
Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity—Pathogenesis and Involvement of the Extracellular Matrix


Abstract
1. Introduction
2. Materials and Methods
3. Obesity as a Heterogenous Disorder
4. Distinctive Characteristics of the Cardiovascular System in the Course of Obesity
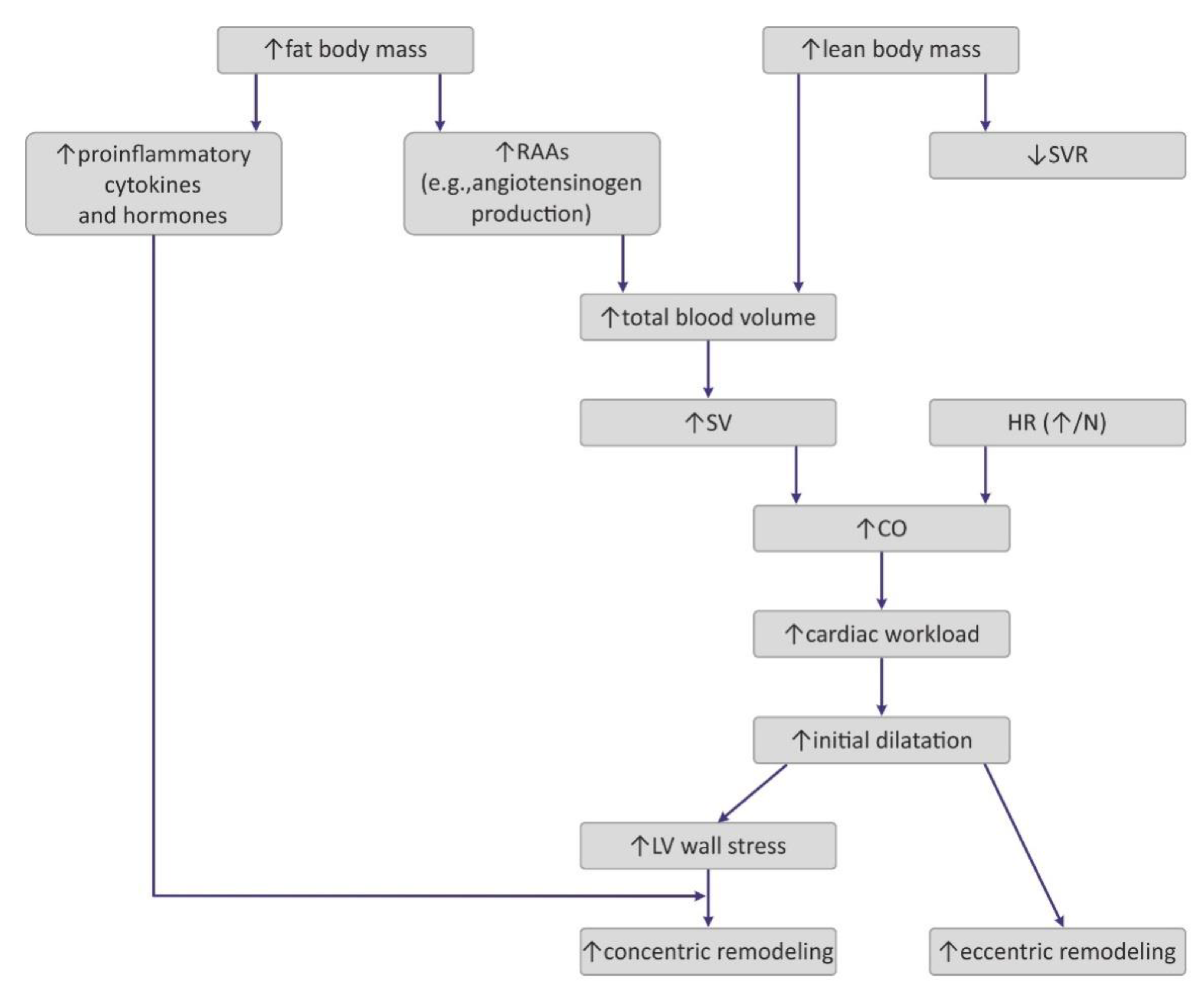
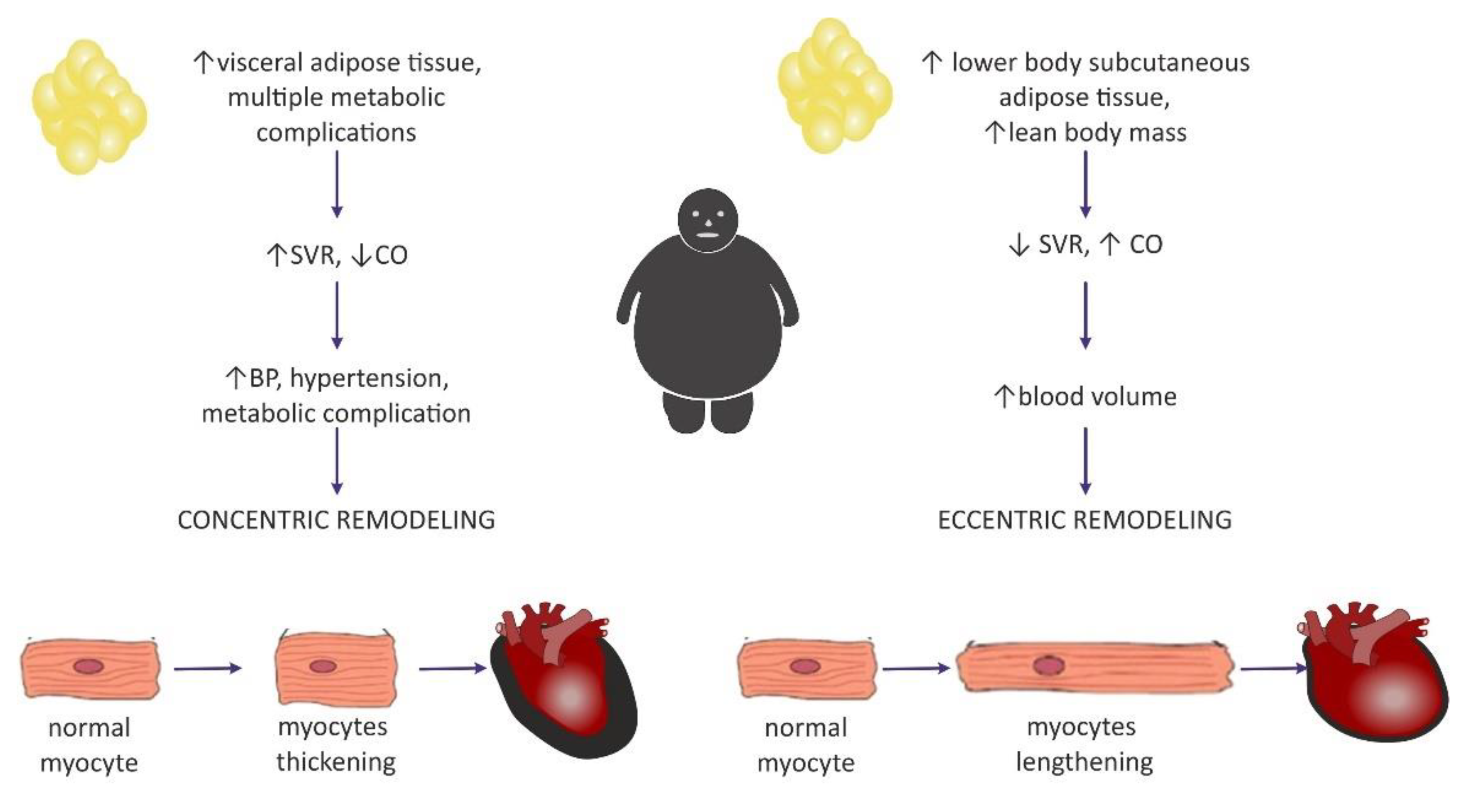
4.1. Hemodynamic Changes Observed in Obesity
4.2. The Impact of Obesity on Myocardial Geometry and the Ejection Fraction
5. Myocardial Extracellular Matrix
5.1. Collagen
5.2. Metalloproteinases (MMPs)
5.3. Tissue Inhibitors of Metalloproteinase (TIMPs)
6. Fundamentals of Heart Fibrosis
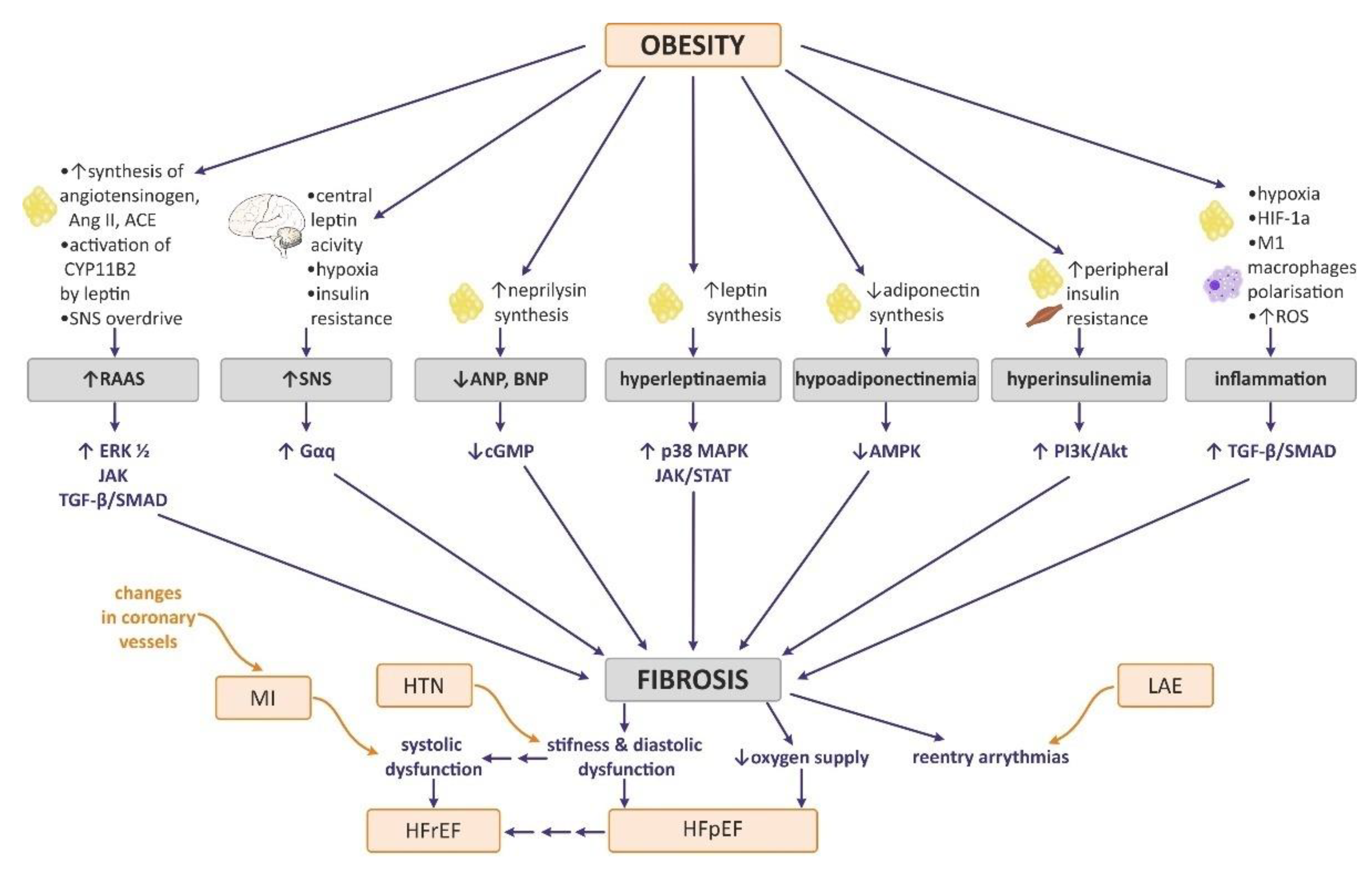
7. The Effect of Obesity on Myocardial ECM Expression, Heart Fibrosis, and Cardiac Hypertrophy
7.1. Neurohormonal Changes in Obesity Attributable to Heart Remodeling
7.1.1. Sympathetic Nervous System (SNS)
7.1.2. Renin–Angiotensin–Aldosterone System (RAAs)
7.1.3. Natriuretic Peptides
7.1.4. Hyperinsulinemia and Insulin Resistance
7.2. Influence of Obesity-Related Tissue Inflammation and Hypoxia on Myocardial ECM Expression and Heart Fibrosis
7.3. The Effect of Selected Adipokines on Myocardial ECM Expression and Heart Fibrosis
7.3.1. Leptin
7.3.2. Adiponectin
8. Heart Remodeling in Selected Cardiovascular Diseases
8.1. Hypertension
8.2. Myocardial Infarction(MI)
8.3. Heart Failure (HF)
8.4. Atrial Fibrillation (AF)
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mahajan, R.; Lau, D.H.; Sanders, P. Impact of Obesity on Cardiac Metabolism, Fibrosis, and Function. Trends Cardiovasc. Med. 2015, 25, 119–126. [Google Scholar] [CrossRef]
- Yumuk, V.; Frühbeck, G.; Oppert, J.M.; Woodward, E.; Toplak, H. An EASO Position Statement on Multidisciplinary Obesity Management in Adults. Obes. Facts 2014, 7, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Kachur, S.; Lavie, C.J.; de Schutter, A.; Milani, R.V.; Ventura, H.O. Obesity and Cardiovascular Diseases. Minerva Med. 2017, 108, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative Stress and Inflammation as Central Mediators of Atrial Fibrillation in Obesity and Diabetes. Cardiovasc. Diabetol. 2017, 16, 120. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Pandey, A.; Lau, D.H.; Alpert, M.A.; Sanders, P. Obesity and Atrial Fibrillation Prevalence, Pathogenesis, and Prognosis: Effects of Weight Loss and Exercise. J. Am. Coll. Cardiol. 2017, 70, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.F.; Kachur, S.M.; Lavie, C.J. Hypertension in Obesity. Curr. Opin. Cardiol. 2020, 35, 389–396. [Google Scholar] [CrossRef]
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure with Preserved Ejection Fraction. Circulation 2017, 136, 6–19. [Google Scholar] [CrossRef]
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806. [Google Scholar] [CrossRef]
- Alpert, M.A.; Karthikeyan, K.; Abdullah, O.; Ghadban, R. Obesity and Cardiac Remodeling in Adults: Mechanisms and Clinical Implications. Prog. Cardiovasc. Dis. 2018, 61, 114–123. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular Pathways Underlying Cardiac Remodeling during Pathophysiological Stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular Matrix Remodeling and Cardiac Fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Fan, D.; Basu, R.; Kandalam, V.; Kassiri, Z. Tissue Inhibitor of Metalloproteinases (TIMPs) in Heart Failure. Heart Fail. Rev. 2012, 17, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS Guideline for the Management of Overweight and Obesity in Adults. Circulation 2014, 129, S102–S138. [Google Scholar] [CrossRef] [PubMed]
- Bessesen, D.H. Update on Obesity. J. Clin. Endocrinol. Metab. 2008, 93, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Weir, C.B.; Jan, A. BMI Classification Percentile and Cut Off Points; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef]
- Jan, A.; Weir, C.B. BMI Classification Percentile and Cut Off Points; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kaila, B.; Raman, M. Obesity: A Review of Pathogenesis and Management Strategies. Can. J. Gastroenterol. 2008, 22, 61–68. [Google Scholar] [CrossRef]
- Pi-Sunyer, F.X. The Obesity Epidemic: Pathophysiology and Consequences of Obesity. Obes. Res. 2002, 10, 97S–104S. [Google Scholar] [CrossRef]
- Mayoral, L.P.C.; Andrade, G.M.; Mayoral, E.P.C.; Huerta, T.H.; Canseco, S.P.; Rodal Canales, F.J.; Cabrera-Fuentes, H.A.; Cruz, M.M.; Pérez Santiago, A.D.; Alpuche, J.J.; et al. Obesity Subtypes, Related Biomarkers & Heterogeneity. Indian J. Med. Res. 2020, 151, 11–21. [Google Scholar]
- Lee, D.C.; Shook, R.P.; Drenowatz, C.; Blair, S.N. Physical Activity and Sarcopenic Obesity: Definition, Assessment, Prevalence and Mechanism. Future Sci. OA 2016, 2, FSO127. [Google Scholar] [CrossRef]
- Sellayah, D.; Cagampang, F.R.; Cox, R.D. On the Evolutionary Origins of Obesity: A New Hypothesis. Endocrinology 2014, 155, 1573–1588. [Google Scholar] [CrossRef]
- Neeland, I.J.; Poirier, P.; Després, J.P. Cardiovascular and Metabolic Heterogeneity of Obesity: Clinical Challenges and Implications for Management. Circulation 2018, 137, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.R.; Guarnieri, A.R.; Gozdiff, A.; Helsley, R.N.; Owens, A.P.; Tranter, M. Mechanisms Linking Adipose Tissue Inflammation to Cardiac Hypertrophy and Fibrosis. Clin. Sci. 2019, 133, 2329–2344. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac Remodeling in Obesity. Physiol. Rev. 2008, 88, 389–419. [Google Scholar] [CrossRef]
- de Simone, G.; Izzo, R.; de Luca, N.; Gerdts, E. Left Ventricular Geometry in Obesity: Is It What We Expect? Nutr. Metab. Cardiovasc. Dis. 2013, 23, 905–912. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C. Obesity and Heart Failure with Preserved Ejection Fraction: A Paradox or Something Else? Heart Fail. Rev. 2019, 24, 379–385. [Google Scholar] [CrossRef]
- Murdolo, G.; Angeli, F.; Reboldi, G.; di Giacomo, L.; Aita, A.; Bartolini, C.; Vedecchia, P. Left Ventricular Hypertrophy and Obesity: Only a Matter of Fat? High Blood Press. Cardiovasc. Prev. 2015, 22, 29–41. [Google Scholar] [CrossRef]
- Hatem, S.N.; Sanders, P. Epicardial Adipose Tissue and Atrial Fibrillation. Cardiovasc. Res. 2014, 102, 205–213. [Google Scholar] [CrossRef]
- Krishnan, A.; Chilton, E.; Raman, J.; Saxena, P.; McFarlane, C.; Trollope, A.F.; Kinobe, R.; Chilton, L. Are Interactions between Epicardial Adipose Tissue, Cardiac Fibroblasts and Cardiac Myocytes Instrumental in Atrial Fibrosis and Atrial Fibrillation? Cells 2021, 10, 2501. [Google Scholar] [CrossRef]
