
Chronic Hyperglycaemia Inhibits Tricarboxylic Acid Cycle in Rat Cardiomyoblasts Overexpressing Glucose Transporter Type 4

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Overexpression of GLUT4 in Hyperglycaemic H9C2 as a Possible Model for Mimicking the Onset of DC
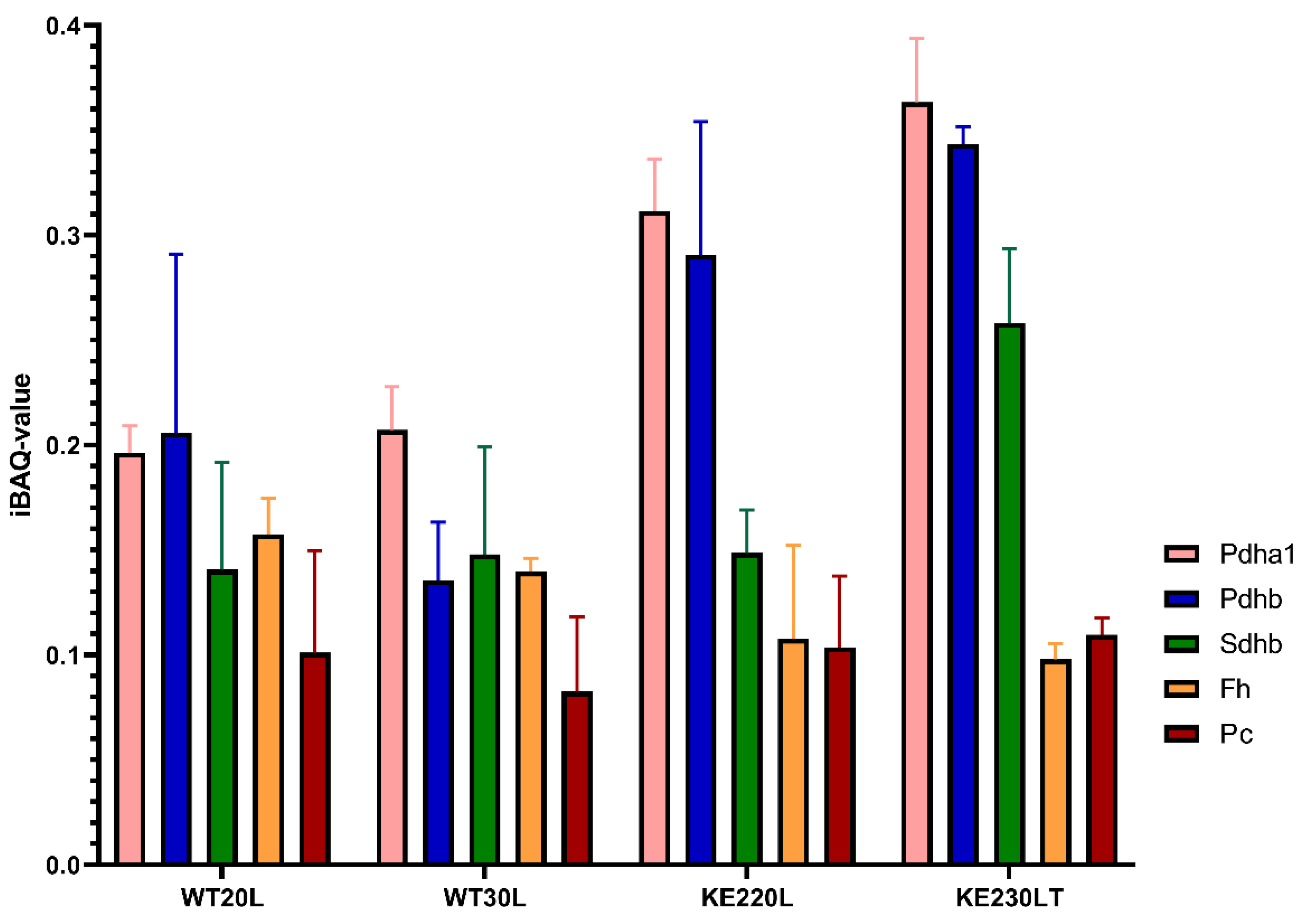
2.2. Glucose Overflow Might Inhibit the Last Steps of the TCA Cycle
2.3. GLUT4 Overexpression Followed by Hyperglycaemia—Effects on Glycolysis
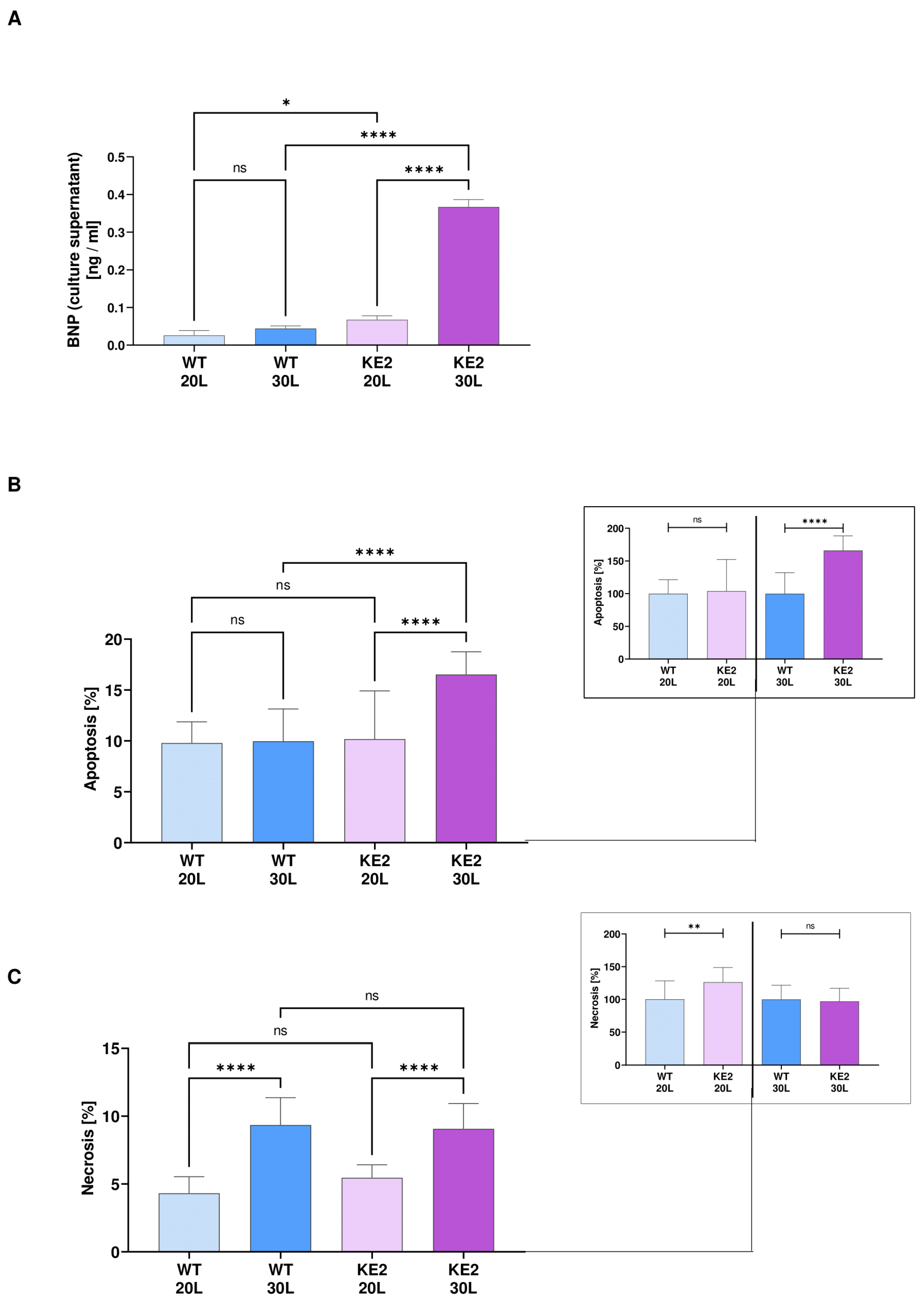
2.4. The Accumulation of Fumarate in H9C2KE2 Cells Induces Oxidative Stress
2.5. The Accumulation of Fumarate in H9C2KE2 Cells Activates Cell Death
3. Discussion
4. Conclusion
5. Materials and Methods
5.1. Plasmid Constructions
5.2. Cell Culture
5.3. Hyperglycaemic Stage Measurements
5.4. Glyoxalase-1 Activity Assay
5.5. Mass Spectrometry
5.6. Immunoblotting Analysis
5.7. Quantitative Real Time PCR Analysis (qRT-PCR)
5.8. Measurement of Ros Generation by Flow Cytometry
5.9. Measurement of Apoptosis and Necrosis by Flow Cytometry
5.10. Pyruvate Assay
5.11. Fumarate Assay
5.12. Brain Natriuretic Peptide (BNP) Measurement
5.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rubler, S.; Dlugash, J.; Yuceoglij, Y.; Kumral, T.; Branwood, A.; Grishman, A. New Type of Cardiomyopathy Associated with Diabetic Glomeruloscierosis. Am. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Dandamudi, S.; Slusser, J.; Mahoney, D.W.; Redfield, M.M.; Rodeheffer, R.J.; Chen, H.H. The Prevalence of Diabetic Cardiomyopathy: A Population Based Study in Olmsted County, Minnesota. J. Card. Fail. 2014, 20, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Goldstein, B.; Thornalley, P.J.; Rabbani, N.; Tschoepe, D. Intracellular Accumulation of Methylglyoxal by Glyoxalase 1 Knock Down Alters Collagen Homoeostasis in L6 Myoblasts. Int. J. Mol. Sci. 2017, 18, 480. [Google Scholar] [CrossRef]
- Villanueva, M.; Michie, C.; Parent, S.; Kanaan, G.N.; Rafatian, G.; Kanda, P.; Ye, B.; Liang, W.; Harper, M.-E.; Davis, D.R. Glyoxalase 1 Prevents Chronic Hyperglycemia Induced Heart-Explant Derived Cell Dysfunction. Theranostics 2019, 9, 5720–5730. [Google Scholar] [CrossRef]
