DM-specific rash | MDA-5 |
|
| NXP-2 |
| |||
| Mi-2 |
| |||
| TIF1-γ |
| |||
| SAE-1 |
| |||
| Immune-mediated necrotizing myopathy (IMNM) | All ages 64% female | Severe symmetrical weakness Axial weakness Muscle atrophy Dysphagia Highly elevated CK (>10× upper limit) Subacute onset Early fatty infiltration muscle-MRI | HMGCR |
|
| SRP |
| |||
| Seronegative |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamperman, R.G.; van der Kooi, A.J.; de Visser, M.; Aronica, E.; Raaphorst, J. Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review. Int. J. Mol. Sci. 2022, 23, 4301. https://doi.org/10.3390/ijms23084301
Kamperman RG, van der Kooi AJ, de Visser M, Aronica E, Raaphorst J. Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review. International Journal of Molecular Sciences. 2022; 23(8):4301. https://doi.org/10.3390/ijms23084301
Chicago/Turabian StyleKamperman, Renske G., Anneke J. van der Kooi, Marianne de Visser, Eleonora Aronica, and Joost Raaphorst. 2022. "Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review" International Journal of Molecular Sciences 23, no. 8: 4301. https://doi.org/10.3390/ijms23084301
APA StyleKamperman, R. G., van der Kooi, A. J., de Visser, M., Aronica, E., & Raaphorst, J. (2022). Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review. International Journal of Molecular Sciences, 23(8), 4301. https://doi.org/10.3390/ijms23084301