
Neurotropic RNA Virus Modulation of Immune Responses within the Central Nervous System

Abstract
1. Introduction
2. Several RNA Viruses Cause Neurotropic Disease
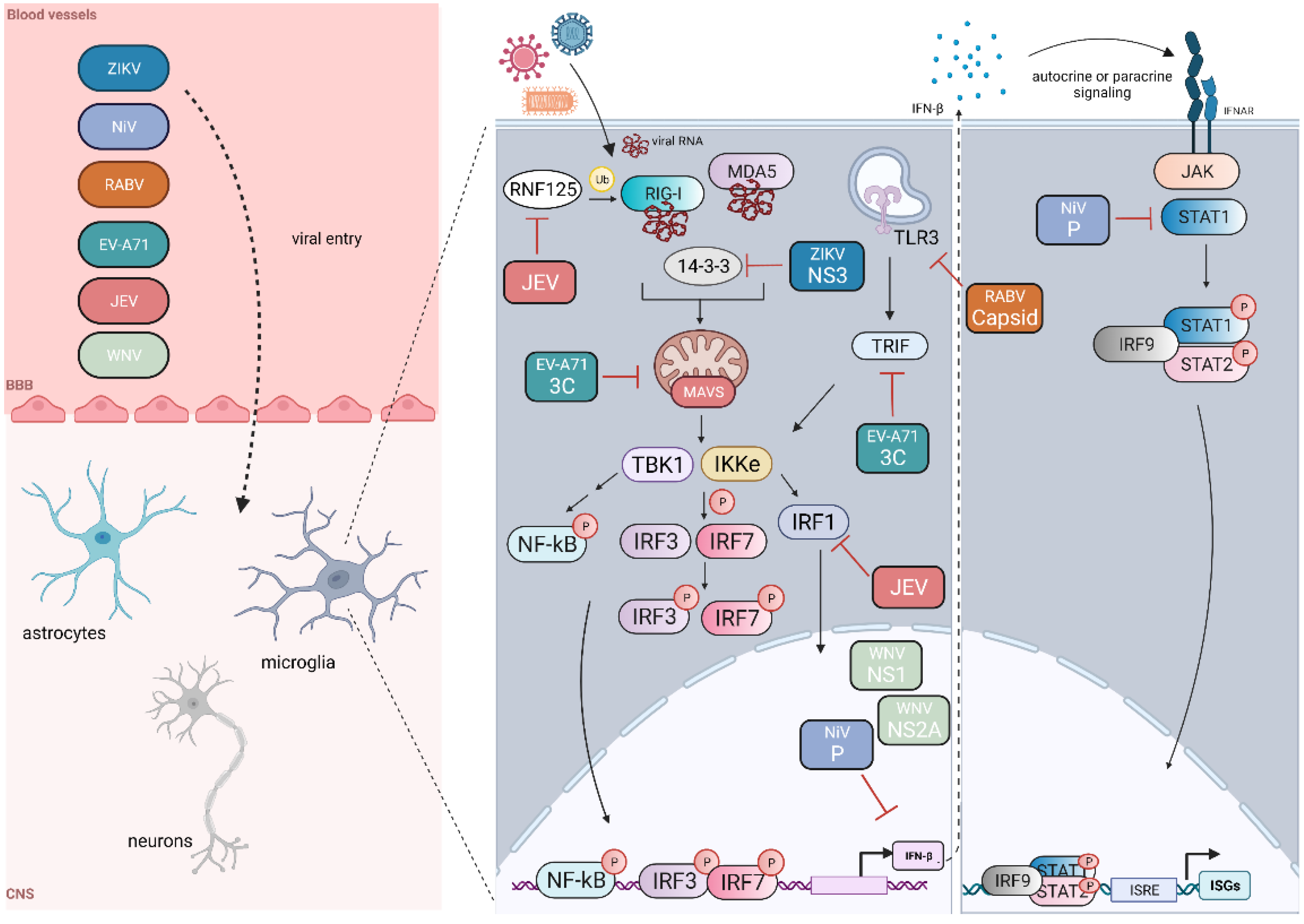
3. Immune Modulation Strategies
3.1. Regulation of Interferon Induction and Response
3.2. Sequestration of Host Immune Proteins
3.3. Post-Translational Modifications and Interactions with Modifying Enzymes
3.4. Maintenance of Blood–Brain Barrier Integrity
3.5. Modulation of Autophagy
3.6. Targeting Host Immune Proteins for Cleavage and Degradation
3.7. Regulation of microRNAs and Host or Viral Gene Expression
3.8. Escape of Adaptive Immune Responses and Molecules
3.9. Amino Acid Mutations Driven by Adaptation
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffin, D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003, 3, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, G. Immune response and blood–brain barrier dysfunction during viral neuroinvasion. Innate Immun. 2021, 27, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Koury, J.; Kaul, M. Innate Immune Sensing of Viruses and Its Consequences for the Central Nervous System. Viruses 2021, 13, 170. [Google Scholar] [CrossRef]
- Ludlow, L.E.; Lo, M.K.; Rodriguez, J.J.; Rota, P.A.; Horvath, C.M. Henipavirus V Protein Association with Polo-Like Kinase Reveals Functional Overlap with STAT1 Binding and Interferon Evasion. J. Virol. 2008, 82, 6259–6271. [Google Scholar] [CrossRef][Green Version]
- Carty, M.; Reinert, L.; Paludan, S.R.; Bowie, A.G. Innate antiviral signalling in the central nervous system. Trends Immunol. 2014, 35, 79–87. [Google Scholar] [CrossRef]
- Jeffries, A.M.; Marriott, I. Cytosolic DNA Sensors and CNS Responses to Viral Pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 576263. [Google Scholar] [CrossRef]
- Welsch, J.C.; Charvet, B.; Dussurgey, S.; Allatif, O.; Aurine, N.; Horvat, B.; Gerlier, D.; Mathieu, C. Type I Interferon Receptor Signaling Drives Selective Permissiveness of Astrocytes and Microglia to Measles Virus during Brain Infection. J. Virol. 2019, 93, e00618-19. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Peltier, D.C.; Lazear, H.M.; Farmer, J.R.; Diamond, M.S.; Miller, D.J. Neurotropic Arboviruses Induce Interferon Regulatory Factor 3-Mediated Neuronal Responses That Are Cytoprotective, Interferon Independent, and Inhibited by Western Equine Encephalitis Virus Capsid. J. Virol. 2013, 87, 1821–1833. [Google Scholar] [CrossRef]
- Satterfield, B.A.; Borisevich, V.; Foster, S.L.; Rodriguez, S.E.; Cross, R.W.; Fenton, K.A.; Agans, K.N.; Basler, C.; Geisbert, T.W.; Mire, C.E. Antagonism of STAT1 by Nipah virus P gene products modulates disease course but not lethal outcome in the ferret model. Sci. Rep. 2019, 9, 16710. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Alpha/Beta Interferon Protects against Lethal West Nile Virus Infection by Restricting Cellular Tropism and Enhancing Neuronal Survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [PubMed]