- Engelbrecht, B.; Stratmann, B.; Hess, C.; Tschoepe, D.; Gawlowski, T. Impact of GLO1 knock down on GLUT4 trafficking and glucose uptake in L6 myoblasts. PLoS ONE 2013, 8, e65195. [Google Scholar] [CrossRef]
- Engelbrecht, B.; Mattern, Y.; Scheibler, S.; Tschoepe, D.; Gawlowski, T.; Stratmann, B. Methylglyoxal impairs GLUT4 trafficking and leads to increased glucose uptake in L6 myoblasts. Horm. Metab. Res. 2014, 46, 77–84. [Google Scholar] [CrossRef]
- Wende, A.R.; Schell, J.C.; Ha, C.M.; Pepin, M.E.; Khalimonchuk, O.; Schwertz, H.; Pereira, R.O.; Brahma, M.K.; Tuinei, J.; Contreras-Ferrat, A.; et al. Maintaining Myocardial Glucose Utilization in Diabetic Cardiomyopathy Accelerates Mitochondrial Dysfunction. Diabetes 2020, 69, 2094–2111. [Google Scholar] [CrossRef]
- Liang, Q.; Wang, B.; Pang, L.; Wang, Y.; Zheng, M.; Wang, Q.; Yan, J.; Xu, J. Application of citrate as a tricarboxylic acid (TCA) cycle intermediate, prevents diabetic-induced heart damages in mice. Iran. J. Basic Med. Sci. 2016, 19, 43–48. [Google Scholar]
- Choi, I.; Son, H.; Baek, J.H. Tricarboxylic Acid (TCA) Cycle Intermediates: Regulators of Immune Responses. Life 2021, 11, 69. [Google Scholar] [CrossRef]
- Mathew, A.V.; Jaiswal, M.; Ang, L.; Michailidis, G.; Pennathur, S.; Pop-Busui, R. Impaired Amino Acid and TCA Metabolism and Cardiovascular Autonomic Neuropathy Progression in Type 1 Diabetes. Diabetes 2020, 68, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Hesselink, M.K.C. Reduced tricarboxylic acid cycle flux in type 2 diabetes mellitus? Diabetologia 2018, 51, 1694–1697. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gaster, M. Reduced TCA Flux in Diabetic Myotubes: Determined by Single Defects? Biochem. Res. Int. 2012, 2012, 716056. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zordoky, B.; El-Kadi, A.O.S. H9c2 cell line is a valuable in vitro model to study the drug metabolizing enzymes in the heart. J. Pharmacol. Toxicol. Methods 2007, 56, 317–322. [Google Scholar] [CrossRef]
- Stratmann, B.; Mattern, Y.; de Carvalho, T.S.; Tschoepe, D. Glucose suicide mechanisms induced by methylglyoxal. Diabetologia 2021, 64, 204. [Google Scholar] [CrossRef]