- Patel, K.H.K.; Hwang, T.; Se Liebers, C.; Ng, F.S. Epicardial Adipose Tissue as a Mediator of Cardiac Arrhythmias. Am. J. Physiol.-Heart Circ. Physiol. 2022, 322, H129–H144. [Google Scholar] [CrossRef] [PubMed]
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; Leprince, P.; Dutour, A.; Clément, K.; et al. Human Epicardial Adipose Tissue Induces Fibrosis of the Atrial Myocardium through the Secretion of Adipo-Fibrokines. Eur. Heart J. 2015, 36, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Teshima, Y.; Kondo, H.; Kaku, H.; Kira, S.; Ikebe, Y.; Saito, S.; Fukui, A.; Shinohara, T.; Yufu, K.; et al. Association of Fibrotic Remodeling and Cytokines/Chemokines Content in Epicardial Adipose Tissue with Atrial Myocardial Fibrosis in Patients with Atrial Fibrillation. Heart Rhythm 2018, 15, 1717–1727. [Google Scholar] [CrossRef]
- Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of Lipotoxicity in the Cardiovascular System. Curr. Hypertens. Rep. 2012, 14, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, E.; Schwenk, R.W.; Glatz, J.F.C.; Luiken, J.J.F.P.; van Eys, G.J.J.M. High Fat Diet Induced Diabetic Cardiomyopathy. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Karbowska, J.; Kochan, Z. Leptin as a Mediator between Obesity and Cardiac Dysfunction. Postępy Hig. I Med. Doświadczalnej 2012, 66, 267–274. [Google Scholar] [CrossRef]
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and Mechanisms of Myocardial Lipotoxicity in Obesity. J. Intern. Med. 2018, 284, 478–491. [Google Scholar] [CrossRef]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic Heart Disease in Obese Rats: Implications for Human Obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef]
- Bielawska, A.E.; Shapiro, J.P.; Jiang, L.; Melkonyan, H.S.; Piot, C.; Wolfe, C.L.; Tomei, L.D.; Hannun, Y.A.; Umansky, S.R. Ceramide Is Involved in Triggering of Cardiomyocyte Apoptosis Induced by Ischemia and Reperfusion. Am. J. Pathol. 1997, 151, 1257–1263. [Google Scholar]
- Csonka, C.; Sárközy, M.; Pipicz, M.; Dux, L.; Csont, T. Modulation of Hypercholesterolemia-Induced Oxidative/Nitrative Stress in the Heart. Oxidative Med. Cell. Longev. 2016, 2016, 3863726. [Google Scholar] [CrossRef] [PubMed]
- Alpert, M.A.; Omran, J.; Bostick, B.P. Effects of Obesity on Cardiovascular Hemodynamics, Cardiac Morphology, and Ventricular Function. Curr. Obes. Rep. 2016, 5, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Ebong, I.A.; Goff, D.C.; Rodriguez, C.J.; Chen, H.; Bertoni, A.G. Mechanisms of Heart Failure in Obesity. Obes. Res. Clin. Pract. 2014, 8, e540–e548. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H.; Commerford, P.J.; Gersh, B.J.; Pfeffer, M.A. Controversies in Ventricular Remodelling. Lancet 2006, 367, 356–367. [Google Scholar] [CrossRef]
- Neeland, I.J.; Gupta, S.; Ayers, C.R.; Turer, A.T.; Rame, J.E.; Das, S.R.; Berry, J.D.; Khera, A.; McGuire, D.K.; Vega, G.L.; et al. Relation of Regional Fat Distribution to Left Ventricular Structure and Function. Circ. Cardiovasc. Imaging 2013, 6, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J.; Pu, H.; He, W.; Zhou, X.; Tong, N.; Peng, L. Cardiac Remodeling and Subclinical Left Ventricular Dysfunction in Adults with Uncomplicated Obesity: A Cardiovascular Magnetic Resonance Study. Quant. Imaging Med. Surg. 2022, 12, 2035–2050. [Google Scholar] [CrossRef] [PubMed]
- Hammond, I.W.; Devereux, R.B.; Alderman, M.H.; Laragh, J.H. Relation of Blood Pressure and Body Build to Left Ventricular Mass in Normotensive and Hypertensive Employed Adults. J. Am. Coll. Cardiol. 1988, 12, 996–1004. [Google Scholar] [CrossRef]
- Lauer, M.S.; Anderson, K.M.; Kannel, W.B.; Levy, D. The Impact of Obesity on Left Ventricular Mass and Geometry: The Framingham Heart Study. JAMA J. Am. Med. Assoc. 1991, 266, 231–236. [Google Scholar] [CrossRef]
- Jongjirasiri, S.; Waeosak, P.; Laothamatas, J.; Sritara, C.; Saengruang-Orn, S. Effect of Obesity on Left Ventricular Mass: Results from 320 Multi-Slices Computed Tomography. J. Med. Assoc. Thail. 2017, 100, 219–229. [Google Scholar]
- Bombelli, M.; Facchetti, R.; Sega, R.; Carugo, S.; Fodri, D.; Brambilla, G.; Giannattasio, C.; Grassi, G.; Mancia, G. Impact of Body Mass Index and Waist Circumference on the Long-Term Risk of Diabetes Mellitus, Hypertension, and Cardiac Organ Damage. Hypertension 2011, 58, 1029–1035. [Google Scholar] [CrossRef]
- Lee, T.C.; Jin, Z.; Homma, S.; Nakanishi, K.; Elkind, M.S.V.; Rundek, T.; Tugcu, A.; Matsumoto, K.; Sacco, R.L.; di Tullio, M.R. Changes in Left Ventricular Mass and Geometry in the Older Adults: Role of Body Mass and Central Obesity. J. Am. Soc. Echocardiogr. 2019, 32, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Woodiwiss, A.J.; Libhaber, C.D.; Majane, O.H.I.; Libhaber, E.; Maseko, M.; Norton, G.R. Obesity Promotes Left Ventricular Concentric Rather than Eccentric Geometric Remodeling and Hypertrophy Independent of Blood Pressure. Am. J. Hypertens. 2008, 21, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Cuspidi, C.; Rescaldani, M.; Sala, C.; Grassi, G. Left-Ventricular Hypertrophy and Obesity: A Systematic Review and Meta-Analysis of Echocardiographic Studies. J. Hypertens. 2014, 32, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Antonetti, I.; Bisognano, J.D.; Sloand, J. Obesity-Related Cardiorenal Syndrome. J. Clin. Hypertens. 2010, 12, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Pascual, D.A.; Soria, F.; Vicente, T.; Hernández, A.M.; Tébar, F.J.; Valdés, M. Effects of Isolated Obesity on Systolic and Diastolic Left Ventricular Function. Heart 2003, 89, 1152–1156. [Google Scholar] [CrossRef]
- Khan, M.F.; Movahed, M.R. Obesity Cardiomyopathy and Systolic Function: Obesity Is Not Independently Associated with Dilated Cardiomyopathy. Heart Fail. Rev. 2013, 18, 207–217. [Google Scholar] [CrossRef]
- Oh, A.; Okazaki, R.; Sam, F.; Valero-Muñoz, M. Heart Failure With Preserved Ejection Fraction and Adipose Tissue: A Story of Two Tales. Front. Cardiovasc. Med. 2019, 6, 110. [Google Scholar] [CrossRef]
- Wong, C.Y.; O’Moore-Sullivan, T.; Leano, R.; Hukins, C.; Jenkins, C.; Marwick, T.H. Association of Subclinical Right Ventricular Dysfunction with Obesity. J. Am. Coll. Cardiol. 2006, 47, 611–616. [Google Scholar] [CrossRef]