- Delhaye, S.; Paul, S.; Blakqori, G.; Minet, M.; Weber, F.; Staeheli, P.; Michiels, T. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. USA 2006, 103, 7835–7840. [Google Scholar] [CrossRef] [PubMed]
- Preéhaud, C.; Meégret, F.; Lafage, M.; Lafon, M. Virus Infection Switches TLR-3-Positive Human Neurons To Become Strong Producers of Beta Interferon. J. Virol. 2005, 79, 12893–12904. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Shrestha, B.; Sen, G.C.; Diamond, M.S. A Role for Ifit2 in Restricting West Nile Virus Infection in the Brain. J. Virol. 2013, 87, 8363–8371. [Google Scholar] [CrossRef]
- Kim, M.-J.; Hwang, S.-Y.; Imaizumi, T.; Yoo, J.-Y. Negative Feedback Regulation of RIG-I-Mediated Antiviral Signaling by Interferon-Induced ISG15 Conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018, 37, 351–366. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Javed, A.O.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef]
- Scott, M.S.; Calafell, S.J.; Thomas, D.Y.; Hallett, M.T. Refining Protein Subcellular Localization. PLoS Comput. Biol. 2005, 1, e66. [Google Scholar] [CrossRef]
- Nazmi, A.; Dutta, K.; Basu, A. RIG-I Mediates Innate Immune Response in Mouse Neurons Following Japanese Encephalitis Virus Infection. PLoS ONE 2011, 6, e21761. [Google Scholar] [CrossRef]
- De Miranda, J.; Yaddanapudi, K.; Hornig, M.; Lipkin, W.I. Astrocytes recognize intracellular polyinosinic-polycytidylic acid via MDA-5. FASEB J. 2008, 23, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Ménager, P.; Roux, P.; Mégret, F.; Bourgeois, J.-P.; Le Sourd, A.-M.; Danckaert, A.; Lafage, M.; Préhaud, C.; Lafon, M. Toll-Like Receptor 3 (TLR3) Plays a Major Role in the Formation of Rabies Virus Negri Bodies. PLoS Pathog. 2009, 5, e1000315. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.K.; Morrison, D.K. Unlocking the code of 14-3-3. J. Cell Sci. 2004, 117, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Riedl, W.; Acharya, D.; Lee, J.-H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 2019, 26, 493–503.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Loo, Y.-M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The Mitochondrial Targeting Chaperone 14-3-3ε Regulates a RIG-I Translocon that Mediates Membrane Association and Innate Antiviral Immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.K.; Miller, D.; Aljofan, M.; Mungall, B.A.; Rollin, P.E.; Bellini, W.J.; Rota, P.A. Characterization of the antiviral and inflammatory responses against Nipah virus in endothelial cells and neurons. Virology 2010, 404, 78–88. [Google Scholar] [CrossRef]
- Konat, G.W.; Kielian, T.; Marriott, I. The role of Toll-like receptors in CNS response to microbial challenge. J. Neurochem. 2006, 99, 1–12. [Google Scholar] [CrossRef]