- Wang, X.; McLennan, S.V.; Allen, T.J.; Tsoutsman, T.; Semsarian, C.; Twigg, S.M. Adverse effects of high glucose and free fatty acid on cardiomyocytes are mediated by connective tissue growth factor. Am. J. Physiol. Cell Physiol. 2009, 297, C1490–C1500. [Google Scholar] [CrossRef]
- Doust, J.; Lehman, R.; Glasziou, P. The role of BNP testing in heart failure. Am. Fam. Physician 2006, 74, 1893–1898. [Google Scholar]
- Barany, T.; Simon, A.; Szabo, G.; Benko, R.; Mezei, Z.; Molnar, L.; Becker, D.; Merkely, B.; Zima, E.; Horvath, E.M. Oxidative Stress-Related Parthanatos of Circulating Mononuclear Leukocytes in Heart Failure. Oxid. Med. Cell. Longev. 2017, 2017, 1249614. [Google Scholar] [CrossRef]
- Talha, S.; Bouitbir, J.; Charles, A.L.; Zoll, J.; Goette-Di Marco, P.; Meziani, F.; Piquard, F.; Geny, B. Pretreatment with brain natriuretic peptide reduces skeletal muscle mitochondrial dysfunction and oxidative stress after ischemia-reperfusion. J. Appl. Physiol. 2013, 114, 172–179. [Google Scholar] [CrossRef]
- Peng, Q.; Hu, W.T.; Su, H.; Yang, Q.; Cheng, X.S. Levels of B-type natriuretic peptide in chronic heart failure patients with and without diabetes mellitus. Exp. Ther. Med. 2013, 5, 229–232. [Google Scholar] [CrossRef][Green Version]
- Huang, C.; Somwar, R.; Patel, N.; Niu, W.; Torok, D.; Klip, A. Sustained exposure of L6 myotubes to high glucose and insulin decreases insulin-stimulated GLUT4 translocation but upregulates GLUT4 activity. Diabetes 2002, 51, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Morisco, C.; Stassi, G.; Notte, A.; Farina, F.; Sgaramella, G.; de Rienzo, A.; Roncarati, R.; Trimarco, B.; Lembo, G. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation 1999, 99, 3071–3078. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, M.; van Niekerk, G.; Fourie, C.; du Plessis, M.; Engelbrecht, A.M. The impact of mitochondria on cancer treatment resistance. Cell. Oncol. 2021, 44, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, J.; Beckmann, D.; Mulac, D.; Muller, L.; Esselen, M.; Dufer, M. Nrf2 Activation Protects Mouse Beta Cells from Glucolipotoxicity by Restoring Mitochondrial Function and Physiological Redox Balance. Oxid. Med. Cell. Longev. 2019, 2019, 7518510. [Google Scholar] [CrossRef]