- Krebber, M.M.; van Dijk, C.G.M.; Vernooij, R.W.M.; Brandt, M.M.; Emter, C.A.; Rau, C.D.; Fledderus, J.O.; Duncker, D.J.; Verhaar, M.C.; Cheng, C.; et al. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Extracellular Matrix Remodeling during Left Ventricular Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 6742. [Google Scholar] [CrossRef]
- Linssen, P.B.C.; Brunner-La Rocca, H.P.; Schalkwijk, C.G.; Beulens, J.W.J.; Elders, P.J.M.; van der Heijden, A.A.; Slieker, R.C.; Stehouwer, C.D.A.; Henry, R.M.A. Serum Matrix Metalloproteinases and Left Atrial Remodeling—The Hoorn Study. Int. J. Mol. Sci. 2020, 21, 4944. [Google Scholar] [CrossRef]
- Stritzke, J.; Markus, M.R.P.; Duderstadt, S.; Lieb, W.; Luchner, A.; Döring, A.; Keil, U.; Hense, H.W.; Schunkert, H. The Aging Process of the Heart: Obesity Is the Main Risk Factor for Left Atrial Enlargement During Aging. The MONICA/KORA (Monitoring of Trends and Determinations in Cardiovascular Disease/Cooperative Research in the Region of Augsburg) Study. J. Am. Coll. Cardiol. 2009, 54, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Heeneman, S.; Cleutjens, J.P.; Faber, B.C.; Creemers, E.E.; van Suylen, R.J.; Lutgens, E.; Cleutjens, K.B.; Daemen, M.J. The Dynamic Extracellular Matrix: Intervention Strategies during Heart Failure and Atherosclerosis. J. Pathol. 2003, 200, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Tung, Y.C.; Jung, S.M.; Chu, Y.; Lin, P.J.; Kao, W.W.Y.; Chu, P.H. Lumican-Null Mice Are Susceptible to Aging and Isoproterenol-Induced Myocardial Fibrosis. Biochem. Biophys. Res. Commun. 2017, 482, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Tyagi, S.C.; Cleutjens, J.P.M. Collagen Network of the Myocardium: Function, Structural Remodeling and Regulatory Mechanisms. J. Mol. Cell. Cardiol. 1994, 26, 279–292. [Google Scholar] [CrossRef]
- Chute, M.; Aujla, P.; Jana, S.; Kassiri, Z. The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis. J. Cardiovasc. Dev. Dis. 2019, 6, 35. [Google Scholar] [CrossRef]
- Spinale, F.G. Myocardial Matrix Remodeling and the Matrix Metalloproteinases: Influence on Cardiac Form and Function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef]
- Fedak, P.W.M.; Verma, S.; Weisel, R.D.; Li, R.-K. Cardiac Remodeling and Failure. Cardiovasc. Pathol. 2005, 14, 49–60. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac Fibroblasts, Fibrosis and Extracellular Matrix Remodeling in Heart Disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef]
- Ju, H.; Dixon, I.M.C. Cardiac Extracellular Matrix and Its Role in the Development of Heart Failure. In Mechanisms of Heart Failure; Springer: Boston, MA, USA, 1995. [Google Scholar]
- Miles, C.; Westaby, J.; Ster, I.C.; Asimaki, A.; Boardman, P.; Joshi, A.; Papadakis, M.; Sharma, S.; Behr, E.R.; Sheppard, M.N. Morphometric Characterization of Collagen and Fat in Normal Ventricular Myocardium. Cardiovasc. Pathol. 2020, 48, 107224. [Google Scholar] [CrossRef]
- Prockop, D.J.; Kivirikko, K.I. Collagens: Molecular Biology, Diseases, and Potentials for Therapy. Annu. Rev. Biochem. 1995, 64, 403–434. [Google Scholar] [CrossRef] [PubMed]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and Heart Failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac Fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G.; Gunasinghe, H.; Sprunger, P.D.; Baskin, J.M.; Bradham, W.C. Extracellular Degradative Pathways in Myocardial Remodeling and Progression to Heart Failure. Proc. J. Card. Fail. 2002, 8, S332–S338. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Schroen, B.; Vermeersch, P.; Milting, H.; Gao, F.; Kassner, A.; Gillijns, H.; Herijgers, P.; Flameng, W.; Carmeliet, P.; et al. Increased Cardiac Expression of Tissue Inhibitor of Metalloproteinase-1 and Tissue Inhibitor of Metalloproteinase-2 Is Related to Cardiac Fibrosis and Dysfunction in the Chronic Pressure-Overloaded Human Heart. Circulation 2005, 112, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Brower, G.L.; Gardner, J.D.; Forman, M.F.; Murray, D.B.; Voloshenyuk, T.; Levick, S.P.; Janicki, J.S. The Relationship between Myocardial Extracellular Matrix Remodeling and Ventricular Function. Eur. J. Cardio-Thorac. Surg. 2006, 30, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Badenhorst, D.; Maseko, M.; Tsotetsi, O.J.; Naidoo, A.; Brooksbank, R.; Norton, G.R.; Woodiwiss, A.J. Cross-Linking Influences the Impact of Quantitative Changes in Myocardial Collagen on Cardiac Stiffness and Remodelling in Hypertension in Rats. Cardiovasc. Res. 2003, 57, 632–641. [Google Scholar] [CrossRef]
- Norton, G.R.; Tsotetsi, J.; Trifunovic, B.; Hartford, C.; Candy, G.P.; Woodiwiss, A.J. Myocardial Stiffness Is Attributed to Alterations in Cross-Linked Collagen Rather than Total Collagen or Phenotypes in Spontaneously Hypertensive Rats. Circulation 1997, 96, 1991–1998. [Google Scholar] [CrossRef]
- Woodiwiss, A.J.; Tsotetsi, O.J.; Sprott, S.; Lancaster, E.J.; Mela, T.; Chung, E.S.; Meyer, T.E.; Norton, G.R. Reduction in Myocardial Collagen Cross-Linking Parallels Left Ventricular Dilatation in Rat Models of Systolic Chamber Dysfunction. Circulation 2001, 103, 155–160. [Google Scholar] [CrossRef]
- Iimoto, D.S.; Covell, J.W.; Harper, E. Increase in Cross-Linking of Type I and Type III Collagens Associated with Volume-Overload Hypertrophy. Circ. Res. 1988, 63, 399–408. [Google Scholar] [CrossRef]
- Herrmann, K.L.; McCulloch, A.D.; Omens, J.H. Glycated Collagen Cross-Linking Alters Cardiac Mechanics in Volume-Overload Hypertrophy. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H1277–H1284. [Google Scholar] [CrossRef] [PubMed]
- Avendano, G.F.; Agarwal, R.K.; Bashey, R.I.; Lyons, M.M.; Soni, B.J.; Jyothirmayi, G.N.; Regan, T.J. Effects of Glucose Intolerance on Myocardial Function and Collagen-Linked Glycation. Diabetes 1999, 48, 1443–1447. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Masurekar, M.R.; Vatner, D.E.; Jyothirmayi, G.N.; Regan, T.J.; Vatner, S.F.; Meggs, L.G.; Malhotra, A. Glycation End-Product Cross-Link Breaker Reduces Collagen and Improves Cardiac Function in Aging Diabetic Heart. Am. J. Physiol.-Heart Circ. Physiol. 2003, 285, H2587–H2591. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Egan, J.; Vasan, S.; Jyothirmayi, G.N.; Masurekar, M.R.; Lopez, S.; Williams, C.; Torres, R.L.; Wagle, D.; Ulrich, P.; et al. An Advanced Glycation Endproduct Cross-Link Breaker Can Reverse Age-Related Increases in Myocardial Stiffness. Proc. Natl. Acad. Sci. USA 2000, 97, 2809–2813. [Google Scholar] [CrossRef]
