
The Transition of Photoreceptor Guanylate Cyclase Type 1 to the Active State

 ,
, 
Abstract
:1. Introduction
2. Results
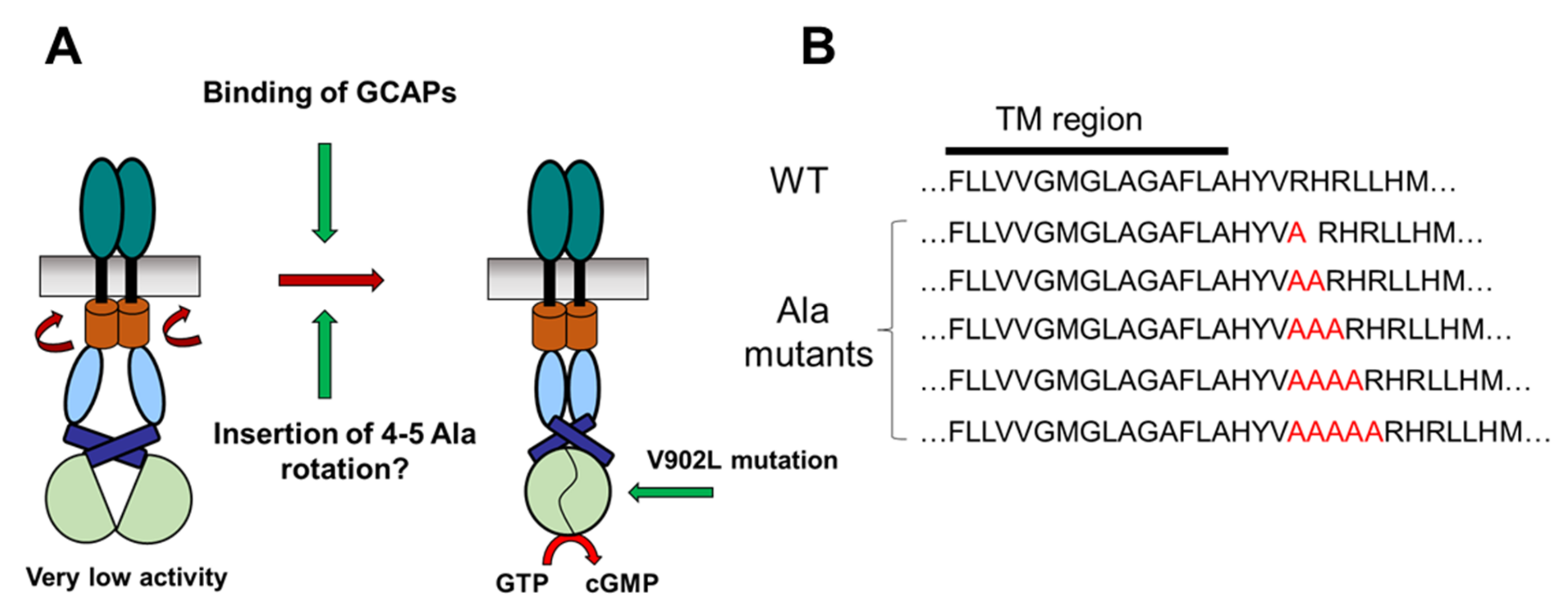
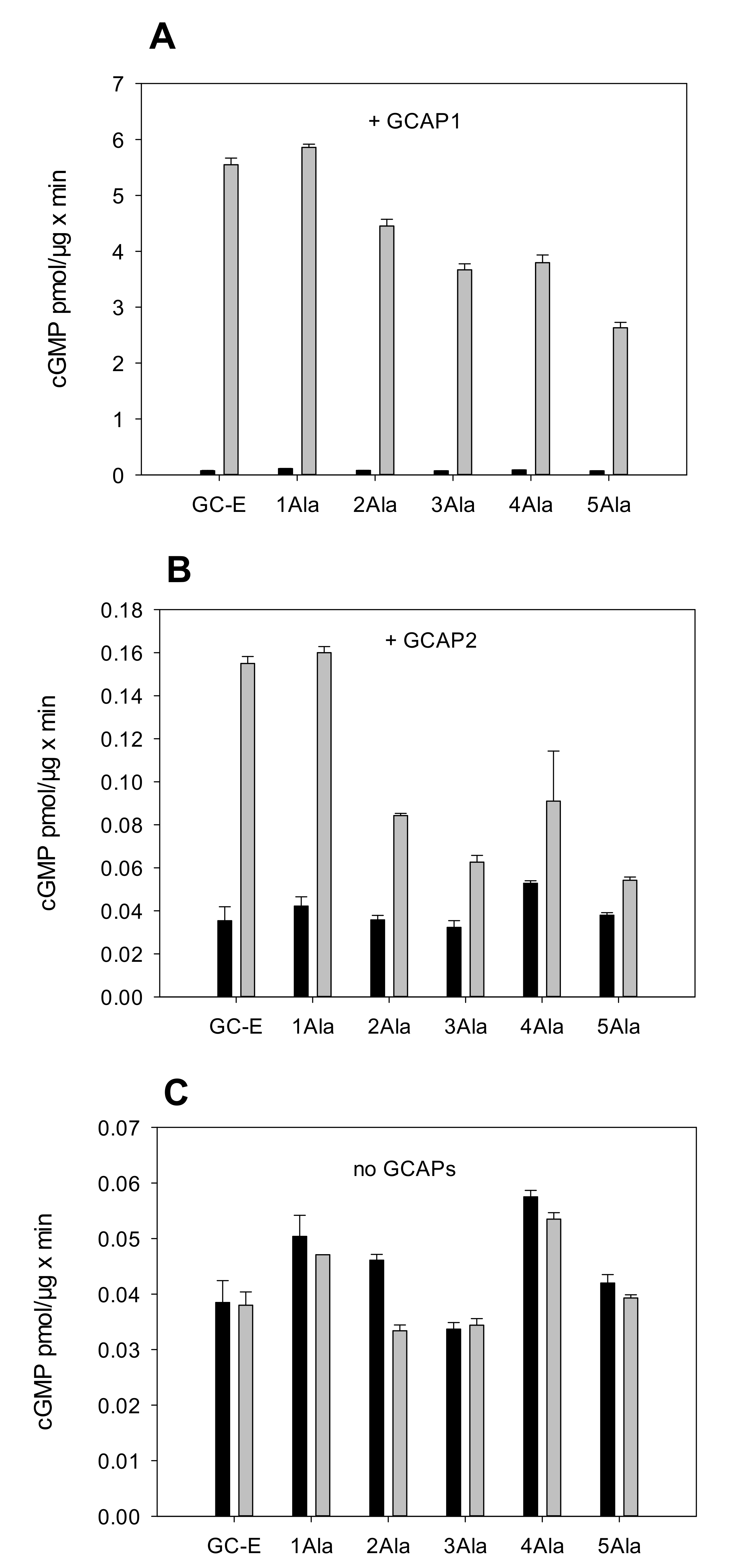
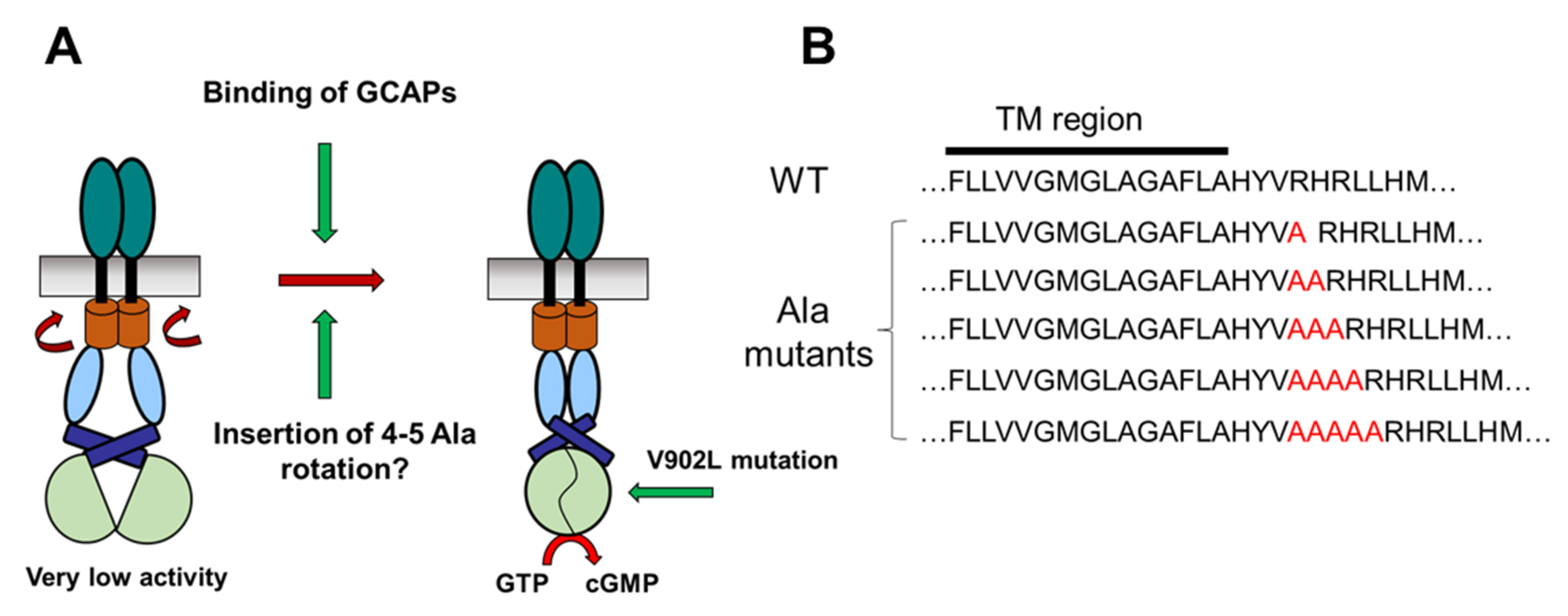
2.1. Test of the Rotation Model in the GC-E Activation Process
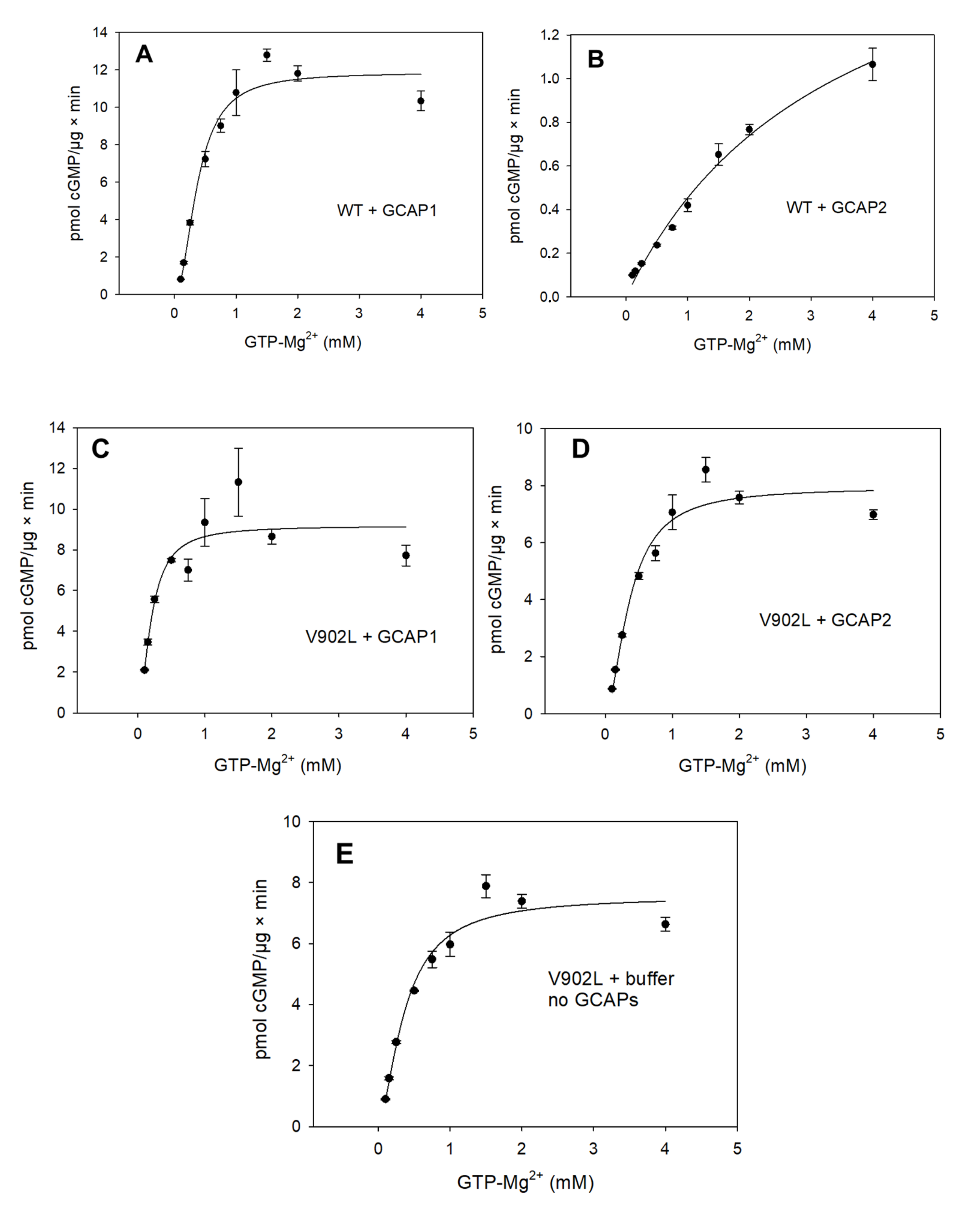
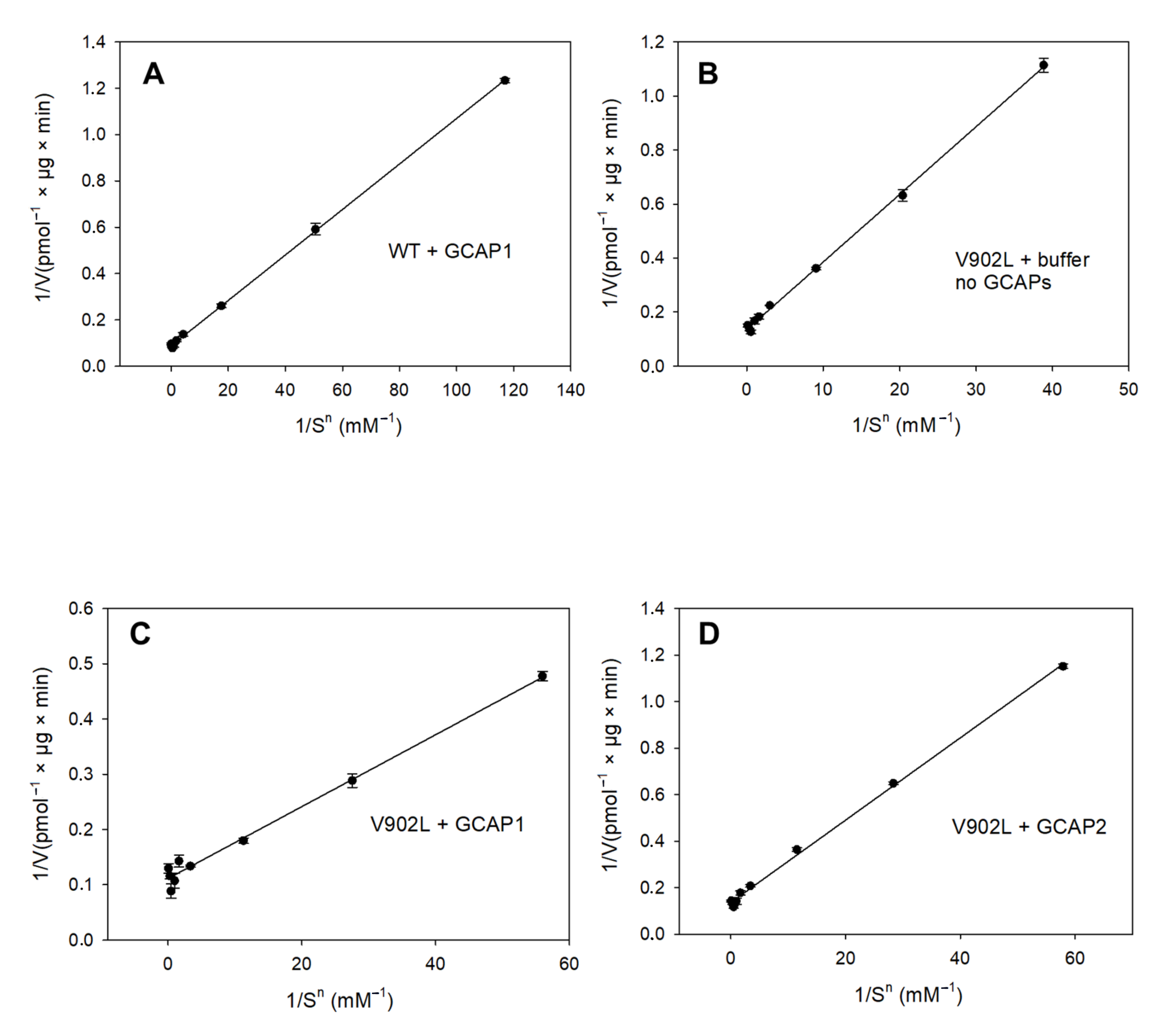
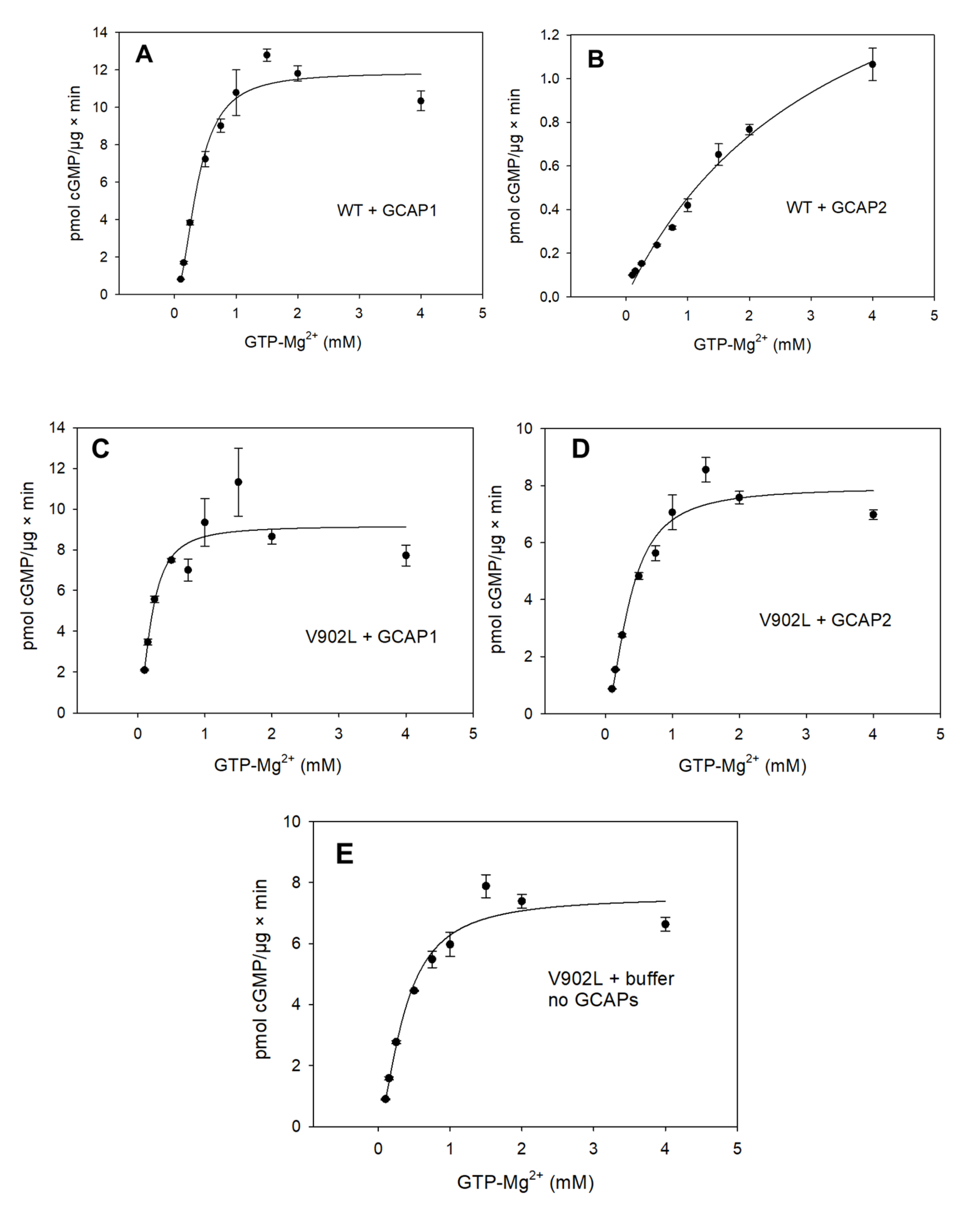
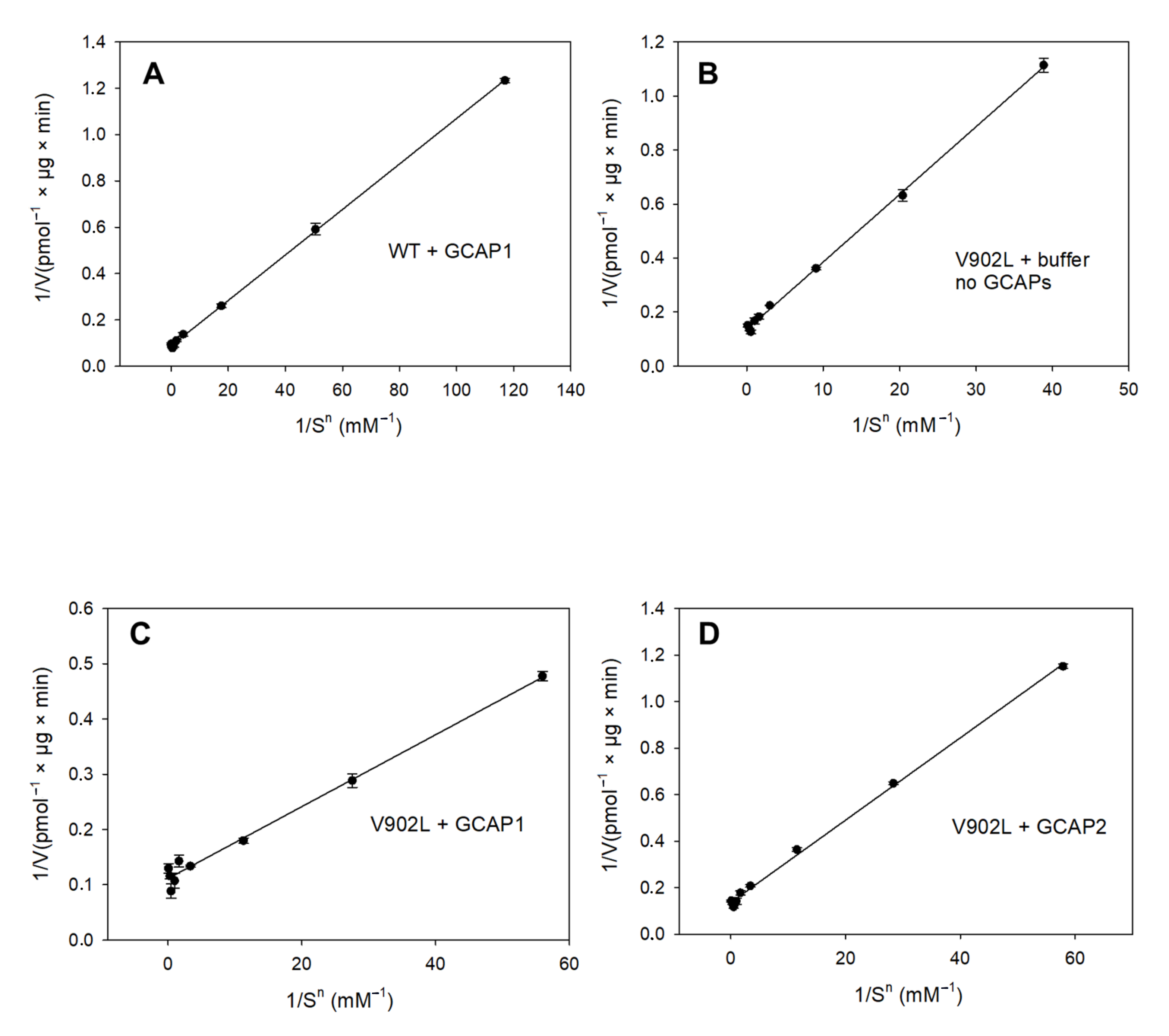
2.2. Kinetic Analysis of the Constitutively Active GC-E Mutant V902L
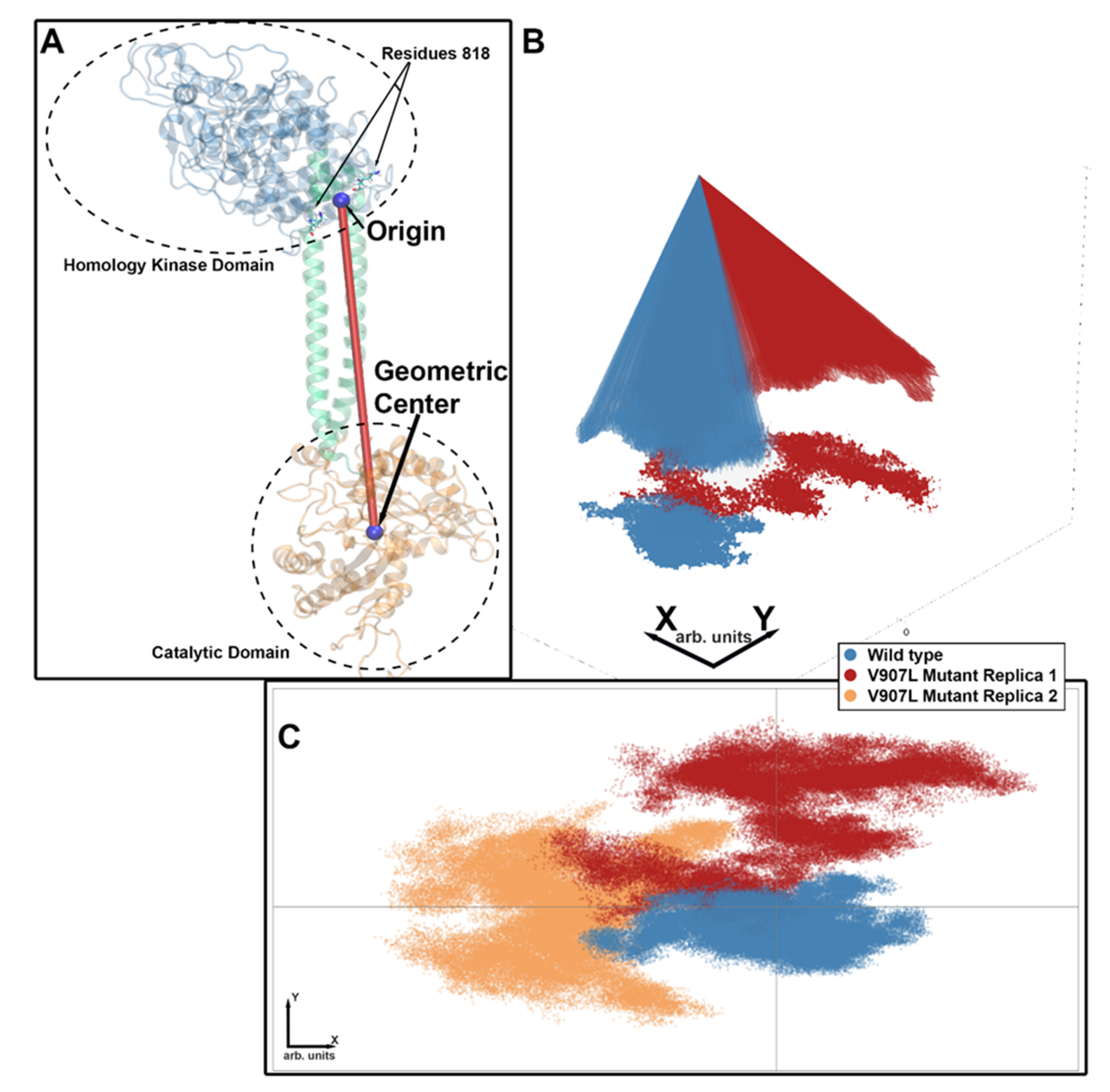
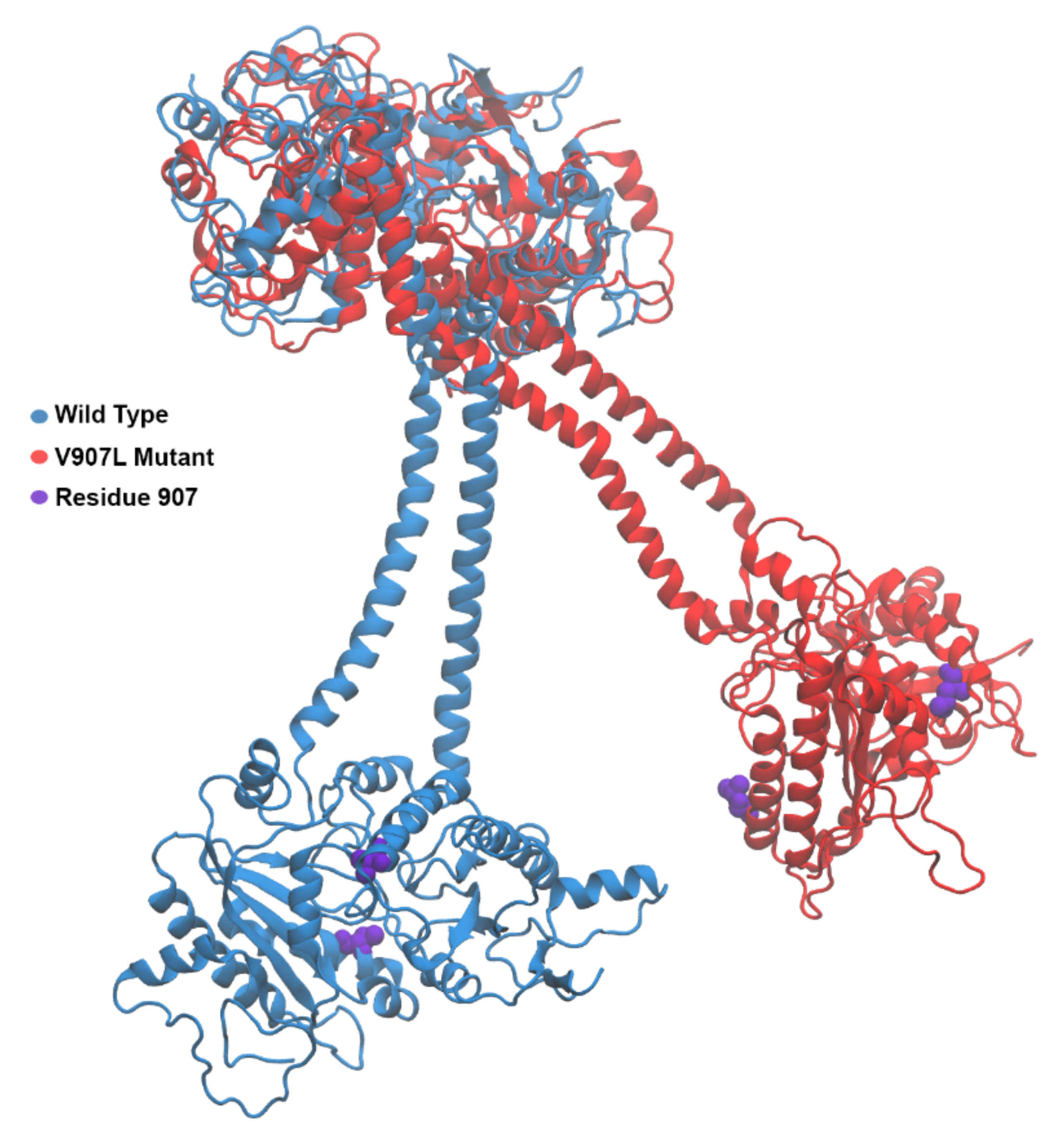
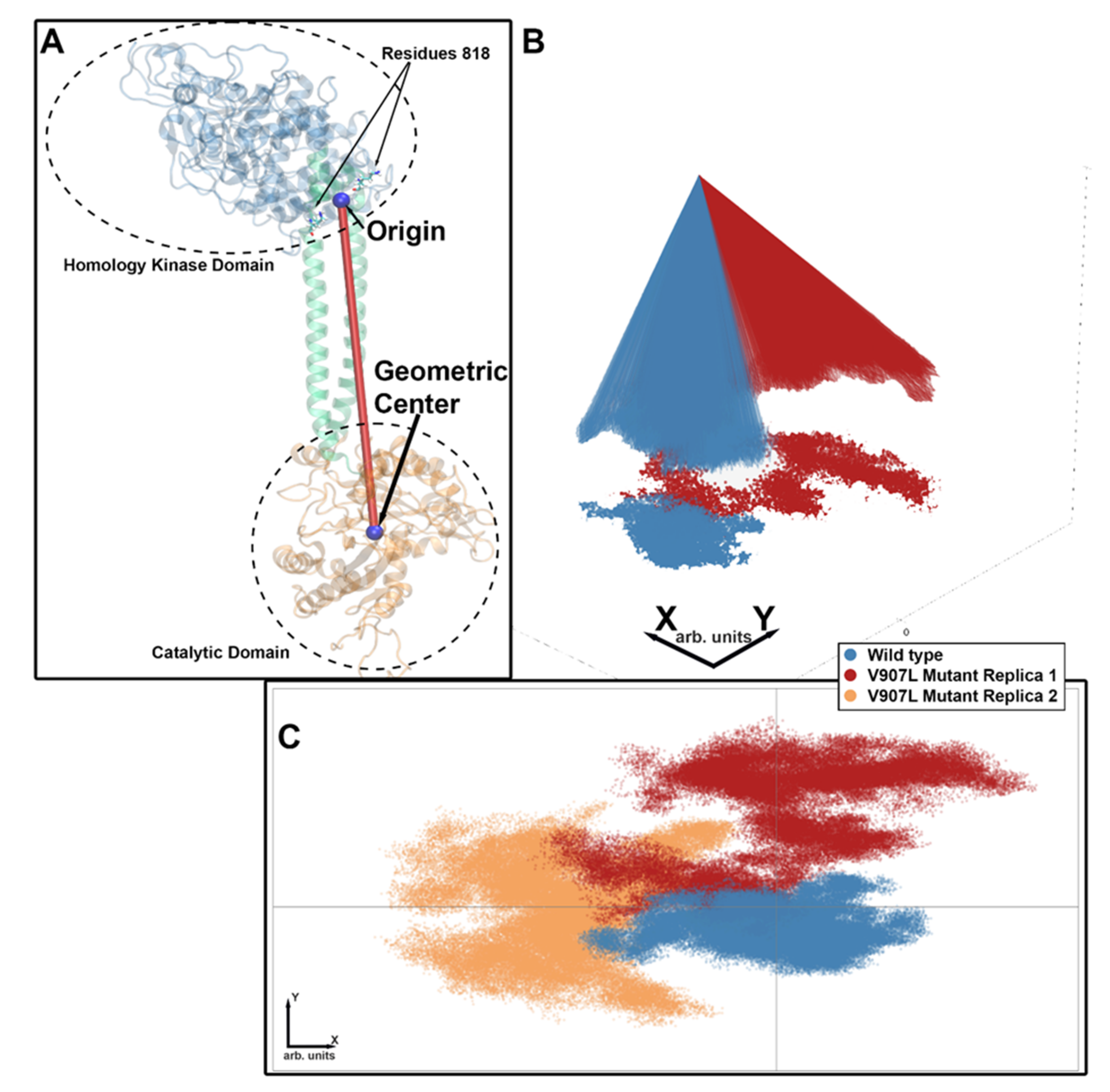
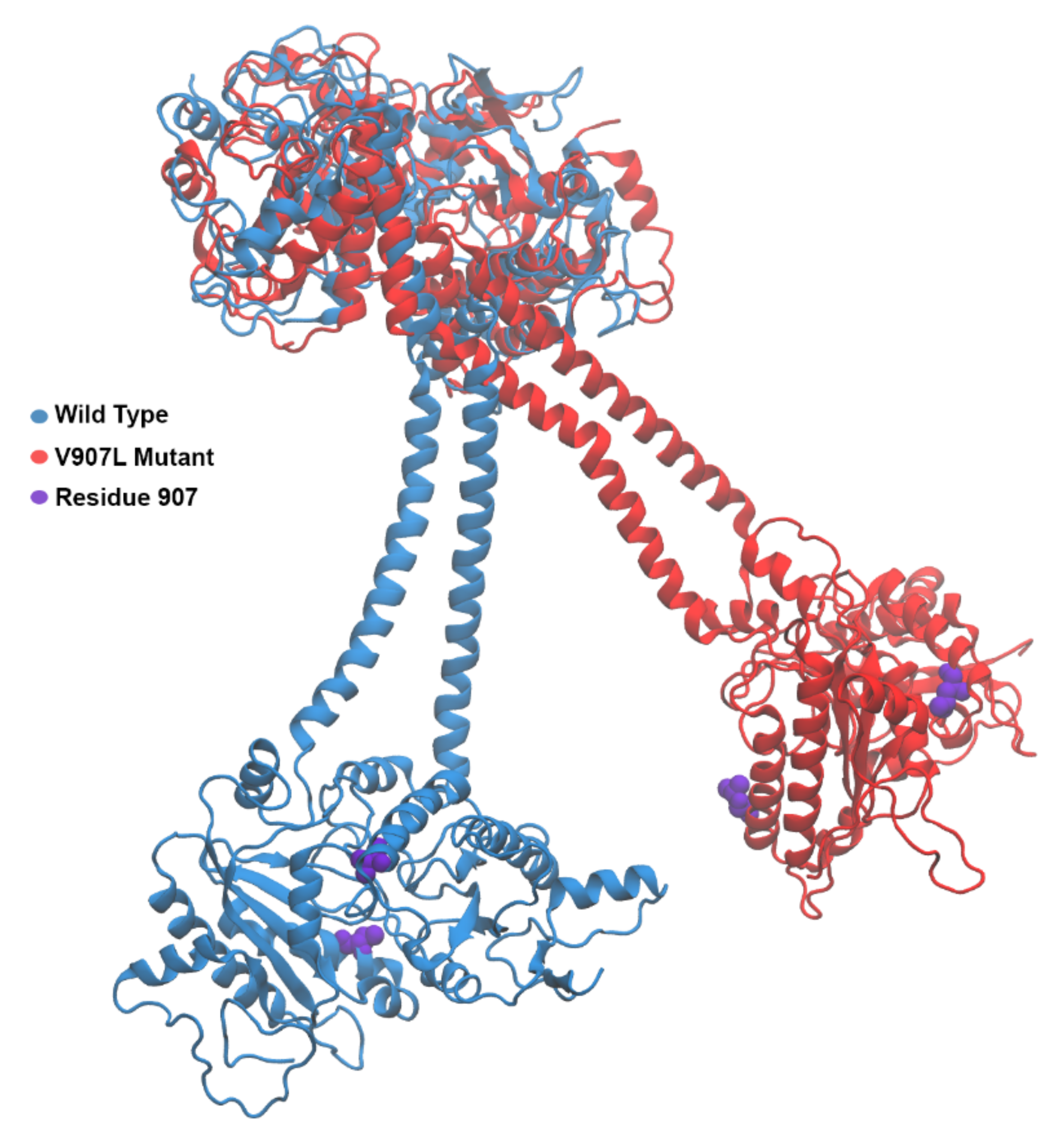
2.3. Molecular-Dynamics Simulations
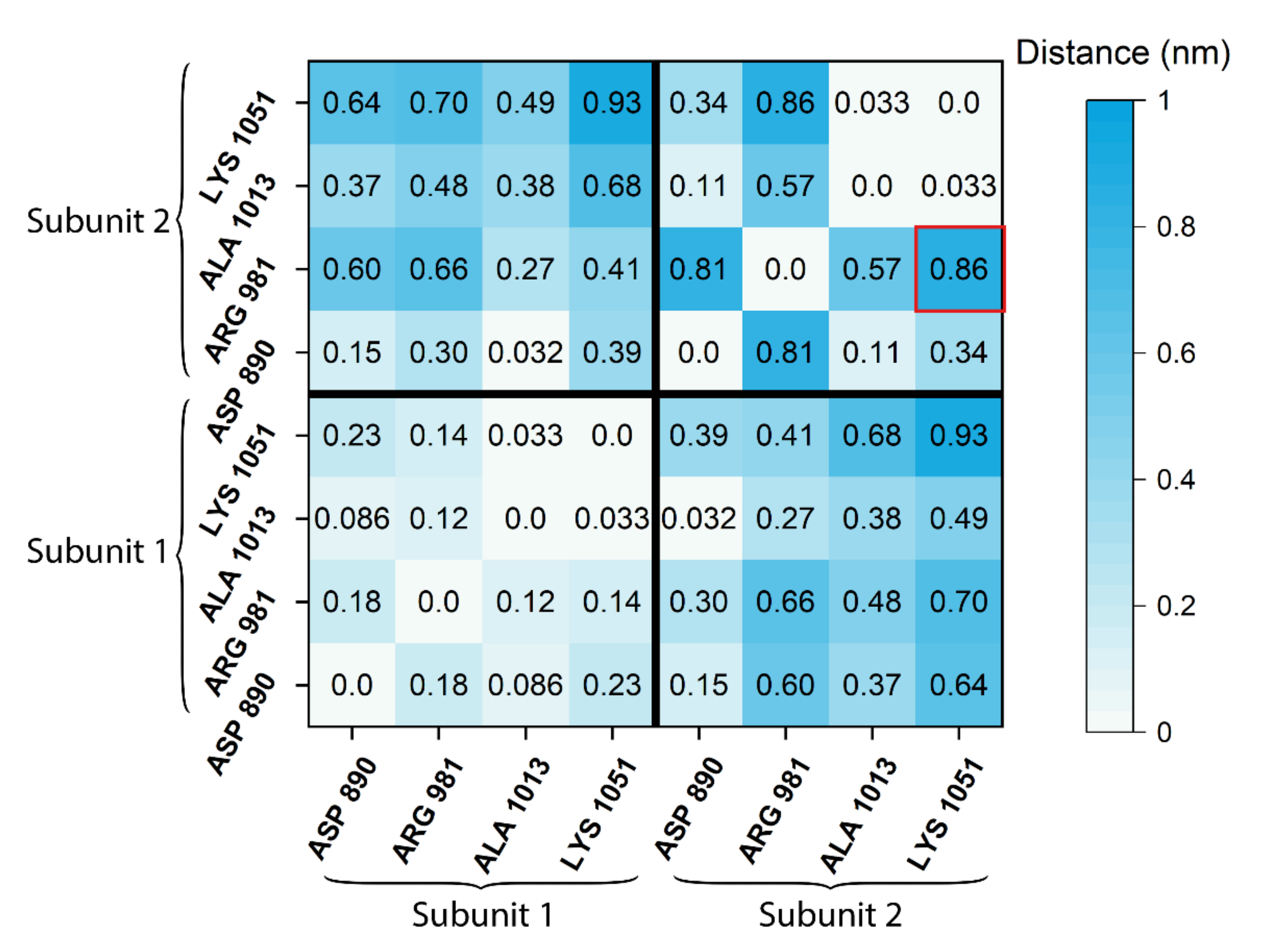
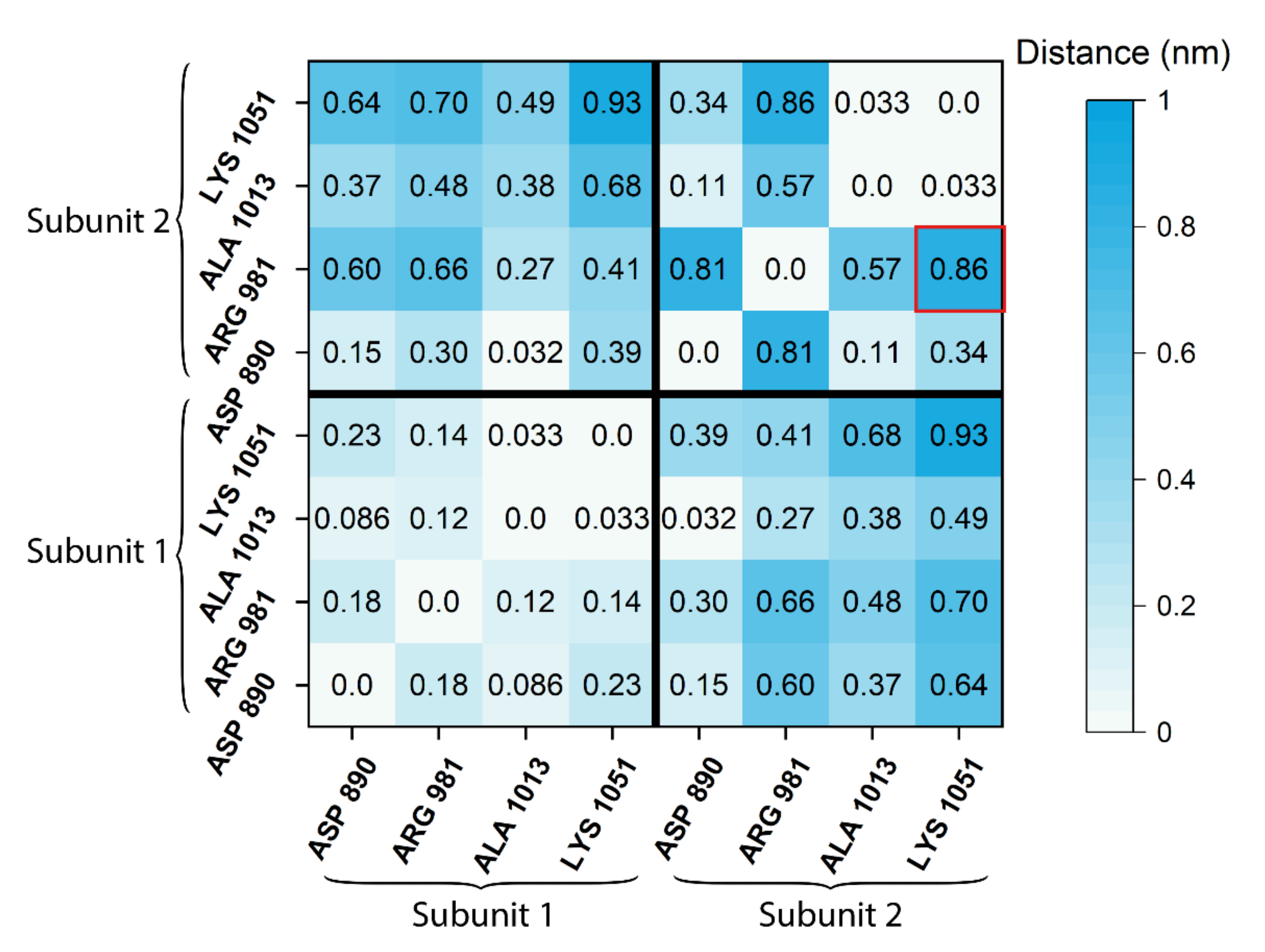
2.4. Difference Distance Matrix
3. Discussion
4. Materials and Methods
4.1. Cloning of Polyalanine GC-E Mutants
4.2. Heterologous Expression of GC-E, Polyalanine Mutants, and V902L Mutant
4.3. Electrophoresis and Western Blotting
4.4. Guanylate Cyclase Assay and Enzyme Kinetics
4.5. Expression and Purification of GCAP1 and GCAP2
4.6. Expression of the Catalytic Domain of GC
4.7. Chemiluminescence Detection and Quantification of Polyalanine Mutants
4.8. Quantification of WT GC-E and V902L Mutant
4.9. Molecular Dynamics Simulation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pichlo, M.; Bungert-Plümke, S.; Weyand, I.; Seifert, R.; Bönigk, W.; Strünker, T.; Kashikar, N.D.; Goodwin, N.; Müller, A.; Pelzer, P.; et al. High density and ligand affinity confer ultrasensitive signal detection by a guanylyl cyclase chemoreceptor. J. Cell Biol. 2014, 206, 541–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.K.; Duda, T.; Makino, C.L. Integrative Signaling Networks of Membrane Guanylate Cyclases: Biochemistry and Physiology. Front. Mol. Neurosci. 