
COVID-19: Are We Facing Secondary Pellagra Which Cannot Simply Be Cured by Vitamin B3?

Abstract

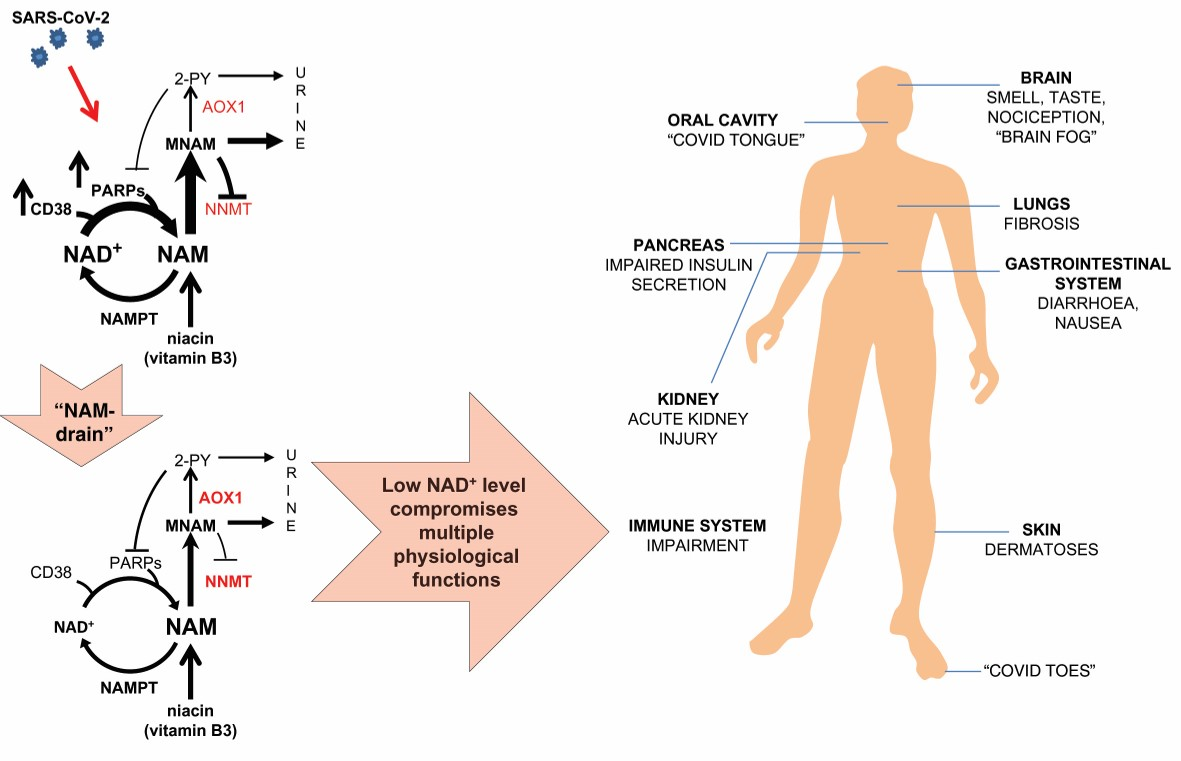{kind=link}
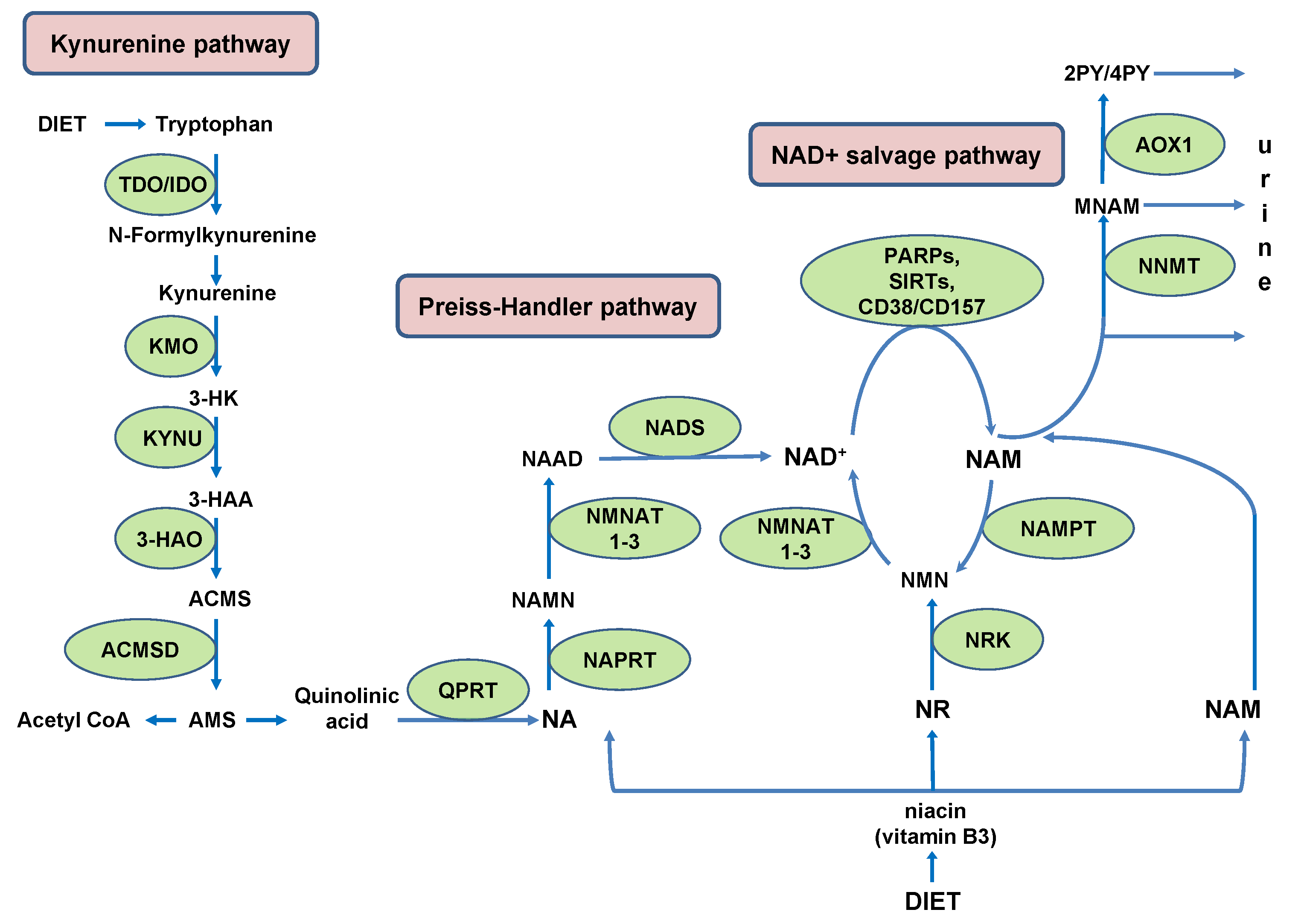{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. NAD+ Biosynthesis with Some Sex- and Species-Related Differences
3. The Role of NAD+ in Immune Response to SARS-CoV-2
3.1. PARPs in Immune Response to SARS-CoV-2
3.2. Sirtuins and COVID-19
3.3. CD38 in COVID-19
4. Direct Contribution of Reduced NAD+ Level to a Range of COVID-19 Pellagra-like Symptoms
4.1. Mucocutaneous and Gastrointestinal Manifestations
4.2. Smell, Taste and Nociception Disturbances Related to Deficient Oxytocin Secretion
4.3. Insulin Secretion Impairment Due to Deficient CD38-Mediated Ca2+ Mobilization
5. Effects of the “NAM Drain”, Other Than NAD+ Deficiency, in COVID-19
6. Concluding Remarks and Open Questions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 2 March 2022).
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.D.; Harris, C.; Cain, J.K.; Hummer, C.; Goyal, H.; Perisetti, A. Pulmonary and Extra-Pulmonary Clinical Manifestations of COVID-19. Front. Med. 2020, 7, 526. [Google Scholar] [CrossRef] [PubMed]
- Sarkesh, A.; Daei Sorkhabi, A.; Sheykhsaran, E.; Alinezhad, F.; Mohammadzadeh, N.; Hemmat, N.; Bannazadeh Baghi, H. Extrapulmonary Clinical Manifestations in COVID-19 Patients. Am. J. Trop. Med. Hyg. 2020, 103, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Scorza, F.A.; Scorza, C.A.; Fiorini, A.C. Extrapulmonary onset manifestations of COVID-19. Clinics 2021, 76, e2900. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; Mariette, X. Systemic and organ-specific immune-related manifestations of COVID-19. Nat. Rev. Rheumatol. 2021, 17, 315–332.e4. [Google Scholar] [CrossRef]
- Higgins, V.; Sohaei, D.; Diamandis, E.P.; Prassas, I. COVID-19: From an acute to chronic disease? Potential long-term health consequences. Crit. Rev. Clin. Lab. Sci. 2021, 58, 297–310. [Google Scholar] [CrossRef]
- Heer, C.D.; Sanderson, D.J.; Voth, L.S.; Alhammad, Y.M.O.; Schmidt, M.S.; Trammell, S.A.J.; Perlman, S.; Cohen, M.S.; Fehr, A.R.; Brenner, C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: An actionable component of innate immunity. J. Biol. Chem. 2020, 295, 17986–17996. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, e140327. [Google Scholar] [CrossRef]
- Blasco, H.; Bessy, C.; Plantier, L.; Lefevre, A.; Piver, E.; Bernard, L.; Marlet, J.; Stefic, K.; Benz-de Bretagne, I.; Cannet, P.; et al. The specific metabolome profiling of patients infected by SARS-CoV-2 supports the key role of tryptophan-nicotinamide pathway and cytosine metabolism. Sci. Rep. 2020, 10, 16824. [Google Scholar] [CrossRef]
- Migaud, M.; Gandotra, S.; Chand, H.S.; Gillespie, M.N.; Thannickal, V.J.; Langley, R.J. Metabolomics to Predict Antiviral Drug Efficacy in COVID-19. Am. J. Respir. Cell Mol. Biol. 2020, 63, 396–398. [Google Scholar] [CrossRef]
- Lionetto, L.; Ulivieri, M.; Capi, M.; De Bernardini, D.; Fazio, F.; Petrucca, A.; Pomes, L.M.; De Luca, O.; Gentile, G.; Casolla, B.; et al. Increased kynurenine-to-tryptophan ratio in the serum of patients infected with SARS-CoV-2: An observational cohort study. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166042. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40.e7. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Pérez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD+ homeostasis in human health and disease. EMBO Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Summers, K.L.; Meinitzer, A.; Zelzer, S.; Almer, G.; Prassl, R.; Schnedl, W.J.; Reininghaus, E.; Paulmichl, K.; Weghuber, D.; et al. Obesity-related dysregulation of the tryptophan-kynurenine metabolism: Role of age and parameters of the metabolic syndrome. Obesity 2014, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, J.; Schwartz, R.A.; Hegyi, V. Pellagra: Dermatitis, dementia, and diarrhea. Int. J. Dermatol. 2004, 43, 1–5. [Google Scholar] [CrossRef]
- Cao, S.; Wang, X.; Cestodio, K. Pellagra, an Almost-Forgotten Differential Diagnosis of Chronic Diarrhea: More Prevalent Than We Think. Nutr. Clin. Pract. 2020, 35, 860–863. [Google Scholar] [CrossRef]