- Zheng, L.; Cardaci, S.; Jerby, L.; MacKenzie, E.D.; Sciacovelli, M.; Johnson, T.I.; Gaude, E.; King, A.; Leach, J.D.; Edrada-Ebel, R.; et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 2015, 6, 6001. [Google Scholar] [CrossRef]
- Xie, H.; Xu, G.; Aa, J.; Gu, S.; Gao, Y. Modulation of Perturbed Cardiac Metabolism in Rats under High-Altitude Hypoxia by Combination Treatment With L-carnitine and Trimetazidine. Front. Physiol. 2021, 12, 671161. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, C.; Liu, C.; Wang, Y.; Zhang, W.; Xing, Y. Trimetazidine and lcarnitine prevent heart aging and cardiac metabolic impairment in rats via regulating cardiac metabolic substrates. Exp. Gerontol. 2019, 119, 120–127. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Bulló, M.; Papandreou, C.; García-Gavilán, J.; Ruiz-Canela, M.; Guasch-Ferré, J.L.M.; Toledo, E.; Clish, C.; Corella, D.; Estruch, R.; Ros, E.; et al. Tricarboxylic acid cycle related-metabolites and risk of atrial fibrillation and heart failure. Metab. Clin. Exp. 2021, 125, 154915. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Koumanov, F.; Jin, B.; Yang, J.; Holman, G.D. Insulin signaling meets vesicle traffic of GLUT4 at a plasma-membrane-activated fusion step. Cell Metab. 2005, 2, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Nihonmatsu-Kikuchi, N.; Itokawa, M.; Rabbani, N.; Thornalley, P.J. Measurement of glyoxalase activities Biochem. Soc. Trans. 2014, 42, 491–494. [Google Scholar]
- Peischard, S.; Ho, H.T.; Piccini, I.; Strutz-Seebohm, N.; Ropke, A.; Liashkovich, I.; Gosain, H.; Rieger, B.; Klingel, K.; Eggers, B.; et al. The first versatile human iPSC-based model of ectopic virus induction allows new insights in RNA-virus disease. Sci. Rep. 2020, 10, 16804. [Google Scholar] [CrossRef] [PubMed]
- Krumpochova, P.; Bruyneela, B.; Molenaarb, D.; Koukoua, A.; Wuhrera, M.; Niessena, W.M.A.; Gierab, M. Amino acid analysis using chromatography–mass spectrometry: An inter platform comparison study. J. Pharm. Biomed. Anal. 2015, 114, 398–407. [Google Scholar] [CrossRef]