2016, 9, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.; Burns, M.E.; Sokal, I.; Dizhoor, A.M.; Baehr, W.; Palczewski, K.; Baylor, D.A.; Chen, J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc. Natl. Acad. Sci. USA 2001, 98, 9948–9953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.Y.; Lange, C.; Helten, A.; Höppner-Heitmann, D.; Duda, T.; Sharma, R.K.; Koch, K.W. Regulatory modes of rod outer segment membrane guanylate cyclase differ in catalytic efficiency and Ca(2+)-sensitivity. Eur. J. Biochem. 2003, 270, 3814–3821. [Google Scholar] [CrossRef] [PubMed]
- Peshenko, I.V.; Olshevskaya, E.V.; Savchenko, A.B.; Karan, S.; Palczewski, K.; Baehr, W.; Dizhoor, A.M. Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (RetGC) from mouse photoreceptors. Biochemistry 2011, 50, 5590–5600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, C.L.; Wen, X.H.; Olshevskaya, E.V.; Peshenko, I.V.; Savchenko, A.B.; Dizhoor, A.M. Enzymatic relay mechanism stimulates cyclic GMP synthesis in ro; photoresponse: Biochemical and physiological study in guanylyl cyclase activating protein 1 knockout mice. PLoS ONE 2012, 7, e47637. [Google Scholar] [CrossRef]
- Koch, K.W.; Dell’Orco, D. A calcium-relay mechanism in vertebrate phototransduction. ACS Chem. Neurosci. 2013, 4, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Zägel, P.; Dell’Orco, D.; Koch, K.W. The dimerization domain in outer segment guanylate cyclase is a Ca2⁺-sensitive control switch module. Biochemistry 2013, 52, 5065–5074. [Google Scholar] [CrossRef]