- Fukuwatari, T.; Ohta, M.; Kimtjra, N.; Sasaki, R.; Shibata, K. Conversion ratio of tryptophan to niacin in Japanese women fed a purified diet conforming to the Japanese Dietary Reference Intakes. J. Nutr. Sci. Vitaminol. 2004, 50, 385–391. [Google Scholar] [CrossRef]
- Palzer, L.; Bader, J.J.; Angel, F.; Witzel, M.; Blaser, S.; McNeil, A.; Wandersee, M.K.; Leu, N.A.; Lengner, C.J.; Cho, C.E.; et al. Alpha-Amino-Beta-Carboxy-Muconate-Semialdehyde Decarboxylase Controls Dietary Niacin Requirements for NAD+ Synthesis. Cell Rep. 2018, 25, 1359–1370.e4. [Google Scholar] [CrossRef]
- Yoshino, J. ACMSD: A Novel Target for Modulating NAD+ Homeostasis. Trends Endocrinol. Metab. 2019, 30, 229–232. [Google Scholar] [CrossRef]
- Koshiguchi, M.; Hirai, S.; Egashira, Y. PGC1α regulates ACMSD expression through cooperation with HNF4α. Amino Acids 2018, 50, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Toda, S. Effects of sex hormones on the metabolism of tryptophan to niacin and to serotonin in male rats. Biosci. Biotechnol. Biochem. 1997, 61, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Su, X.; Quinn, W.J., III; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of evolutionary and kinetic drivers of NAD-dependent signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef]
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef]
- Slominska, E.M.; Kowalik, K.; Smolenski, R.T.; Szolkiewicz, M.; Rutkowski, P.; Rutkowski, B.; Swierczynski, J. Accumulation of poly(ADP-ribose) polymerase inhibitors in children with chronic renal failure. Pediatr. Nephrol. 2006, 21, 800–806. [Google Scholar] [CrossRef]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Novak Kujundžić, R.; Prpić, M.; Đaković, N.; Dabelić, N.; Tomljanović, M.; Mojzeš, A.; Fröbe, A.; Trošelj, K.G. Nicotinamide N-Methyltransferase in Acquisition of Stem Cell Properties and Therapy Resistance in Cancer. Int. J. Mol. Sci. 2021, 22, 5681. [Google Scholar] [CrossRef]
- Slominska, E.M.; Rutkowski, P.; Smolenski, R.T.; Szutowicz, A.; Rutkowski, B.; Swierczynski, J. The age-related increase in N-methyl-2-pyridone-5-carboxamide (NAD catabolite) in human plasma. Mol. Cell Biochem. 2004, 267, 25–30. [Google Scholar] [CrossRef]
- Garattini, E.; Fratelli, M.; Terao, M. Mammalian aldehyde oxidases: Genetics, evolution and biochemistry. Cell Mol. Life Sci. 2008, 65, 1019–1048. [Google Scholar] [CrossRef] [PubMed]
- Teo, H.K.; Lee, B.J.; Lau, A.J. Regulation of Human Aldehyde Oxidase Expression by Estrogen Receptor Agonists. FASEB J. 2020, 30, 1. [Google Scholar] [CrossRef]
- Kücükgöze, G.; Leimkühler, S. Direct comparison of the four aldehyde oxidase enzymes present in mouse gives insight into their substrate specificities. PLoS ONE 2018, 13, e0191819. [Google Scholar] [CrossRef]
- Terao, M.; Garattini, E.; Romão, M.J.; Leimkühler, S. Evolution, expression, and substrate specificities of aldehyde oxidase enzymes in eukaryotes. J. Biol. Chem. 2020, 295, 5377–5389. [Google Scholar] [CrossRef] [PubMed]
- Terao, M.; Romão, M.J.; Leimkühler, S.; Bolis, M.; Fratelli, M.; Coelho, C.; Santos-Silva, T.; Garattini, E. Structure and function of mammalian aldehyde oxidases. Arch. Toxicol. 2016, 90, 753–780. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No Love Lost Between Viruses and Interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Crosse, K.M.; Monson, E.A.; Beard, M.R.; Helbig, K.J. Interferon-Stimulated Genes as Enhancers of Antiviral Innate Immune Signaling. J. Innate Immun. 2018, 10, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, N.; Jin, R.; Feng, Y.; Wang, S.; Gao, S.; Gao, R.; Wu, G.; Tian, D.; Tan, W.; et al. Immune suppression in the early stage of COVID-19 disease. Nat. Commun. 2020, 11, 5859. [Google Scholar] [CrossRef]
- Van Eijk, L.E.; Binkhorst, M.; Bourgonje, A.R.; Offringa, A.K.; Mulder, D.J.; Bos, E.M.; Kolundzic, N.; Abdulle, A.E.; van der Voort, P.H.; Olde Rikkert, M.G.; et al. COVID-19: Immunopathology, pathophysiological mechanisms, and treatment options. J. Pathol. 2021, 254, 307–331. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, M.; Fu, Z.; Zhao, L. The Pathogenic Features of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): Possible Mechanisms for Immune Evasion? Front. Immunol. 2021, 12, 693579. [Google Scholar] [CrossRef]