- Peltier, D.C.; Simms, A.; Farmer, J.R.; Miller, D.J. Human Neuronal Cells Possess Functional Cytoplasmic and TLR-Mediated Innate Immune Pathways Influenced by Phosphatidylinositol-3 Kinase Signaling. J. Immunol. 2010, 184, 7010–7021. [Google Scholar] [CrossRef]
- Dong, L.; Li, Y.Z.; An, H.T.; Wang, Y.L.; Chen, S.H.; Qian, Y.J.; Wang, K.; Zhen, J.L.; Fan, Z.; Gong, X.L.; et al. The E3 Ubiquitin Ligase c-Cbl Inhibits Microglia-Mediated CNS Inflammation by Regulating PI3K/Akt/NF-κB Pathway. CNS Neurosci. Ther. 2016, 22, 661–669. [Google Scholar] [CrossRef]
- Ndoja, A.; Reja, R.; Lee, S.-H.; Webster, J.D.; Ngu, H.; Rose, C.M.; Kirkpatrick, D.S.; Modrusan, Z.; Chen, Y.-J.J.; Dugger, D.L.; et al. Ubiquitin Ligase COP1 Suppresses Neuroinflammation by Degrading c/EBPβ in Microglia. Cell 2020, 182, 1156–1169.e12. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Chang, J.-H.; Xiao, Y.; Brittain, G.C.; Yu, J.; Zhou, X.; Wang, Y.-H.; Cheng, X.; Li, P.; et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat. Immunol. 2011, 12, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.-H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, Ø.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, J.; Zou, Q.; Hu, H.; Cheng, X.; Sun, S.-C. Peli1 negatively regulates type I interferon induction and antiviral immunity in the CNS. Cell Biosci. 2015, 5, 1–8. [Google Scholar] [CrossRef][Green Version]
- Luo, H.; Winkelmann, E.R.; Zhu, S.; Ru, W.; Mays, E.; Silvas, J.A.; Vollmer, L.L.; Gao, J.; Peng, B.-H.; Bopp, N.E.; et al. Peli1 facilitates virus replication and promotes neuroinflammation during West Nile virus infection. J. Clin. Investig. 2018, 128, 4980–4991. [Google Scholar] [CrossRef]
- Daniels, B.; Snyder, A.G.; Olsen, T.M.; Orozco, S.L.; Oguin, T.H.; Tait, S.W.G.; Martinez, J.; Gale, M.; Loo, Y.-M.; Oberst, A. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell 2017, 169, 301–313.e11. [Google Scholar] [CrossRef]
- Rastogi, M.; Sharma, N.; Singh, S.K. Flavivirus NS1: A multifaceted enigmatic viral protein. Virol. J. 2016, 13, 131. [Google Scholar] [CrossRef]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef]
- Pryor, M.J.; Wright, P.J. Glycosylation Mutants of Dengue Virus NS1 Protein. J. Gen. Virol. 1994, 75, 1183–1187. [Google Scholar] [CrossRef]
- Muylaert, I.R.; Chambers, T.J.; Galler, R.; Rice, C.M. Mutagenesis of the N-Linked Glycosylation Sites of the Yellow Fever Virus NS1 Protein: Effects on Virus Replication and Mouse Neurovirulence. Virology 1996, 222, 159–168. [Google Scholar] [CrossRef]
- Dahm, T.; Rudolph, H.; Schwerk, C.; Schroten, H.; Tenenbaum, T. Neuroinvasion and Inflammation in Viral Central Nervous System Infections. Mediat. Inflamm. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Daniels, B.P.; Shrestha, B.; Proenca-Modena, J.L.; Lew, E.D.; Lazear, H.M.; Gorman, M.J.; Lemke, G.; Klein, R.S.; Diamond, M.S. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat. Med. 2015, 21, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Nunez, N.V.; Gaudin, R. A viral journey to the brain: Current considerations and future developments. PLoS Pathog. 2020, 16, e1008434. [Google Scholar] [CrossRef] [PubMed]