- Peshenko, I.V.; Olshevskaya, E.V.; Dizhoor, A.M. Evaluating the role of retinal membrane guanylyl cyclase 1 (RetGC1) domains in binding guanylyl cyclase-activating proteins (GCAPs). J. Biol. Chem. 2015, 290, 6913–6924. [Google Scholar] [CrossRef] [Green Version]
- Sulmann, S.; Kussrow, A.; Bornhop, D.J.; Koch, K.W. Label-free quantification of calcium-sensor targeting to photoreceptor guanylate cyclase and rhodopsin kinase by backscattering interferometry. Sci. Rep. 2017, 7, 45515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.W.; Dell’Orco, D. Protein and Signaling Networks in Vertebrate Photoreceptor Cells. Front. Mol. Neurosci. 2015, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azadi, S.; Molday, L.L.; Molday, R.S. RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. USA 2010, 107, 21158–21163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peshenko, I.V.; Olshevskaya, E.V.; Azadi, S.; Molday, L.L.; Molday, R.S.; Dizhoor, A.M. Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 2011, 50, 9511–9519. [Google Scholar] [CrossRef] [Green Version]
- Misono, K.S.; Philo, J.S.; Arakawa, T.; Ogata, C.M.; Qiu, Y.; Ogawa, H.; Young, H.S. Structure, signaling mechanism and regulation of the natriuretic peptide receptor guanylate cyclase. FEBS J. 2011, 278, 1818–1829. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, I.N. Activation of transmembrane cell-surface receptors via a common mechanism? The “rotation model”. Bioessays 2015, 37, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Parat, M.; Blanchet, J.; De Léan, A. Role of juxtamembrane and transmembrane domains in the mechanism of natriuretic peptide receptor A activation. Biochemistry 2010, 49, 4601–4610. [Google Scholar] [CrossRef]