- Yamamoto, K.; Masuyama, T.; Sakata, Y.; Nishikawa, N.; Mano, T.; Yoshida, J.; Miwa, T.; Sugawara, M.; Yamaguchi, Y.; Ookawara, T.; et al. Myocardial Stiffness Is Determined by Ventricular Fibrosis, but Not by Compensatory or Excessive Hypertrophy in Hypertensive Heart. Cardiovasc. Res. 2002, 55, 76–82. [Google Scholar] [CrossRef]
- Burgess, M.L.; Buggy, J.; Price, R.L.; Abel, F.L.; Terracio, L.; Samarel, A.M.; Borg, T.K. Exercise- and Hypertension-Induced Collagen Changes Are Related to Left Ventricular Function in Rat Hearts. Am. J. Physiol.-Heart Circ. Physiol. 1996, 270, H151–H159. [Google Scholar] [CrossRef]
- Mukherjee, D.; Sen, S. Collagen Phenotypes during Development and Regression of Myocardial Hypertrophy in Spontaneously Hypertensive Rats. Circ. Res. 1990, 67, 1474–1480. [Google Scholar] [CrossRef]
- Marijianowski, M.M.H.; Teeling, P.; Mann, J.; Becker, A.E. Dilated Cardiomyopathy Is Associated with an Increase in the Type I/Type III Collagen Ratio: A Quantitative Assessment. J. Am. Coll. Cardiol. 1995, 25, 1263–1272. [Google Scholar] [CrossRef]
- Eid, R.A.; Alkhateeb, M.A.; El-Kott, A.F.; Eleawa, S.M.; Zaki, M.S.A.; Alaboodi, S.A.; Salem Al-Shudiefat, A.A.R.; Aldera, H.; Alnamar, N.M.; Alassiri, M.; et al. A High-Fat Diet Rich in Corn Oil Induces Cardiac Fibrosis in Rats by Activating JAK2/STAT3 and Subsequent Activation of ANG II/TGF-1β/Smad3 Pathway: The Role of ROS and IL-6 Trans-Signaling. J. Food Biochem. 2019, 43, e12952. [Google Scholar] [CrossRef]
- Nachar, W.; Merlet, N.; Maafi, F.; Shi, Y.; Mihalache-Avram, T.; Mecteau, M.; Ferron, M.; Rhéaume, E.; Tardif, J.C. Cardiac Inflammation and Diastolic Dysfunction in Hypercholesterolemic Rabbits. PLoS ONE 2019, 14, e0220707. [Google Scholar] [CrossRef]
- Wei, S.; Chow, L.T.C.; Shum, I.O.L.; Qin, L.; Sanderson, J.E. Left and Right Ventricular Collagen Type I/III Ratios and Remodeling Post-Myocardial Infarction. J. Card. Fail. 1999, 5, 117–126. [Google Scholar] [CrossRef]
- Shimizu, M.; Umeda, K.; Sugihara, N.; Yoshio, H.; Ino, H.; Takeda, R.; Okada, Y.; Nakanishi, I. Collagen Remodelling in Myocardia of Patients with Diabetes. J. Clin. Pathol. 1993, 46, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Woessner, J.F. Matrix Metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Membrane-Type Matrix Metalloproteinases: Their Functions and Regulations. Matrix Biol. 2015, 44–46, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, B.; Wallon, U.M.; Overall, C.M. Extracellular Matrix Binding Properties of Recombinant Fibronectin Type II-like Modules of Human 72-KDa Gelatinase/Type IV Collagenase: High Affinity Binding to Native Type I Collagen but Not Native Type IV Collagen. J. Biol. Chem. 1995, 270, 11555–11566. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, J.; Solomonov, I.; Zehorai, E.; Sagi, I. Multilevel Regulation of Matrix Metalloproteinases in Tissue Homeostasis Indicates Their Molecular Specificity In Vivo. Matrix Biol. 2015, 44–46, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases: Structure, Function, and Biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef]
- Fedak, P.W.M.; Altamentova, S.M.; Weisel, R.D.; Nili, N.; Ohno, N.; Verma, S.; Lee, T.Y.J.; Kiani, C.; Mickle, D.A.G.; Strauss, B.H.; et al. Matrix Remodeling in Experimental and Human Heart Failure: A Possible Regulatory Role for TIMP-3. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H626–H634. [Google Scholar] [CrossRef]
- Lambert, E.; Dassé, E.; Haye, B.; Petitfrère, E. TIMPs as Multifacial Proteins. Crit. Rev. Oncol./Hematol. 2004, 49, 187–198. [Google Scholar] [CrossRef]
- Timms, P.M.L.; Wright, A.; Maxwell, P.; Campbell, S.; Dawnay, A.B.; Srikanthan, V. Plasma Tissue Inhibitor of Metalloproteinase-1 Levels Are Elevated in Essential Hypertension and Related to Left Ventricular Hypertrophy. Am. J. Hypertens. 2002, 15, 269–272. [Google Scholar] [CrossRef]
- Chaturvedi, R.R.; Herron, T.; Simmons, R.; Shore, D.; Kumar, P.; Sethia, B.; Chua, F.; Vassiliadis, E.; Kentish, J.C. Passive Stiffness of Myocardium from Congenital Heart Disease and Implications for Diastole. Circulation 2010, 121, 979–988. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; de Keulenaer, G.; Bauersachs, J.; Brutsaert, D.; Cleland, J.G.; Diez, J.; Du, X.; Ford, P.; Heinzel, F.R.; Lipson, K.E.; et al. Towards Better Definition, Quantification and Treatment of Fibrosis in Heart Failure. A Scientific Roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Sales, E.; Jeddi, S.; Alipour, M.R. Trans-Chalcone Inhibits Transforming Growth Factor-Β1 and Connective Tissue Growth Factor-Dependent Collagen Expression in the Heart of High-Fat Diet-Fed Rats. Arch. Physiol. Biochem. 2020, 14, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Warbrick, I.; Rabkin, S.W. Hypoxia-Inducible Factor 1-Alpha (HIF-1α) as a Factor Mediating the Relationship between Obesity and Heart Failure with Preserved Ejection Fraction. Obes. Rev. 2019, 20, 701–712. [Google Scholar] [CrossRef]
- Takawale, A.; Sakamuri, S.S.V.P.; Kassiri, Z. Extracellular Matrix Communication and Turnover in Cardiac Physiology and Pathology. Compr. Physiol. 2015, 5, 687–719. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming Growth Factor (TGF)-β Signaling in Cardiac Remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.F. Signaling Cross-Talk between TGF-β/BMP and Other Pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef]
- Zhu, X.Y.; Daghini, E.; Rodriguez-Porcel, M.; Chade, A.R.; Napoli, C.; Lerman, A.; Lerman, L.O. Redox-Sensitive Myocardial Remodeling and Dysfunction in Swine Diet-Induced Experimental Hypercholesterolemia. Atherosclerosis 2007, 193, 62–69. [Google Scholar] [CrossRef]
- Czarzasta, K.; Koperski, L.; Segiet, A.; Janiszewski, M.; Kuch, M.; Gornicka, B.; Cudnoch-Jedrzejewska, A. The Role of High Fat Diet in the Regulation of MAP Kinases Activity in Left Ventricular Fibrosis. Acta Histochem. 2019, 121, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovich, I. Cell Surface-Localized Matrix Metalloproteinase-9 Proteolytically Activates TGF-Beta and Promotes Tumor Invasion and Angiogenesis. Genes Dev. 2000, 15, 163–176. [Google Scholar] [CrossRef]
- Bergman, M.R.; Teerlink, J.R.; Mahimkar, R.; Li, L.; Zhu, B.Q.; Nguyen, A.; Dahi, S.; Karliner, J.S.; Lovett, D.H. Cardiac Matrix Metalloproteinase-2 Expression Independently Induces Marked Ventricular Remodeling and Systolic Dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H1847–H1860. [Google Scholar] [CrossRef] [PubMed]