- Cain, M.D.; Salimi, H.; Diamond, M.S.; Klein, R.S. Mechanisms of Pathogen Invasion into the Central Nervous System. Neuron 2019, 103, 771–783. [Google Scholar] [CrossRef] [PubMed]
- MacGibeny, M.A.; Koyuncu, O.O.; Wirblich, C.; Schnell, M.; Enquist, L.W. Retrograde axonal transport of rabies virus is unaffected by interferon treatment but blocked by emetine locally in axons. PLoS Pathog. 2018, 14, e1007188. [Google Scholar] [CrossRef]
- Tiong, V.; Shu, M.-H.; Wong, W.F.; Abubakar, S.; Chang, L.-Y. Nipah Virus Infection of Immature Dendritic Cells Increases Its Transendothelial Migration Across Human Brain Microvascular Endothelial Cells. Front. Microbiol. 2018, 9, 2747. [Google Scholar] [CrossRef]
- Wong, K.T.; Shieh, W.J.; Kumar, S.; Norain, K.; Abdullah, W.; Guarner, J.; Goldsmith, C.S.; Chua, K.B.; Lam, S.K.; Tan, C.T.; et al. Nipah virus infection: Pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am. J. Pathol. 2002, 161, 2153–2167. [Google Scholar] [CrossRef]
- Ferren, M.; Horvat, B.; Mathieu, C. Measles Encephalitis: Towards New Therapeutics. Viruses 2019, 11, 1017. [Google Scholar] [CrossRef]
- Willoughby, R.E.; Tieves, K.S.; Hoffman, G.M.; Ghanayem, N.S.; Amlie-Lefond, C.M.; Schwabe, M.J.; Chusid, M.J.; Rupprecht, C.E. Survival after Treatment of Rabies with Induction of Coma. N. Engl. J. Med. 2005, 352, 2508–2514. [Google Scholar] [CrossRef]
- Roy, A.; Hooper, D.C. Immune evasion by rabies viruses through the maintenance of blood-brain barrier integrity. J. NeuroVirology 2008, 14, 401–411. [Google Scholar] [CrossRef]
- Long, T.; Zhang, B.; Fan, R.; Wu, Y.; Mo, M.; Luo, J.; Chang, Y.; Tian, Q.; Mei, M.; Jiang, H.; et al. Phosphoprotein Gene of Wild-Type Rabies Virus Plays a Role in Limiting Viral Pathogenicity and Lowering the Enhancement of BBB Permeability. Front. Microbiol. 2020, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Daniels, B.P.; Pinto, A.K.; Huang, A.C.; Vick, S.C.; Doyle, S.E.; Gale, M., Jr.; Klein, R.S.; Diamond, M.S. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl. Med. 2015, 7, 284ra59. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M.; Diamond, M.S. Toll-Like Receptor 3 Has a Protective Role against West Nile Virus Infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.-Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef]
- Bar-Yosef, T.; Damri, O.; Agam, G. Dual Role of Autophagy in Diseases of the Central Nervous System. Front. Cell. Neurosci. 2019, 13, 196. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar]