- Dell’Orco, D.; Dal Cortivo, G. Normal GCAPs partly compensate for altered cGMP signaling in retinal dystrophies associated with mutations in GUCA1A. Sci. Rep. 2019, 9, 20105. [Google Scholar] [CrossRef]
- Avesani, A.; Marino, V.; Zanzoni, S.; Koch, K.W.; Dell’Orco, D. Molecular properties of human guanylate cyclase-activating protein 2 (GCAP2) and its retinal dystrophy-associated variant G157R. J. Biol. Chem. 2021, 296, 100619. [Google Scholar] [CrossRef]
- Kitiratschky, V.B.; Behnen, P.; Kellner, U.; Heckenlively, J.R.; Zrenner, E.; Jagle, H.; Kohl, S.; Wissinger, B.; Koch, K.W. Mutations in the GUCA1A gene involved in hereditary cone dystrophies impair calcium-mediated regulation of guanylate cyclase. Hum. Mutat. 2009, 30, E782–E796. [Google Scholar] [CrossRef]
- Dizhoor, A.M.; Boikov, S.G.; Olshevskaya, E.V. Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1. Possible role in causing human autosomal dominant cone degeneration. J. Biol. Chem. 1998, 273, 17311–17314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokal, I.; Li, N.; Surgucheva, I.; Warren, M.J.; Payne, A.M.; Bhattacharya, S.S.; Baehr, W.; Palczewski, K. GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol. Cell 1998, 2, 129–133. [Google Scholar] [CrossRef]
- Biasi, A.; Marino, V.; Dal Cortivo, G.; Maltese, P.E.; Modarelli, A.M.; Bertelli, M.; Colombo, L.; Dell’Orco, D. A Novel GUCA1A Variant Associated with Cone Dystrophy Alters cGMP Signaling in Photoreceptors by Strongly Interacting with and Hyperactivating Retinal Guanylate Cyclase. Int. J. Mol. Sci. 2021, 22, 10809. [Google Scholar] [CrossRef] [PubMed]