- Zavadzkas, J.A.; Mukherjee, R.; Rivers, W.T.; Patel, R.K.; Meyer, E.C.; Black, L.E.; McKinney, R.A.; Marshall Oelsen, J.; Stroud, R.E.; Spinale, F.G. Direct Regulation of Membrane Type 1 Matrix Metalloproteinase Following Myocardial Infarction Causes Changes in Survival, Cardiac Function, and Remodeling. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1656–H1666. [Google Scholar] [CrossRef]
- Czarzasta, K.; Koperski, Ł.; Fus, Ł.; Wojno, O.; Górnicka, B.; Cudnoch-Jędrzejewska, A. The Effects of a High-Fat Diet on Left Ventricular Fibrosis. Kardiol. Pol. 2018, 76, 802–804. [Google Scholar] [CrossRef]
- Leopoldo, A.S.; Sugizaki, M.M.; Lima-Leopoldo, A.P.; do Nascimento, A.F.; Luvizotto, R.D.A.M.; de Campos, D.H.S.; Okoshi, K.; Pai-Silva, M.D.; Padovani, C.R.; Cicogna, A.C. Cardiac Remodeling in a Rat Model of Diet-Induced Obesity. Can. J. Cardiol. 2010, 26, 423–429. [Google Scholar] [CrossRef]
- Nascimento, A.R.; Machado, M.; de Jesus, N.; Gomes, F.; Lessa, M.A.; Bonomo, I.T.; Tibiriçá, E. Structural and Functional Microvascular Alterations in a Rat Model of Metabolic Syndrome Induced by a High-Fat Diet. Obesity 2013, 21, 2046–2054. [Google Scholar] [CrossRef]
- Martins, F.; Campos, D.H.S.; Pagan, L.U.; Martinez, P.F.; Okoshi, K.; Okoshi, M.P.; Padovani, C.R.; de Souza, A.S.; Cicogna, A.C.; de Oliveira, S.A. High-Fat Diet Promotes Cardiac Remodeling in an Experimental Model of Obesity. Arq. Bras. De Cardiol. 2015, 105, 479–486. [Google Scholar] [CrossRef]
- Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef]
- Zeng, H.; Vaka, V.R.; He, X.; Booz, G.W.; Chen, J. High-fat Diet Induces Cardiac Remodelling and Dysfunction: Assessment of the Role Played by SIRT3 Loss. J. Cell. Mol. Med. 2015, 19, 1847–1856. [Google Scholar] [CrossRef]
- Aurich, A.-C.; Niemann, B.; Pan, R.; Gruenler, S.; Issa, H.; Silber, R.-E.; Rohrbach, S. Age-Dependent Effects of High Fat-Diet on Murine Left Ventricles: Role of Palmitate. Basic Res. Cardiol. 2013, 108, 369. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-T.; Liu, C.-F.; Tsai, T.-H.; Chen, Y.-L.; Chang, H.-W.; Tsai, C.-Y.; Leu, S.; Zhen, Y.-Y.; Chai, H.-T.; Chung, S.-Y.; et al. Effect of Obesity Reduction on Preservation of Heart Function and Attenuation of Left Ventricular Remodeling, Oxidative Stress and Inflammation in Obese Mice. J. Transl. Med. 2012, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.C.T.; Lima-Leopoldo, A.P.; Leopoldo, A.S.; de Campos, D.H.S. do Nascimento, A.F.; de Oliveira Junior, S.A.; Padovani, C.R.; Cicogna, A.C. Influence of Term of Exposure to High-Fat Diet-Induced Obesity on Myocardial Collagen Type I and III. Arq. Bras. De Cardiol. 2013, 102, 157–164. [Google Scholar] [CrossRef]
- da Silva-Bertani, D.C.T.; Vileigas, D.F.; Mota, G.A.F.; de Souza, S.L.B.; de Tomasi, L.C.; de Campos, D.H.S.; de Deus, A.F.; Freire, P.P.; Alves, C.A.B.; Padovani, C.R.; et al. A Redução Do Colágeno Tipo I Está Associada Ao Aumento Da Atividade Da Metaloproteinase-2 e Da Expressão Proteica de Leptina No Miocárdio de Ratos Obesos. Arq. Bras. De Cardiol. 2020, 115, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yan, F.; Li, J.; Zhang, C.; Su, H.; Bu, P. SIRT3 Ablation Deteriorates Obesity-Related Cardiac Remodeling by Modulating ROS-NF-ΚB-MCP-1 Signaling Pathway. J. Cardiovasc. Pharm. 2020, 76, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Hubesch, G.; Hanthazi, A.; Acheampong, A.; Chomette, L.; Lasolle, H.; Hupkens, E.; Jespers, P.; Vegh, G.; Wembonyama, C.W.M.; Verhoeven, C.; et al. A Preclinical Rat Model of Heart Failure With Preserved Ejection Fraction with Multiple Comorbidities. Front. Cardiovasc. Med. 2022, 8, 809885. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Jaffré, F.; Friedman, A.E.; Hu, Z.; MacKman, N.; Blaxall, B.C. β-Adrenergic Receptor Stimulation Transactivates Protease-Activated Receptor 1 via Matrix Metalloproteinase 13 in Cardiac Cells. Circulation 2012, 125, 2993–3003. [Google Scholar] [CrossRef]
- Turner, N.A.; Porter, K.E.; Smith, W.H.T.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic Β2-Adrenergic Receptor Stimulation Increases Proliferation of Human Cardiac Fibroblasts via an Autocrine Mechanism. Cardiovasc. Res. 2003, 57, 784–792. [Google Scholar] [CrossRef]
- Nguyen, M.N.; Kiriazis, H.; Ruggiero, D.; Gao, X.M.; Su, Y.; Jian, A.; Han, L.P.; McMullen, J.R.; Du, X.J. Spontaneous Ventricular Tachyarrhythmias in Β2-Adrenoceptor Transgenic Mice in Relation to Cardiac Interstitial Fibrosis. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, H946–H957. [Google Scholar] [CrossRef]
- Frigolet, M.E.; Torres, N.; Tovar, A.R. The Renin-Angiotensin System in Adipose Tissue and Its Metabolic Consequences during Obesity. J. Nutr. Biochem. 2013, 24, 2003–2015. [Google Scholar] [CrossRef] [PubMed]
- Schütten, M.T.J.; Houben, A.J.H.M.; de Leeuw, P.W.; Stehouwer, C.D.A. The Link between Adipose Tissue Renin-Angiotensin-Aldosterone System Signaling and Obesity-Associated Hypertension. Physiology 2017, 32, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; di Gioia, C.R.T.; Bombardi, C.; Catalucci, D.; Corradi, B.; Gualazzi, M.G.; Leopizzi, M.; Mancini, M.; Zerbini, G.; Condorelli, G.; et al. MiR-133a Regulates Collagen 1A1: Potential Role of MiR-133a in Myocardial Fibrosis in Angiotensin II-Dependent Hypertension. J. Cell. Physiol. 2012, 227, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, M.; Liu, L.; Zhu, D.; Tian, G. Highlight Article: Telmisartan Improves Myocardial Remodeling by Inhibiting Leptin Autocrine Activity and Activating PPARγ. Exp. Biol. Med. 2020, 245, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Maeda, N.; Sonoda, M.; Ohashi, K.; Hibuse, T.; Nishizawa, H.; Nishida, M.; Hiuge, A.; Kurata, A.; Kihara, S.; et al. Adiponectin Protects against Angiotensin II-Induced Cardiac Fibrosis through Activation of PPAR-α. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.J.; Borthwick, G.M.; Oakenfull, R.; Robson, A.; Arthur, H.M. Angiotensin II-Induced Cardiomyocyte Hypertrophy in Vitro Is TAK1-Dependent and Smad2/3-Independent. Hypertens. Res. 2012, 35, 393–398. [Google Scholar] [CrossRef]
- Volpe, M.; Carnovali, M.; Mastromarino, V. The Natriuretic Peptides System in the Pathophysiology of Heart Failure: From Molecular Basis to Treatment. Clin. Sci. 2016, 130, 57–77. [Google Scholar] [CrossRef]