- Ricci, D.; Etna, M.P.; Rizzo, F.; Sandini, S.; Severa, M.; Coccia, E.M. Innate Immune Response to SARS-CoV-2 Infection: From Cells to Soluble Mediators. Int. J. Mol. Sci. 2021, 22, 7017. [Google Scholar] [CrossRef]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; on behalf of the HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Zhu, H.; Tang, Y.D.; Zhan, G.; Su, C.; Zheng, C. The Critical Role of PARPs in Regulating Innate Immune Responses. Front. Immunol. 2021, 12, 712556. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Patel, S.; Majumdar, A. Role of NRF2 and Sirtuin activators in COVID-19. Clin. Immunol. 2021, 233, 108879. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Faini, A.C.; Malavasi, F. CD38 in the age of COVID-19: A medical perspective. Physiol. Rev. 2021, 101, 1457–1486. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Daugherty, M.D.; Young, J.M.; Kerns, J.A.; Malik, H.S. Rapid evolution of PARP genes suggests a broad role for ADP-ribosylation in host-virus conflicts. PLoS Genet. 2014, 10, e1004403. [Google Scholar] [CrossRef]
- Hoch, N.C. Host ADP-ribosylation and the SARS-CoV-2 macrodomain. Biochem. Soc. Trans. 2021, 49, 1711–1721. [Google Scholar] [CrossRef]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Athmer, J.; Channappanavar, R.; Phillips, J.M.; Meyerholz, D.K.; Perlman, S. The Nsp3 macrodomain promotes virulence in mice with coronavirus-induced encephalitis. J. Virol. 2015, 89, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, M.E.; Chen, Y.; Kuny, C.; Maejima, T.; Lease, R.; Ferraris, D.; Aikawa, M.; Sullivan, C.S.; Perlman, S.; Fehr, A.R. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 2019, 15, e1007756. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L.; McPherson, R.L.; Griffin, D.E. Macrodomain ADP-ribosylhydrolase and the pathogenesis of infectious diseases. PLoS Pathog. 2018, 14, e1006864. [Google Scholar] [CrossRef] [PubMed]
- Alhammad, Y.M.O.; Fehr, A.R. The Viral Macrodomain Counters Host Antiviral ADP-Ribosylation. Viruses 2020, 12, 384. [Google Scholar] [CrossRef]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2016, 7, e01721-16. [Google Scholar] [CrossRef]
- Alhammad, Y.M.O.; Kashipathy, M.M.; Roy, A.; Gagné, J.P.; McDonald, P.; Gao, P.; Nonfoux, L.; Battaile, K.P.; Johnson, D.K.; Holmstrom, E.D.; et al. The SARS-CoV-2 Conserved Macrodomain Is a Mono-ADP-Ribosylhydrolase. J. Virol. 2021, 95, e01969-20. [Google Scholar] [CrossRef] [PubMed]
- Nchioua, R.; Kmiec, D.; Müller, J.A.; Conzelmann, C.; Groß, R.; Swanson, C.M.; Neil, S.J.D.; Stenger, S.; Sauter, D.; Münch, J.; et al. SARS-CoV-2 Is Restricted by Zinc Finger Antiviral Protein despite Preadaptation to the Low-CpG Environment in Humans. mBio 2020, 11, e01930-20. [Google Scholar] [CrossRef]
- Ficarelli, M.; Neil, S.J.D.; Swanson, C.M. Targeted Restriction of Viral Gene Expression and Replication by the ZAP Antiviral System. Annu. Rev. Virol. 2021, 8, 265–283. [Google Scholar] [CrossRef]
- Kerns, J.A.; Emerman, M.; Malik, H.S. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 2008, 4, e21. [Google Scholar] [CrossRef]
- Kmiec, D.; Lista, M.J.; Ficarelli, M.; Swanson, C.M.; Neil, S.J.D. S-farnesylation is essential for antiviral activity of the long ZAP isoform against RNA viruses with diverse replication strategies. PLoS Pathog. 2021, 17, e1009726. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K. Poly(ADP-ribose): An organizer of cellular architecture. J. Cell Biol. 2014, 205, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Braczyk, K.; Gonçalves-Carneiro, D.; Ong, H.; Dawidziak, D.M.; Zawada, K.; Ong, H.; Wan, Y.; Zadrozny, K.K.; Ganser-Pornillos, B.K.; et al. Poly(ADP-ribose) potentiates ZAP antiviral activity. PLoS Pathog. 2022, 18, e1009202. [Google Scholar] [CrossRef]
- Carter-O’Connell, I.; Vermehren-Schmaedick, A.; Jin, H.; Morgan, R.K.; David, L.L.; Cohen, M.S. Combining Chemical Genetics with Proximity-Dependent Labeling Reveals Cellular Targets of Poly(ADP-ribose) Polymerase 14 (PARP14). ACS Chem. Biol. 2018, 13, 2841–2848. [Google Scholar] [CrossRef]
- Russo, L.C.; Tomasin, R.; Matos, I.A.; Manucci, A.C.; Sowa, S.T.; Dale, K.; Caldecott, K.W.; Lehtiö, L.; Schechtman, D.; Meotti, F.C.; et al. The SARS-CoV-2 Nsp3 macrodomain reverses PARP9/DTX3L-dependent ADP-ribosylation induced by interferon signaling. J. Biol. Chem. 2021, 297, 101041. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Jividen, K.; Spencer, A.; Dworak, N.; Ni, L.; Oostdyk, L.T.; Chatterjee, M.; Kuśmider, B.; Reon, B.; Parlak, M.; et al. Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9. Mol. Cell 2017, 66, 503–516.e5. [Google Scholar] [CrossRef]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAK-STAT 2013, 2, e23931. [Google Scholar] [CrossRef]