- Marino, V.; Dal Cortivo, G.; Oppici, E.; Maltese, P.E.; D’Esposito, F.; Manara, E.; Ziccardi, L.; Falsini, B.; Magli, A.; Bertelli, M.; et al. A novel p.(Glu111Val) missense mutation in GUCA1A associated with cone-rod dystrophy leads to impaired calcium sensing and perturbed second messenger homeostasis in photoreceptors. Hum. Mol. Genet. 2018, 27, 4204–4217. [Google Scholar] [CrossRef]
- Vocke, F.; Weisschuh, N.; Marino, V.; Malfatti, S.; Jacobson, S.G.; Reiff, C.M.; Dell’Orco, D.; Koch, K.W. Dysfunction of cGMP signalling in photoreceptors by a macular dystrophy-related mutation in the calcium sensor GCAP1. Hum. Mol. Genet. 2017, 26, 133–144. [Google Scholar] [CrossRef]
- Peshenko, I.V.; Cideciyan, A.V.; Sumaroka, A.; Olshevskaya, E.V.; Scholten, A.; Abbas, S.; Koch, K.W.; Jacobson, S.G.; Dizhoor, A.M. A G86R mutation in the calcium-sensor protein GCAP1 alters regulation of retinal guanylyl cyclase and causes dominant cone-rod degeneration. J. Biol. Chem. 2019, 294, 3476–3488. [Google Scholar] [CrossRef] [Green Version]
- Wimberg, H.; Lev, D.; Yosovich, K.; Namburi, P.; Banin, E.; Sharon, D.; Koch, K.W. Photoreceptor Guanylate Cyclase (GUCY2D) Mutations Cause Retinal Dystrophies by Severe Malfunction of Ca2+-Dependent Cyclic GMP Synthesis. Front. Mol. Neurosci. 2018, 11, 348. [Google Scholar] [CrossRef]
- Sharon, D.; Wimberg, H.; Kinarty, Y.; Koch, K.W. Genotype-functional-phenotype correlations in photoreceptor guanylate cyclase (GC-E) encoded by GUCY2D. Prog. Retin. Eye Res. 2018, 63, 69–91. [Google Scholar] [CrossRef]
- Rehkamp, A.; Tänzler, D.; Tüting, C.; Kastritis, P.L.; Iacobucci, C.; Ihling, C.H.; Kipping, C.; Koch, K.-W.; Sinz, A. First 3D-Structural Data of Full-Length Guanylyl Cyclase 1 in Rod-Outer-Segment Preparations of Bovine Retina by Cross-Linking/Mass Spectrometry. J. Mol. Biol. 2021, 433, 166947. [Google Scholar] [CrossRef]