- Standeven, K.F.; Hess, K.; Carter, A.M.; Rice, G.I.; Cordell, P.A.; Balmforth, A.J.; Lu, B.; Scott, D.J.; Turner, A.J.; Hooper, N.M.; et al. Neprilysin, Obesity and the Metabolic Syndrome. Int. J. Obes. 2011, 35, 1031–1040. [Google Scholar] [CrossRef]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac Natriuretic Peptides. Nat. Rev. Cardiol. 2020, 17, 1031–1040. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The Glucose Fatty-Acid Cycle Its Role in Insulin Sensitivity and the Metabolic Disturbances of Diabetes Mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- de Luca, C.; Olefsky, J.M. Inflammation and Insulin Resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Yilmaz, G.; Chen, X.; Harmancey, R. Dietary Fat and Sugar Differentially Affect β-Adrenergic Stimulation of Cardiac ERK and AKT Pathways in C57BL/6 Male Mice Subjected to High-Calorie Feeding. J. Nutr. 2020, 150, 1041–1050. [Google Scholar] [CrossRef]
- Schiekofer, S.; Shiojima, I.; Sato, K.; Galasso, G.; Oshima, Y.; Walsh, K. Microarray Analysis of Akt1 Activation in Transgenic Mouse Hearts Reveals Transcript Expression Profiles Associated with Compensatory Hypertrophy and Failure. Physiol. Genom. 2006, 27, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.-C.; Hsieh, P.-S. The Role of Adipocyte Hypertrophy and Hypoxia in the Development of Obesity-Associated Adipose Tissue Inflammation and Insulin Resistance. In Adiposity—Omics and Molecular Understanding; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Andrei, A.M.; Berbecaru-Iovan, A.; Din-Anghel, F.R.I.; Stanciulescu, C.E.; Berbecaru-Iovan, S.; Banita, I.M.; Pisoschi, C.G. Interplay between Hypoxia, Inflammation and Adipocyte Remodeling in the Metabolic Syndrome. In Hypoxia and Human Diseases; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia Signaling in Human Diseases and Therapeutic Targets. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Misra, S.; Fu, A.A.; Misra, K.D.; Shergill, U.M.; Leof, E.B.; Mukhopadhyay, D.D. Hypoxia Induces a Phenotypic Switch of Fibroblasts to Myofibroblasts through a MMP-2/TIMP Mediated Pathway: Implications for Venous Neointimal Hyperplasia in Hemodialysis Access. J. Vasc. Interv. Radiol. 2010, 21, 896–902. [Google Scholar] [CrossRef]
- Wasim, M.; Awan, F.R.; Najam, S.S.; Khan, A.R.; Khan, H.N. Role of Leptin Deficiency, Inefficiency, and Leptin Receptors in Obesity. Biochem. Genet. 2016, 54, 565–572. [Google Scholar] [CrossRef]
- Pemberton, C.J. Leptin-Induced Cardiac Hypertrophy: RhoAing a Lipid Raft down a Protective P38 MAPK Signalling Stream? Cardiovasc. Res. 2008, 77, 4–5. [Google Scholar] [CrossRef][Green Version]
- Poetsch, M.S.; Strano, A.; Guan, K. Role of Leptin in Cardiovascular Diseases. Front. Endocrinol. 2020, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin Levels in Human and Rodent: Measurement of Plasma Leptin and Ob RNA in Obese and Weight-Reduced Subjects. Nat. Med. 1995, 1, 1116–1155. [Google Scholar] [CrossRef] [PubMed]
- Lönnqvist, F.; Arner, P.; Nordfors, L.; Schalling, M. Overexpression of the Obese (Ob) Gene in Adipose Tissue of Human Obese Subjects. Nat. Med. 1995, 1, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Ok, M.; Lee, S.K. Leptin as a Key between Obesity and Cardiovascular Disease. J. Obes. Metab. Syndr. 2020, 29, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martínez, E.; Jurado-López, R.; Cervantes-Escalera, P.; Cachofeiro, V.; Miana, M. Leptin, a Mediator of Cardiac Damage Associated with Obesity. Horm. Mol. Biol. Clin. Investig. 2014, 18, 3–14. [Google Scholar] [CrossRef]
- Palanivel, R.; Eguchi, M.; Shuralyova, I.; Coe, I.; Sweeney, G. Distinct Effects of Short- and Long-Term Leptin Treatment on Glucose and Fatty Acid Uptake and Metabolism in HL-1 Cardiomyocytes. Metab. Clin. Exp. 2006, 55, 1067–1075. [Google Scholar] [CrossRef]
- Zeidan, A.; Javadov, S.; Karmazyn, M. Essential Role of Rho/ROCK-Dependent Processes and Actin Dynamics in Mediating Leptin-Induced Hypertrophy in Rat Neonatal Ventricular Myocytes. Cardiovasc. Res. 2006, 72, 101–111. [Google Scholar] [CrossRef]
- Zeidan, A.; Javadov, S.; Chakrabarti, S.; Karmazyn, M. Leptin-Induced Cardiomyocyte Hypertrophy Involves Selective Caveolae and RhoA/ROCK-Dependent P38 MAPK Translocation to Nuclei. Cardiovasc. Res. 2008, 77, 64–72. [Google Scholar] [CrossRef]
- Madani, S.; de Girolamo, S.; Muñoz, D.M.; Li, R.K.; Sweeney, G. Direct Effects of Leptin on Size and Extracellular Matrix Components of Human Pediatric Ventricular Myocytes. Cardiovasc. Res. 2006, 69, 716–725. [Google Scholar] [CrossRef]
- Schram, K.; de Girolamo, S.; Madani, S.; Munoz, D.; Thong, F.; Sweeney, G. Leptin Regulates MMP-2, TIMP-1 and Collagen Synthesis via P38 MAPK in HL-1 Murine Cardiomyocytes. Cell. Mol. Biol. Lett. 2010, 15, 551–563. [Google Scholar] [CrossRef]
- Schram, K.; Wong, M.M.C.; Palanivel, R.; No, E.K.; Dixon, I.M.C.; Sweeney, G. Increased Expression and Cell Surface Localization of MT1-MMP Plays a Role in Stimulation of MMP-2 Activity by Leptin in Neonatal Rat Cardiac Myofibroblasts. J. Mol. Cell. Cardiol. 2008, 44, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Schram, K.; Ganguly, R.; No, E.K.; Fang, X.; Thong, F.S.L.; Sweeney, G. Regulation of MT1-MMP and MMP-2 by Leptin in Cardiac Fibroblasts Involves Rho/ROCK-Dependent Actin Cytoskeletal Reorganization and Leads to Enhanced Cell Migration. Endocrinology 2011, 152, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a Mouse Model of Obesity-Related Fibrotic Cardiomyopathy That Recapitulates Features of Human Heart Failure with Preserved Ejection Fraction. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H934–H949. [Google Scholar] [CrossRef]
- Hall, M.E.; Maready, M.W.; Hall, J.E.; Stec, D.E. Rescue of Cardiac Leptin Receptors in Db/Db Mice Prevents Myocardial Triglyceride Accumulation. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E316–E325. [Google Scholar] [CrossRef]
- Dong, F.; Yang, X.; Sreejayan, N.; Ren, J. Chromium (D-Phenylalanine)3 Improves Obesity-Induced Cardiac Contractile Defect in Ob/Ob Mice. Obesity 2007, 15, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Cavalera, M.; Wang, J.; Frangogiannis, N.G. Obesity, Metabolic Dysfunction, and Cardiac Fibrosis: Pathophysiological Pathways, Molecular Mechanisms, and Therapeutic Opportunities. Transl. Res. 2014, 164, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin Stimulates Glucose Utilization and Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kadowaki, T. Physiological and Pathophysiological Roles of Adiponectin and Adiponectin Receptors in the Integrated Regulation of Metabolic and Cardiovascular Diseases. Int. J. Obes. 2008, 32, S13–S18. [Google Scholar] [CrossRef]