- Cao, Z.; Xia, H.; Rajsbaum, R.; Xia, X.; Wang, H.; Shi, P.Y. Ubiquitination of SARS-CoV-2 ORF7a promotes antagonism of interferon response. Cell Mol. Immunol. 2021, 18, 746–748. [Google Scholar] [CrossRef]
- Wickenhagen, A.; Sugrue, E.; Lytras, S.; Kuchi, S.; Noerenberg, M.; Turnbull, M.L.; Loney, C.; Herder, V.; Allan, J.; Jarmson, I.; et al. A prenylated dsRNA sensor protects against severe COVID-19. Science 2021, 374, eabj3624. [Google Scholar] [CrossRef]
- Frick, D.N.; Virdi, R.S.; Vuksanovic, N.; Dahal, N.; Silvaggi, N.R. Molecular Basis for ADP-Ribose Binding to the Mac1 Domain of SARS-CoV-2 Nsp3. Biochemistry 2020, 59, 2608–2615. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.A.P.M.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak-Yin Tsang, O.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Brodin, P. Immune determinants of COVID-19 disease presentation and severity. Nat. Med. 2021, 27, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 2009, 394, 101–109. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2α promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef]
- Budayeva, H.G.; Rowland, E.A.; Cristea, I.M. Intricate Roles of Mammalian Sirtuins in Defense against Viral Pathogens. J. Virol. 2015, 90, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Preyat, N.; Leo, O. Sirtuin deacylases: A molecular link between metabolism and immunity. J. Leukoc. Biol. 2013, 93, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, E.; Budayeva, H.G.; Miteva, Y.V.; Ricci, D.P.; Silhavy, T.J.; Shenk, T.; Cristea, I.M. Sirtuins are evolutionarily conserved viral restriction factors. mBio 2014, 5, e02249-14. [Google Scholar] [CrossRef]
- Yao, C.; Bora, S.A.; Parimon, T.; Zaman, T.; Friedman, O.A.; Palatinus, J.A.; Surapaneni, N.S.; Matusov, Y.P.; Cerro Chiang, G.; Kassar, A.G.; et al. Cell-Type-Specific Immune Dysregulation in Severely Ill COVID-19 Patients. Cell Rep. 2021, 34, 108590. [Google Scholar] [CrossRef]
- Bordoni, V.; Tartaglia, E.; Sacchi, A.; Fimia, G.M.; Cimini, E.; Casetti, R.; Notari, S.; Grassi, G.; Marchioni, L.; Bibas, M.; et al. The unbalanced p53/SIRT1 axis may impact lymphocyte homeostasis in COVID-19 patients. Int. J. Infect. Dis. 2021, 105, 49–53. [Google Scholar] [CrossRef]
- Miller, R.; Wentzel, A.R.; Richards, G.A. COVID-19: NAD+ deficiency may predispose the aged, obese and type2 diabetics to mortality through its effect on SIRT1 activity. Med. Hypotheses 2020, 144, 110044. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Garbers, C.; Rose-John, S. ADAM17: A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011, 32, 380–387. [Google Scholar] [CrossRef]
- Cardellini, M.; Menghini, R.; Martelli, E.; Casagrande, V.; Marino, A.; Rizza, S.; Porzio, O.; Mauriello, A.; Solini, A.; Ippoliti, A.; et al. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SIRT1. Diabetes 2009, 58, 2396–2401. [Google Scholar] [CrossRef]
- Zipeto, D.; Palmeira, J.D.F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 576745. [Google Scholar] [CrossRef]
- Kang, B.N.; Tirumurugaan, K.G.; Deshpande, D.A.; Amrani, Y.; Panettieri, R.A.; Walseth, T.F.; Kannan, M.S. Transcriptional regulation of CD38 expression by tumor necrosis factor-α in human airway smooth muscle cells: Role of NF-κB and sensitivity to glucocorticoids. FASEB J. 2006, 20, 1000–1002. [Google Scholar] [CrossRef] [PubMed]
- Matalonga, J.; Glaria, E.; Bresque, M.; Escande, C.; Carbó, J.M.; Kiefer, K.; Vicente, R.; León, T.E.; Beceiro, S.; Pascual-García, M.; et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep. 2017, 18, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Musso, T.; Deaglio, S.; Franco, L.; Calosso, L.; Badolato, R.; Garbarino, G.; Dianzani, U.; Malavasi, F. CD38 expression and functional activities are up-regulated by IFN-γ on human monocytes and monocytic cell lines. J. Leukoc. Biol. 2001, 69, 605–612. [Google Scholar]