- Lange, C.; Duda, T.; Beyermann, M.; Sharma, R.K.; Koch, K.W. Regions in vertebrate photoreceptor guanylyl cyclase ROS-GC1 involved in Ca(2+)-dependent regulation by guanylyl cyclase-activating protein GCAP-1. FEBS Lett. 1999, 460, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Krylov, D.M.; Hurley, J.B. Identification of proximate regions in a complex of retinal guanylyl cyclase 1 and guanylyl cyclase-activating protein-1 by a novel mass spectrometry-based method. J. Biol. Chem. 2001, 276, 30648–30654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.B.; Garbers, D.L. Two eye guanylyl cyclases are expressed in the same photoreceptor cells and form homomers in preference to heteromers. J. Biol. Chem. 1997, 272, 13738–13742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, C.L.; Hurley, J.H.; Miller, T.R.; Hurley, J.B. Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc. Natl. Acad. Sci. USA 1998, 95, 5993–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ruoho, A.E.; Rao, V.D.; Hurley, J.H. Catalytic mechanism of the adenylyl and guanylyl cyclases: Modeling and mutational analysis. Proc. Natl. Acad. Sci. USA 1997, 94, 13414–13419. [Google Scholar] [CrossRef] [Green Version]
- Schuhmann, F.; Korol, V.; Solov’yov, I.A. Introducing Pep McConst—A user-friendly peptide modeler for biophysical applications. J. Comput. Chem. 2021, 42, 572–580. [Google Scholar] [CrossRef]
- Koch, K.W. Target recognition of guanylate cyclase by guanylate cyclase-activating proteins. Adv. Exp. Med. Biol. 2002, 514, 349–360. [Google Scholar] [CrossRef]
- Scholten, A.; Koch, K.W. Differential calcium signaling by cone specific guanylate cyclase-activating proteins from the zebrafish retina. PLoS ONE 2011, 6, e23117. [Google Scholar] [CrossRef] [Green Version]