- Fang, X.; Palanivel, R.; Cresser, J.; Schram, K.; Ganguly, R.; Thong, F.S.L.; Tuinei, J.; Xu, A.; Abel, E.D.; Sweeney, G. An APPL1-AMPK Signaling Axis Mediates Beneficial Metabolic Effects of Adiponectin in the Heart. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E219–E721. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the Past Two Decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Berg, G.; Schreier, L.; Miksztowicz, V. Circulating and Adipose Tissue Matrix Metalloproteinases in Cardiometabolic Risk Environments: Pathophysiological Aspects. Horm. Mol. Biol. Clin. Investig. 2014, 17, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and Adiponectin Receptors in Insulin Resistance, Diabetes, and the Metabolic Syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Lord, E.; Ledoux, S.; Murphy, B.D.; Beaudry, D.; Palin, M.F. Expression of Adiponectin and Its Receptors in Swine. J. Anim. Sci. 2005, 83. [Google Scholar] [CrossRef] [PubMed]
- Dadson, K.; Chasiotis, H.; Wannaiampikul, S.; Tungtrongchitr, R.; Xu, A.; Sweeney, G. Adiponectin Mediated APPL1-AMPK Signaling Induces Cell Migration, MMP Activation, and Collagen Remodeling in Cardiac Fibroblasts. J. Cell. Biochem. 2014, 115, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Shibata, R.; Ouchi, N.; Ito, M.; Kihara, S.; Shiojima, I.; Pimentel, D.R.; Kumada, M.; Sato, K.; Schiekofer, S.; Ohashi, K.; et al. Adiponectin-Mediated Modulation of Hypertrophic Signals in the Heart. Nat. Med. 2004, 10, 1384–1389. [Google Scholar] [CrossRef]
- Liao, Y.; Takashima, S.; Maeda, N.; Ouchi, N.; Komamura, K.; Shimomura, I.; Hori, M.; Matsuzawa, Y.; Funahashi, T.; Kitakaze, M. Exacerbation of Heart Failure in Adiponectin-Deficient Mice Due to Impaired Regulation of AMPK and Glucose Metabolism. Cardiovasc. Res. 2005, 67, 705–713. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, H.; Xie, X.; Chen, X.; Kosuru, R.; Li, S.; Lian, Q.; Cheung, C.W.; Irwin, M.G.; Ge, R.S.; et al. Adiponectin Facilitates Postconditioning Cardioprotection through Both AMPK-Dependent Nuclear and AMPK-Independent Mitochondrial STAT3 Activation. Oxidative Med. Cell. Longev. 2020, 2020, 4253457. [Google Scholar] [CrossRef]
- Jenke, A.; Schur, R.; Röger, C.; Karadeniz, Z.; Grüger, M.; Holzhauser, L.; Savvatis, K.; Poller, W.; Schultheiss, H.P.; Landmesser, U.; et al. Adiponectin Attenuates Profibrotic Extracellular Matrix Remodeling Following Cardiac Injury by Up-Regulating Matrix Metalloproteinase 9 Expression in Mice. Physiol. Rep. 2017, 5, e13523. [Google Scholar] [CrossRef]
- Yan, C.J.; Li, S.M.; Xiao, Q.; Liu, Y.; Hou, J.; Chen, A.F.; Xia, L.P.; Li, X.C. Influence of Serum Adiponectin Level and SNP +45 Polymorphism of Adiponectin Gene on Myocardial Fibrosis. J. Zhejiang Univ. Sci. B 2013, 14, 721–728. [Google Scholar] [CrossRef]
- Mills, K.T.; Stefanescu, A.; He, J. The Global Epidemiology of Hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef]
- Adeoye, R.I.; Joel, E.B.; Igunnu, A.; Arise, R.O.; Malomo, S.O. A Review of Some Common African Spices with Antihypertensive Potential. J. Food Biochem. 2021, 46, e14003. [Google Scholar] [CrossRef]
- Landsberg, L.; Aronne, L.J.; Beilin, L.J.; Burke, V.; Igel, L.I.; Lloyd-Jones, D.; Sowers, J. Obesity-Related Hypertension: Pathogenesis, Cardiovascular Risk, and Treatment. J. Clin. Hypertens. 2013, 15, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef] [PubMed]
- Mechanic, O.J.; Gavin, M.; Grossman, S.A. Acute Myocardial Infarction; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Yusuf, S.; Hawken, S.; Ôunpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of Potentially Modifiable Risk Factors Associated with Myocardial Infarction in 52 Countries (the INTERHEART Study): Case-Control Study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Albakri, A. Heart Failure with Reduced Ejection Fraction: A Review of Clinical Status and Meta-Analyses of Diagnosis by 3D Echocardiography and Natriuretic Peptides-Guided Heart Failure Therapy. Trends Res. 2018, 1. [Google Scholar] [CrossRef]
- Iyer, R.P.; Jung, M.; Lindsey, M.L. MMP-9 Signaling in the Left Ventricle Following Myocardial Infarction. Am. J. Physiol.-Heart Circ. Physiol. 2016, 311, H190–H198. [Google Scholar] [CrossRef] [PubMed]
- Bektik, E.; Fu, J. Ameliorating the Fibrotic Remodeling of the Heart through Direct Cardiac Reprogramming. Cells 2019, 8, 679. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; Flynn, E.R.; Moak, S.P.; Li, X.; da Silva, A.A.; Wang, Z.; do Carmo, J.M.; Hall, M.E.; Hall, J.E. Interaction of Obesity and Hypertension on Cardiac Metabolic Remodeling and Survival Following Myocardial Infarction. J. Am. Heart Assoc. 2021, 10, e018212. [Google Scholar] [CrossRef]
- Poncelas, M.; Inserte, J.; Vilardosa, Ú.; Rodriguez-Sinovas, A.; Bañeras, J.; Simó, R.; Garcia-Dorado, D. Obesity Induced by High Fat Diet Attenuates Postinfarct Myocardial Remodeling and Dysfunction in Adult B6D2F1 Mice. J. Mol. Cell. Cardiol. 2015, 84, 154–161. [Google Scholar] [CrossRef]
- Neeland, I.J.; Das, S.R.; Simon, D.N.; Diercks, D.B.; Alexander, K.P.; Wang, T.Y.; de Lemos, J.A. The Obesity Paradox, Extreme Obesity, and Long-Term Outcomes in Older Adults with ST-Segment Elevation Myocardial Infarction: Results from the NCDR. Eur. Heart J.-Qual. Care Clin. Outcomes 2017, 3, 183–191. [Google Scholar] [CrossRef]
- Inserte, J.; Aluja, D.; Barba, I.; Ruiz-Meana, M.; Miró, E.; Poncelas, M.; Vilardosa, Ú.; Castellano, J.; Garcia-Dorado, D. High-Fat Diet Improves Tolerance to Myocardial Ischemia by Delaying Normalization of Intracellular PH at Reperfusion. J. Mol. Cell. Cardiol. 2019, 133, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Huisamen, B.; Dietrich, D.; Bezuidenhout, N.; Lopes, J.; Flepisi, B.; Blackhurst, D.; Lochner, A. Early Cardiovascular Changes Occurring in Diet-Induced, Obese Insulin-Resistant Rats. Mol. Cell. Biochem. 2012, 368, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Heaberlin, J.R.; Ma, Y.; Zhang, J.; Ahuja, S.S.; Lindsey, M.L.; Halade, G.V. Obese and Diabetic KKAy Mice Show Increased Mortality but Improved Cardiac Function Following Myocardial Infarction. Cardiovasc. Pathol. 2013, 22, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Tao, J.; del Monte, F.; Lee, K.H.; Li, L.; Picard, M.; Force, T.L.; Franke, T.F.; Hajjar, R.J.; Rosenzweig, A. Akt Activation Preserves Cardiac Function and Prevents Injury after Transient Cardiac Ischemia in Vivo. Circulation 2001, 104, 330–335. [Google Scholar] [CrossRef] [PubMed]