- Funaro, A.; Horenstein, A.L.; Calosso, L.; Morra, M.; Tarocco, R.P.; Franco, L.; De Flora, A.; Malavasi, F. Identification and characterization of an active soluble form of human CD38 in normal and pathological fluids. Int. Immunol. 1996, 8, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal 2012, 5, ra67. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, S.; Tohgo, A.; Noguchi, N.; Koguma, T.; Nata, K.; Sugimoto, T.; Yonekura, H.; Okamoto, H. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J. Biol. Chem. 1993, 268, 26052–26054. [Google Scholar] [CrossRef]
- Shrimp, J.H.; Hu, J.; Dong, M.; Wang, B.S.; MacDonald, R.; Jiang, H.; Hao, Q.; Yen, A.; Lin, H. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J. Am. Chem. Soc. 2014, 136, 5656–5663. [Google Scholar] [CrossRef]
- De Flora, A.; Guida, L.; Franco, L.; Zocchi, E. The CD38/cyclic ADP-ribose system: A topological paradox. Int. J. Biochem. Cell Biol. 1997, 29, 1149–1166. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Hon, C.L. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [PubMed]
- Graeff, R.; Liu, Q.; Kriksunov, I.A.; Hao, Q.; Lee, H.C. Acidic residues at the active sites of CD38 and ADP-ribosyl cyclase determine nicotinic acid adenine dinucleotide phosphate (NAADP) synthesis and hydrolysis activities. J. Biol. Chem. 2006, 281, 28951–28957. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef]
- Joe, Y.; Chen, Y.; Park, J.; Kim, H.J.; Rah, S.Y.; Ryu, J.; Cho, G.J.; Choi, H.S.; Ryter, S.W.; Park, J.W.; et al. Cross-talk between CD38 and TTP Is Essential for Resolution of Inflammation during Microbial Sepsis. Cell Rep. 2020, 30, 1063–1076.e5. [Google Scholar] [CrossRef]
- Kato, I.; Yamamoto, Y.; Fujimura, M.; Noguchi, N.; Takasawa, S.; Okamoto, H. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J. Biol. Chem. 1999, 274, 1869–1872. [Google Scholar] [CrossRef]
- Partida-Sánchez, S.; Cockayne, D.A.; Monard, S.; Jacobson, E.L.; Oppenheimer, N.; Garvy, B.; Kusser, K.; Goodrich, S.; Howard, M.; Harmsen, A.; et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 2001, 7, 1209–1216. [Google Scholar] [CrossRef]
- Jin, D.; Liu, H.X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Weiss, R.; Schilling, E.; Grahnert, A.; Kölling, V.; Dorow, J.; Ceglarek, U.; Sack, U.; Hauschildt, S. Nicotinamide: A vitamin able to shift macrophage differentiation toward macrophages with restricted inflammatory features. Innate Immun. 2015, 21, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Kilgour, M.K.; MacPherson, S.; Zacharias, L.G.; Ellis, A.E.; Sheldon, R.D.; Liu, E.Y.; Keyes, S.; Pauly, B.; Carleton, G.; Allard, B.; et al. 1-Methylnicotinamide is an immune regulatory metabolite in human ovarian cancer. Sci. Adv. 2021, 7, eabe1174. [Google Scholar] [CrossRef]
- Varella Morandi Junqueira-Franco, M.; Ernesto Troncon, L.; Garcia Chiarello, P.; do Rosário Del Lama Unamuno, M.; Afonso Jordao, A.; Vannucchi, H. Intestinal permeability and oxidative stress in patients with alcoholic pellagra. Clin. Nutr. 2006, 25, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Pellagra and alcoholism: A biochemical perspective. Alcohol Alcohol. 2014, 49, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, E.; Bassotti, G.; Tramontana, M.; Hansel, K.; Stingeni, L.; Ardizzone, S.; Genovese, G.; Marzano, A.V.; Maconi, G. Dermatological Manifestations in Inflammatory Bowel Diseases. J. Clin. Med. 2021, 10, 364. [Google Scholar] [CrossRef]
- Atzori, L.; Recalcati, S.; Ferreli, C.; Hoenig, L.J.; Rongioletti, F. COVID-19-related skin manifestations: Update on therapy. Clin. Dermatol. 2021, 39, 920–926. [Google Scholar] [CrossRef]
- Nuno-Gonzalez, A.; Martin-Carrillo, P.; Magaletsky, K.; Martin Rios, M.D.; Herranz Mañas, C.; Artigas Almazan, J.; García Casasola, G.; Perez Castro, E.; Gallego Arenas, A.; Mayor Ibarguren, A.; et al. Prevalence of mucocutaneous manifestations in 666 patients with COVID-19 in a field hospital in Spain: Oral and palmoplantar findings. Br. J. Dermatol. 2021, 184, 184–185. [Google Scholar] [CrossRef]
- Joshi, T.; Ahmed, A.; Cholankeril, G. Gastrointestinal manifestations of coronavirus disease 2019. Curr. Opin. Infect. Dis. 2021, 34, 471–476. [Google Scholar] [CrossRef]
- Baeck, M.; Herman, A. COVID toes: Where do we stand with the current evidence? Int. J. Infect. Dis. 2021, 102, 53–55. [Google Scholar] [CrossRef]
- Díaz Rodríguez, M.; Jimenez Romera, A.; Villarroel, M. Oral manifestations associated with COVID-19. Oral Dis. 2020. [Google Scholar] [CrossRef]