- Koch, K.W.; Helten, A. Guanylate cyclase-based signaling in photoreceptors and retina. In Signal Transduction in the Retina; Taylor and Francis CRC Press: Boca Raton, FL, USA, 2008; pp. 121–143. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Foloppe, N.; MacKerell, A.D. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, K.; Foloppe, N.; Baker, C.M.; Denning, E.J.; Nilsson, L.; MacKerell, A.D. Optimization of the CHARMM Additive Force Field for DNA: Improved Treatment of the BI/BII Conformational Equilibrium. J. Chem. Theory Comput. 2012, 8, 348–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavelites, J.J.; Gao, J.; Bash, P.A. A Molecular Mechanics Force Field for NAD+, NADH, and the Pyrophosphate Groups of Nucleotides. J. Comput. Chem. 1997, 18, 221–239. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Banavali, N.K. All-Atom Empirical Force Field for Nucleic Acids: II. Application to Molecular Dynamics Simulations of DNA and RNA in Solution. J. Comput. Chem. 2000, 21, 105–120. [Google Scholar] [CrossRef]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; MacKerell, A.D. Impact of 20-Hydroxyl Sampling on the Conformational Properties of RNA: Update of the CHARMM All-Atom Additive Force Field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc. 2004, 126, 698–699. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Korol, V.; Husen, P.; Sjulstok, E.; Nielsen, C.; Friis, I.; Frederiksen, A.; Salo, A.B.; Solov’yov, I.A. Introducing VIKING: A Novel Online Platform for Multiscale Modeling. ACS Omega 2019, 5, 1254–1260. [Google Scholar] [CrossRef]