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.-R. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity 2020, 52, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification ofmammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Engelbrecht, B.; Espelage, B.C.; Klusmeier, N.; Tiemann, J.; Gawlowski, T.; Mattern, Y.; Eisenacher, M.; Meyer, H.E.; Rabbani, N.; et al. Glyoxalase 1-knockdown in human aortic endothelial cells—Effect on the proteome and endothelial function estimates. Sci. Rep. 2016, 6, 37737. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Begg, R.; Jesse, H.E.; Van Beilen, J.W.A.; Ali, S.; Svistunenko, D.; McLean, S.; Hellingwerf, K.J.; Sanguinetti, G.; Poole, R.K. Carbon Monoxide Gas Is Not Inert, but Global, in Its Consequences for Bacterial Gene Expression, Iron Acquisition, and Antibiotic Resistance. Antioxid. Redox Signal. 2016, 24, 1013–1028. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, M.; Zeng, W.; Chen, Z.; Lu, G.; Gong, Y.; Finnell, R.H.; Xiao, H.; Qiao, B.; Wang, H. Combined effects of FH (E404D) and ACOX2 (R409H) cause metabolic defects in primary cardiac malignant tumor. Cell Death Discov. 2018, 4, 70. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stratmann, B.; Eggers, B.; Mattern, Y.; Silva de Carvalho, T.; Marcus, K.; Tschoepe, D. Chronic Hyperglycaemia Inhibits Tricarboxylic Acid Cycle in Rat Cardiomyoblasts Overexpressing Glucose Transporter Type 4. Int. J. Mol. Sci. 2022, 23, 7255. https://doi.org/10.3390/ijms23137255
Stratmann B, Eggers B, Mattern Y, Silva de Carvalho T, Marcus K, Tschoepe D. Chronic Hyperglycaemia Inhibits Tricarboxylic Acid Cycle in Rat Cardiomyoblasts Overexpressing Glucose Transporter Type 4. International Journal of Molecular Sciences. 2022; 23(13):7255. https://doi.org/10.3390/ijms23137255
Chicago/Turabian StyleStratmann, Bernd, Britta Eggers, Yvonne Mattern, Tayana Silva de Carvalho, Katrin Marcus, and Diethelm Tschoepe. 2022. "Chronic Hyperglycaemia Inhibits Tricarboxylic Acid Cycle in Rat Cardiomyoblasts Overexpressing Glucose Transporter Type 4" International Journal of Molecular Sciences 23, no. 13: 7255. https://doi.org/10.3390/ijms23137255
APA StyleStratmann, B., Eggers, B., Mattern, Y., Silva de Carvalho, T., Marcus, K., & Tschoepe, D. (2022). Chronic Hyperglycaemia Inhibits Tricarboxylic Acid Cycle in Rat Cardiomyoblasts Overexpressing Glucose Transporter Type 4. International Journal of Molecular Sciences, 23(13), 7255. https://doi.org/10.3390/ijms23137255