- Hathway, R.W. COVID tongue. Br. Dent. J. 2021, 230, 114. [Google Scholar] [CrossRef] [PubMed]
- Lipsker, D. Paraviral eruptions in the era of COVID-19: Do some skin manifestations point to a natural resistance to SARS-CoV-2? Clin. Dermatol. 2020, 38, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Chichakli, H.; Frosch, P.J.; Brinkmeier, T. Symmetrische bullöse akrale Erytheme bei einer 58-jährigen alkoholabhängigen Frau [Symmetrical bullous acral erythema in a 58-year-old female alcoholic]. Hautarzt 2006, 57, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Bakwin, H.; Reardon, H.S.; Winn, J.S.; Tenbrinck, M.S.; Stern, M.L.; Engel, M.G. Relation of lesions of the tongue in children to niacin deficiency. Am. J. Dis. Child. 1947, 74, 657–668. [Google Scholar] [CrossRef]
- Klopfenstein, T.; Zahra, H.; Kadiane-Oussou, N.J.; Lepiller, Q.; Royer, P.Y.; Toko, L.; Gendrin, V.; Zayet, S. New loss of smell and taste: Uncommon symptoms in COVID-19 patients on Nord Franche-Comte cluster, France. Int. J. Infect. Dis. 2020, 100, 117–122. [Google Scholar] [CrossRef]
- Rocke, J.; Hopkins, C.; Philpott, C.; Kumar, N. Is loss of sense of smell a diagnostic marker in COVID-19: A systematic review and meta-analysis. Clin. Otolaryngol. 2020, 45, 914–922. [Google Scholar] [CrossRef]
- Agyeman, A.A.; Chin, K.L.; Landersdorfer, C.B.; Liew, D.; Ofori-Asenso, R. Smell and Taste Dysfunction in Patients With COVID-19: A Systematic Review and Meta-analysis. Mayo Clin. Proc. 2020, 95, 1621–1631. [Google Scholar] [CrossRef]
- Green, R.F. Subclinical pellagra and idiopathic hypogeusia. JAMA 1971, 218, 1303. [Google Scholar] [CrossRef] [PubMed]
- Quintana, D.S.; Rokicki, J.; van der Meer, D.; Alnæs, D.; Kaufmann, T.; Córdova-Palomera, A.; Dieset, I.; Andreassen, O.A.; Westlye, L.T. Oxytocin pathway gene networks in the human brain. Nat. Commun. 2019, 10, 668. [Google Scholar] [CrossRef]
- De Flora, A.; Zocchi, E.; Guida, L.; Franco, L.; Bruzzone, S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N.Y. Acad. Sci. 2004, 1028, 176–191. [Google Scholar] [CrossRef]
- Sinclair, M.S.; Perea-Martinez, I.; Dvoryanchikov, G.; Yoshida, M.; Nishimori, K.; Roper, S.D.; Chaudhari, N. Oxytocin signaling in mouse taste buds. PLoS ONE 2010, 5, e11980. [Google Scholar] [CrossRef] [PubMed]
- Diep, P.T.; Talash, K.; Kasabri, V. Hypothesis: Oxytocin is a direct COVID-19 antiviral. Med. Hypotheses 2020, 145, 110329. [Google Scholar] [CrossRef] [PubMed]
- Imami, A.S.; O’Donovan, S.M.; Creeden, J.F.; Wu, X.; Eby, H.; McCullumsmith, C.B.; Uvnäs-Moberg, K.; McCullumsmith, R.E.; Andari, E. Oxytocin’s anti-inflammatory and proimmune functions in COVID-19: A transcriptomic signature-based approach. Physiol. Genom. 2020, 52, 401–407. [Google Scholar] [CrossRef]
- Thakur, P.; Shrivastava, R.; Shrivastava, V.K. Oxytocin as a Potential Adjuvant Against COVID-19 Infection. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Diep, P.T. TRPV1, Nrf2, and COVID-19: Could Oxytocin Have a Beneficial Role to Play? Int. Arch. Allergy Immunol. 2022, 183, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Carson, D.S.; Berquist, S.W.; Trujillo, T.H.; Garner, J.P.; Hannah, S.L.; Hyde, S.A.; Sumiyoshi, R.D.; Jackson, L.P.; Moss, J.K.; Strehlow, M.C.; et al. Cerebrospinal fluid and plasma oxytocin concentrations are positively correlated and negatively predict anxiety in children. Mol. Psychiatry 2015, 20, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Kagerbauer, S.M.; Gempt, J.; Podtschaske, A.; Hapfelmeier, A.; Schneider, G. Oxytocin levels in saliva correlate better than plasma levels with concentrations in the cerebrospinal fluid of patients in neurocritical care. J. Neuroendocrinol. 2018, 30, e12596. [Google Scholar] [CrossRef]
- Elabd, C.; Cousin, W.; Upadhyayula, P.; Chen, R.Y.; Chooljian, M.S.; Li, J.; Kung, S.; Jiang, K.P.; Conboy, I.M. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat. Commun. 2014, 5, 4082. [Google Scholar] [CrossRef]
- Plasencia, G.; Luedicke, J.M.; Nazarloo, H.P.; Carter, C.S.; Ebner, N.C. Plasma oxytocin and vasopressin levels in young and older men and women: Functional relationships with attachment and cognition. Psychoneuroendocrinology 2019, 110, 104419. [Google Scholar] [CrossRef]