- Schuhmann, F.; Kattnig, D.R.; Solov’yov, I.A. Exploring Post-activation Conformational Changes in Pigeon Cryptochrome 4. J. Phys. Chem. B 2021, 125, 9652–9659. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, L.P.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Vmax (pmol/µg × min) | EC50 (mM) | KM (mM) | Kcat (s−1) | Kcat/KM (103 M−1 × s−1) | Hill Parameter h |
|---|---|---|---|---|---|---|
| WT GC-E + GCAP1 | 11.33 | 0.37 | 0.34 | 0.8 | 2.35 | 2.07 |
| WT GC-E + GCAP2 | - | 3.45 | - | - | - | - |
| V902L + GCAP1 | 9 | 0.20 | 0.20 | 0.8 | 4 | 1.75 |
| V902L + GCAP2 | 7.32 | 0.36 | 0.31 | 0.7 | 2.2 | 1.76 |
| V902L no GCAPs | 7.36 | 0.37 | 0.34 | 0.7 | 2.1 | 1.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahu, M.K.; Schuhmann, F.; Scholten, A.; Solov’yov, I.A.; Koch, K.-W. The Transition of Photoreceptor Guanylate Cyclase Type 1 to the Active State. Int. J. Mol. Sci. 2022, 23, 4030. https://doi.org/10.3390/ijms23074030
Shahu MK, Schuhmann F, Scholten A, Solov’yov IA, Koch K-W. The Transition of Photoreceptor Guanylate Cyclase Type 1 to the Active State. International Journal of Molecular Sciences. 2022; 23(7):4030. https://doi.org/10.3390/ijms23074030
Chicago/Turabian StyleShahu, Manisha Kumari, Fabian Schuhmann, Alexander Scholten, Ilia A. Solov’yov, and Karl-Wilhelm Koch. 2022. "The Transition of Photoreceptor Guanylate Cyclase Type 1 to the Active State" International Journal of Molecular Sciences 23, no. 7: 4030. https://doi.org/10.3390/ijms23074030
APA StyleShahu, M. K., Schuhmann, F., Scholten, A., Solov’yov, I. A., & Koch, K.-W. (2022). The Transition of Photoreceptor Guanylate Cyclase Type 1 to the Active State. International Journal of Molecular Sciences, 23(7), 4030. https://doi.org/10.3390/ijms23074030