- du Toit, E.F.; Smith, W.; Muller, C.; Strijdom, H.; Stouthammer, B.; Woodiwiss, A.J.; Norton, G.R.; Lochner, A. Myocardial Susceptibility to Ischemic-Reperfusion Injury in a Prediabetic Model of Dietary-Induced Obesity. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H2336–H2343. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Kurmani, S.; Squire, I. Acute Heart Failure: Definition, Classification and Epidemiology. Curr. Heart Fail. Rep. 2017, 14, 385–392. [Google Scholar] [CrossRef]
- Kenchaiah, S.; Evans, J.C.; Levy, D.; Wilson, P.W.F.; Benjamin, E.J.; Larson, M.G.; Kannel, W.B.; Vasan, R.S. Obesity and the Risk of Heart Failure. N. Engl. J. Med. 2002, 347, 305–313. [Google Scholar] [CrossRef]
- Davis, B.R.; Kostis, J.B.; Simpson, L.M.; Black, H.R.; Cushman, W.C.; Einhorn, P.T.; Farber, M.A.; Ford, C.E.; Levy, D.; Massie, B.M.; et al. Heart Failure with Preserved and Reduced Left Ventricular Ejection Fraction in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Circulation 2008, 118, 2259–2267. [Google Scholar] [CrossRef]
- Savji, N.; Meijers, W.C.; Bartz, T.M.; Bhambhani, V.; Cushman, M.; Nayor, M.; Kizer, J.R.; Sarma, A.; Blaha, M.J.; Gansevoort, R.T.; et al. The Association of Obesity and Cardiometabolic Traits With Incident HFpEF and HFrEF. JACC Heart Fail. 2018, 6, 701–709. [Google Scholar] [CrossRef]
- van Heerebeek, L. Impact of Comorbidities on Myocardial Remodeling and Dysfunction In Heart Failure with Preserved Ejection Fraction. SOJ Pharm. Pharm. Sci. 2014. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of Cardiac Hypertrophy and Heart Failure: Signaling Pathways and Novel Therapeutic Targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.N.V.; Borlaug, B.A. Heart Failure with Preserved Ejection Fraction. Curr. Probl. Cardiol. 2016, 41, 145–188. [Google Scholar] [CrossRef] [PubMed]
- Zulkifly, H.; Lip, G.Y.H.; Lane, D.A. Epidemiology of Atrial Fibrillation. Int. J. Clin. Pract. 2018, 72, e13070. [Google Scholar] [CrossRef]
- Wanahita, N.; Messerli, F.H.; Bangalore, S.; Gami, A.S.; Somers, V.K.; Steinberg, J.S. Atrial Fibrillation and Obesity—Results of a Meta-Analysis. Am. Heart J. 2008, 155, 310–315. [Google Scholar] [CrossRef]
- McCauley, M.D.; Hong, L.; Sridhar, A.; Menon, A.; Perike, S.; Zhang, M.; da Silva, I.B.; Yan, J.; Bonini, M.G.; Ai, X.; et al. Ion Channel and Structural Remodeling in Obesity-Mediated Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2020, 13, e008296. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Investigators | Animal Model | Details of HFD Regimen | Concomitant Diseases | Indicators of Fibrosis and ECM Alterations | Cardiac Remodeling and Dysfunction |
|---|---|---|---|---|---|
| Wang et al., 2012 [125] | C57BL/6 J mice | D: 22 weeks A: 8 weeks P: 45% | glucose intolerance, hypercholesterolemia, hyperleptinemia, hypoadiponectinemia | ↑Smad3, TGF-β ↓Smad1/5 and BMP-2 ↑MMP-9 | ↑heart weight, HW/TL, LVESD, LVEDD ↓LVEF, FS |
| Aurich et al., 2013 [124] | C57BL/6 J mice | D: 16 weeks A: 3 and 18 months P: 45% | hyperinsulinemia | ↑fibrosis, col I, col III, | ↑LV weight, BNP, cardiomyocyte hypertrophy |
| Guo et al., 2020 [128] | 129S1/SvImJ mice | D: 16 weeks A: 8 weeks P: 60% | ND | ↑fibrosis, col I, col III | ↑heart weight, IVS, LVPW |
| Leopoldo et al., 2010 [119] | Wistar rats | D: 15 weeks A: 30 days P: 45.2% | glucose intolerance, hyperinsulinemia, hyperleptinemia, hypertension | ↑fibrosis | ↑LV mass, LVESD, LV wall systolic stress, PWTd |
| Da Silva et al., 2014 [126] | Wistar rats | D: 15 and 30 weeks A: 30 days P: 49.2% fat | glucose intolerance, hyperinsulinemia, hyperleptinemia | ↑col I (15 wk), ↓col II (30 wk) | ND |
| Martins et al., 2015 [121] | Wistar rats | D: 20 weeks A: 30 days P: 22.7% | glucose intolerance | ↑fibrosis | ↑LAE, MCSAs |
| Eid et al., 2019 [92] | Wistar rats | D: 8 weeks A: ND P: 40% + CO | hyperinsulinemia, insulin resistance | ↑TGF-β1, Smad-3, total collagen, collagen type I/III ratio, cardiomyocyte apoptosis | ↑LVEDD ↓LVESD, LV contractility |
| Jiménez-González et al., 2020 [122] | Wistar rats | D: 7 weeks A: ND P: 35% | insulin resistance | ↑fibrosis | ↑heart weight, HW/TL, cardiac hypertrophy |
| Da Silva-Bertani et al., 2020 [127] | Wistar rats | D: 34 weeks A: 30 days P: 49.2% | glucose intolerance, hyperinsulinemia, insulin resistance, hyperleptinemia | ↓col I ↑MMP-2; ↓TIMP-1, and TIMP-2 | ND |
| Nascimento et al., 2013 [120] | Wistar Kyoto rats | D: 20 weeks A: 4 weeks P: 30% | glucose intolerance, hyperinsulinemia, insulin resistance, hypercholesterolemia, hypertriglyceridemia, hypertension | ↑fibrosis | ↑LVW/BW, cardiomyocyte hypertrophy |
| Czarzasta et al., 2018 [118] | Sprague Dawley rats | D: 12 and 16 weeks A: 4 weeks T: 31% | ND | ↑fibrosis, cardiomyocyte apoptosis | ND |
| Hubesch et al., 2022 [129] | Sprague-Dawley rats | D: 4 and 12 months A: 4 week T:43% | glucose intolerance, hyperlipidemia, hyperleptinemia, hyperadiponectinemia | ↑fibrosis | Concentric hypertrophy, ↑HW, LVSP, LVEDP, RVESP, * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruszewska, J.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity—Pathogenesis and Involvement of the Extracellular Matrix. Int. J. Mol. Sci. 2022, 23, 4195. https://doi.org/10.3390/ijms23084195
Kruszewska J, Cudnoch-Jedrzejewska A, Czarzasta K. Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity—Pathogenesis and Involvement of the Extracellular Matrix. International Journal of Molecular Sciences. 2022; 23(8):4195. https://doi.org/10.3390/ijms23084195
Chicago/Turabian StyleKruszewska, Jagoda, Agnieszka Cudnoch-Jedrzejewska, and Katarzyna Czarzasta. 2022. "Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity—Pathogenesis and Involvement of the Extracellular Matrix" International Journal of Molecular Sciences 23, no. 8: 4195. https://doi.org/10.3390/ijms23084195
APA StyleKruszewska, J., Cudnoch-Jedrzejewska, A., & Czarzasta, K. (2022). Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity—Pathogenesis and Involvement of the Extracellular Matrix. International Journal of Molecular Sciences, 23(8), 4195. https://doi.org/10.3390/ijms23084195