- Weisman, O.; Zagoory-Sharon, O.; Schneiderman, I.; Gordon, I.; Feldman, R. Plasma oxytocin distributions in a large cohort of women and men and their gender-specific associations with anxiety. Psychoneuroendocrinology 2013, 38, 694–701. [Google Scholar] [CrossRef]
- Gerasimenko, M.; Cherepanov, S.M.; Furuhara, K.; Lopatina, O.; Salmina, A.B.; Shabalova, A.A.; Tsuji, C.; Yokoyama, S.; Ishihara, K.; Brenner, C.; et al. Nicotinamide riboside supplementation corrects deficits in oxytocin, sociability and anxiety of CD157 mutants in a mouse model of autism spectrum disorder. Sci. Rep. 2020, 10, 10035. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Cacicedo, J.M.; Ido, Y. Impaired nicotinamide adenine dinucleotide (NAD+) metabolism in diabetes and diabetic tissues: Implications for nicotinamide-related compound treatment. J. Diabetes Investig. 2020, 11, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Zhu, T.; Tang, B.; Yu, S.; Hu, H.; Sun, W.; Pan, R.; Wang, J.; Wang, D.; Yang, L.; et al. Decreased circulating levels of oxytocin in obesity and newly diagnosed type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2014, 99, 4683–4689. [Google Scholar] [CrossRef] [PubMed]
- Gouveri, E.; Katotomichelakis, M.; Gouveris, H.; Danielides, V.; Maltezos, E.; Papanas, N. Olfactory dysfunction in type 2 diabetes mellitus: An additional manifestation of microvascular disease? Angiology 2014, 65, 869–876. [Google Scholar] [CrossRef]
- Yazla, S.; Özmen, S.; Kıyıcı, S.; Yıldız, D.; Haksever, M.; Gencay, S. Evaluation of olfaction and taste function in type 2 diabetic patients with and without peripheral neuropathy. Diabetes Metab. Res. Rev. 2018, 34, e2973. [Google Scholar] [CrossRef]
- Catamo, E.; Tornese, G.; Concas, M.P.; Gasparini, P.; Robino, A. Differences in taste and smell perception between type 2 diabetes mellitus patients and healthy controls. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 193–200. [Google Scholar] [CrossRef]
- Gouveri, E.; Papanas, N. Olfactory Dysfunction: A Complication of Diabetes or a Factor That Complicates Glucose Metabolism? A Narrative Review. J. Clin. Med. 2021, 10, 5637. [Google Scholar] [CrossRef]
- Nersesyan, Y.; Demirkhanyan, L.; Cabezas-Bratesco, D.; Oakes, V.; Kusuda, R.; Dawson, T.; Sun, X.; Cao, C.; Cohen, A.M.; Chelluboina, B.; et al. Oxytocin Modulates Nociception as an Agonist of Pain-Sensing TRPV1. Cell Rep. 2017, 21, 1681–1691. [Google Scholar] [CrossRef]
- Song, W.J.; Hui, C.K.M.; Hull, J.H.; Birring, S.S.; McGarvey, L.; Mazzone, S.B.; Chung, K.F. Confronting COVID-19-associated cough and the post-COVID syndrome: Role of viral neurotropism, neuroinflammation, and neuroimmune responses. Lancet Respir. Med. 2021, 9, 533–544. [Google Scholar] [CrossRef]
- Okamoto, H. Recent advances in physiological and pathological significance of tryptophan-NAD+ metabolites: Lessons from insulin-producing pancreatic β-cells. Adv. Exp. Med. Biol. 2003, 527, 243–252. [Google Scholar] [CrossRef]
- Takasawa, S.; Nata, K.; Yonekura, H.; Okamoto, H. Cyclic ADP-ribose in insulin secretion from pancreatic β cells. Science 1993, 259, 370–373. [Google Scholar] [CrossRef]
- Kato, I.; Takasawa, S.; Akabane, A.; Tanaka, O.; Abe, H.; Takamura, T.; Suzuki, Y.; Nata, K.; Yonekura, H.; Yoshimoto, T.; et al. Regulatory role of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in insulin secretion by glucose in pancreatic β cells. Enhanced insulin secretion in CD38-expressing transgenic mice. J. Biol. Chem. 1995, 270, 30045–30050. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.L.; Islam, M.S.; Efanov, A.M.; Brown, G.; Köhler, M.; Larsson, O.; Berggren, P.O. Insulin exocytosis and glucose-mediated increase in cytoplasmic free Ca2+ concentration in the pancreatic β-cell are independent of cyclic ADP-ribose. J. Biol. Chem. 1996, 271, 19074–19079. [Google Scholar] [CrossRef]
- Johnson, J.D.; Misler, S. Nicotinic acid-adenine dinucleotide phosphate-sensitive calcium stores initiate insulin signaling in human β cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14566–14571. [Google Scholar] [CrossRef] [PubMed]
- Heister, P.M.; Powell, T.; Galione, A. Glucose and NAADP trigger elementary intracellular β-cell Ca2+ signals. Sci. Rep. 2021, 11, 10714. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Hug, C.; Lodish, H.F. Medicine. Visfatin: A new adipokine. Science 2005, 307, 366–367. [Google Scholar] [CrossRef]
- Accili, D. Can COVID-19 cause diabetes? Nat. Metab. 2021, 3, 123–125. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Powers, A.C. Distinguishing the real from the hyperglycaemia: Does COVID-19 induce diabetes? Lancet Diabetes Endocrinol. 2021, 9, 328–329. [Google Scholar] [CrossRef]
- Drucker, D.J. Diabetes, obesity, metabolism, and SARS-CoV-2 infection: The end of the beginning. Cell Metab. 2021, 33, 479–498. [Google Scholar] [CrossRef]
- Imai, S.I. The NAD World 2.0: The importance of the inter-tissue communication mediated by NAMPT/NAD+/SIRT1 in mammalian aging and longevity control. NPJ Syst. Biol. Appl. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef]
- Riederer, M.; Erwa, W.; Zimmermann, R.; Frank, S.; Zechner, R. Adipose tissue as a source of nicotinamide N-methyltransferase and homocysteine. Atherosclerosis 2009, 204, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Roli, L.; Oliva, G.; Manfredini, M.; Trenti, T.; Kaleci, S.; Iannella, R.; Balzano, B.; Coppola, A.; Fiorentino, G.; et al. Homocysteine (Hcy) assessment to predict outcomes of hospitalized COVID-19 patients: A multicenter study on 313 COVID-19 patients. Clin. Chem. Lab. Med. 2021, 59, e354–e357. [Google Scholar] [CrossRef]
- Yang, Z.; Shi, J.; He, Z.; Lü, Y.; Xu, Q.; Ye, C.; Chen, S.; Tang, B.; Yin, K.; Lu, Y.; et al. Predictors for imaging progression on chest CT from coronavirus disease 2019 (COVID-19) patients. Aging 2020, 12, 6037–6048. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD+ metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef]
- Ponticos, M.; Holmes, A.M.; Shi-wen, X.; Leoni, P.; Khan, K.; Rajkumar, V.S.; Hoyles, R.K.; Bou-Gharios, G.; Black, C.M.; Denton, C.P.; et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009, 60, 2142–2155. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, Ł.; Wojnar-Lason, K.; Kaczara, P.; Tworzydło, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef]
- Hong, S.; Moreno-Navarrete, J.M.; Wei, X.; Kikukawa, Y.; Tzameli, I.; Prasad, D.; Lee, Y.; Asara, J.M.; Fernandez-Real, J.M.; Maratos-Flier, E.; et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through SIRT1 protein stabilization. Nat. Med. 2015, 21, 887–894. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Kheterpal, I.; Kennedy, N.; Davis, R.J.; Ye, J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem. 2011, 286, 22227–22234. [Google Scholar] [CrossRef] [PubMed]
- Maltos, A.L.; Portari, G.V.; Moraes, G.V.; Monteiro, M.C.; Vannucchi, H.; da Cunha, D.F. Niacin metabolism and indoleamine 2,3-dioxygenase activation in malnourished patients with flaky paint dermatosis. Nutrition 2015, 31, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Spudich, S.; Nath, A. Nervous system consequences of COVID-19. Science 2022, 375, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Lemle, M.D.; Komaroff, A.L.; Snyder, S.H. Redox imbalance links COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2021, 118, e2024358118. [Google Scholar] [CrossRef]
- Vidugiriene, J.; Leippe, D.; Sobol, M.; Vidugiris, G.; Zhou, W.; Meisenheimer, P.; Gautam, P.; Wennerberg, K.; Cali, J.J. Bioluminescent cell-based NAD(P)/NAD(P)H assays for rapid dinucleotide measurement and inhibitor screening. Assay Drug Dev. Technol. 2014, 12, 514–526. [Google Scholar] [CrossRef]
- Garattini, E.; Terao, M. The role of aldehyde oxidase in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2012, 8, 487–503. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novak Kujundžić, R. COVID-19: Are We Facing Secondary Pellagra Which Cannot Simply Be Cured by Vitamin B3? Int. J. Mol. Sci. 2022, 23, 4309. https://doi.org/10.3390/ijms23084309
Novak Kujundžić R. COVID-19: Are We Facing Secondary Pellagra Which Cannot Simply Be Cured by Vitamin B3? International Journal of Molecular Sciences. 2022; 23(8):4309. https://doi.org/10.3390/ijms23084309
Chicago/Turabian StyleNovak Kujundžić, Renata. 2022. "COVID-19: Are We Facing Secondary Pellagra Which Cannot Simply Be Cured by Vitamin B3?" International Journal of Molecular Sciences 23, no. 8: 4309. https://doi.org/10.3390/ijms23084309
APA StyleNovak Kujundžić, R. (2022). COVID-19: Are We Facing Secondary Pellagra Which Cannot Simply Be Cured by Vitamin B3? International Journal of Molecular Sciences, 23(8), 4309. https://doi.org/10.3390/ijms23084309