- Lin, J.-Y.; Huang, H.-I. Autophagy is induced and supports virus replication in Enterovirus A71-infected human primary neuronal cells. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Barral, P.M.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. RIG-I is cleaved during picornavirus infection. Virology 2009, 391, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Fanunza, E.; Carletti, F.; Quartu, M.; Grandi, N.; Ermellino, L.; Milia, J.; Corona, A.; Capobianchi, M.R.; Ippolito, G.; Tramontano, E. Zika virus NS2A inhibits interferon signaling by degradation of STAT1 and STAT2. Virulence 2021, 12, 1580–1596. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-I.; Lin, J.-Y.; Chen, S.-H. EV71 Infection Induces IFNβ Expression in Neural Cells. Viruses 2019, 11, 1121. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 Protease 2Apro Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Pang, Z.; Zhou, Z.; Li, X.; Liu, S.; Cheng, J.; Liu, P.; Tan, W.; Wang, Z.; Wang, T. Enterovirus D68 Protease 2A pro Targets TRAF3 To Subvert Host Innate Immune Responses. J. Virol. 2021, 95, e01856-20. [Google Scholar] [CrossRef]
- Xiang, Z.; Li, L.; Lei, X.; Zhou, H.; Zhou, Z.; He, B.; Wang, J. Enterovirus 68 3C Protease Cleaves TRIF To Attenuate Antiviral Responses Mediated by Toll-Like Receptor 3. J. Virol. 2014, 88, 6650–6659. [Google Scholar] [CrossRef]
- Cao, D.-D.; Li, L.; Chan, W.-Y. MicroRNAs: Key Regulators in the Central Nervous System and Their Implication in Neurological Diseases. Int. J. Mol. Sci. 2016, 17, 842. [Google Scholar] [CrossRef]
- Rastogi, M.; Srivastava, N.; Singh, S.K. Exploitation of microRNAs by Japanese Encephalitis virus in human microglial cells. J. Med. Virol. 2018, 90, 648–654. [Google Scholar] [CrossRef]
- Mukherjee, S.; Akbar, I.; Bhagat, R.; Hazra, B.; Bhattacharyya, A.; Seth, P.; Roy, D.; Basu, A. Identification and Classification of Hubs in microRNA Target Gene Networks in Human Neural Stem/Progenitor Cells following Japanese Encephalitis Virus Infection. mSphere 2019, 4, e00588-19. [Google Scholar] [CrossRef]
- Zhu, B.; Ye, J.; Nie, Y.; Ashraf, U.; Zohaib, A.; Duan, X.; Fu, Z.F.; Song, Y.; Chen, H.; Cao, S. MicroRNA-15b Modulates Japanese Encephalitis Virus–Mediated Inflammation via Targeting RNF125. J. Immunol. 2015, 195, 2251–2262. [Google Scholar] [CrossRef]
- Hazra, B.; Kumawat, K.L.; Basu, A. The host microRNA miR-301a blocks the IRF1-mediated neuronal innate immune response to Japanese encephalitis virus infection. Sci. Signal. 2017, 10, eaaf5185. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, Q.; Cao, Q.; Chen, H.; Qian, P. Japanese Encephalitis Virus Upregulates the Expression of SOCS3 in Mouse Brain and Raw264.7 Cells. Viruses 2014, 6, 4280–4293. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Kumawat, K.L.; Rastogi, M.; Basu, A.; Singh, S.K. Japanese Encephalitis Virus exploits the microRNA-432 to regulate the expression of Suppressor of Cytokine Signaling (SOCS) 5. Sci. Rep. 2016, 6, 27685. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Gardner, C.L.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Similarities and differences in antagonism of neuron alpha/beta interferon responses by Venezuelan equine encephalitis and Sindbis alphaviruses. J. Virol. 2009, 83, 10036–10047. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Lokugamage, K.; Kamitani, W.; Ikegami, T.; Tseng, C.-T.K.; Makino, S. Severe Acute Respiratory Syndrome Coronavirus nsp1 Suppresses Host Gene Expression, Including That of Type I Interferon, in Infected Cells. J. Virol. 2008, 82, 4471–4479. [Google Scholar] [CrossRef]
- Hixon, A.M.; Frost, J.; Rudy, M.J.; Messacar, K.; Clarke, P.; Tyler, K.L. Understanding Enterovirus D68-Induced Neurologic Disease: A Basic Science Review. Viruses 2019, 11, 821. [Google Scholar] [CrossRef]
- Lafon, M.; Prehaud, C.; Megret, F.; Lafage, M.; Mouillot, G.; Roa, M.; Moreau, P.; Rouas-Freiss, N.; Carosella, E.D. Modulation of HLA-G Expression in Human Neural Cells after Neurotropic Viral Infections. J. Virol. 2005, 79, 15226–15237. [Google Scholar] [CrossRef]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef]
- Domingo, E.; Holland, J.J. RNA VIRUS MUTATIONS AND FITNESS FOR SURVIVAL. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef]
- Mattenberger, F.; Vila-Nistal, M.; Geller, R. Increased RNA virus population diversity improves adaptability. Sci. Rep. 2021, 11, 6824. [Google Scholar] [CrossRef]
- Lamers, M.M.; Mykytyn, A.Z.; Breugem, T.I.; Wang, Y.; Wu, D.C.; Riesebosch, S.; van den Doel, P.B.; Schipper, D.; Bestebroer, T.; Wu, N.C.; et al. Human airway cells prevent SARS-CoV-2 multibasic cleavage site cell culture adaptation. Elife 2021, 10, e66815. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Miazgowicz, K.; McMinn, R.; Seifert, S.N.; Sola, I.; Enjuanes, L.; Carmody, A.; van Doremalen, N.; Munster, V. Adaptive Evolution of MERS-CoV to Species Variation in DPP4. Cell Rep. 2018, 24, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Lu, X.; Chen, Q.; Xu, K.; Chen, Y.; Cheng, M.; Chen, K.; Cheng, L.; Weng, T.; Shi, D.; et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 2020, 6, 76. [Google Scholar] [CrossRef]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef]
- Zhou, Z.; Kang, H.; Li, S.; Zhao, X. Understanding the neurotropic characteristics of SARS-CoV-2: From neurological manifestations of COVID-19 to potential neurotropic mechanisms. J. Neurol. 2020, 267, 2179–2184. [Google Scholar] [CrossRef]
- Alenina, N.; Bader, M. ACE2 in Brain Physiology and Pathophysiology: Evidence from Transgenic Animal Models. Neurochem. Res. 2019, 44, 1323–1329. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Virus | CNS Manifestation |
|---|---|
| PV | Polio (paralysis) |
| EV-D68 | Acute flaccid myelitis, encephalitis |
| EV-A71 | Meningitis, acute flaccid paralysis, hand-foot-and-mouth disease (non-CNS) |
| Coxsackievirus A16 | Encephalitis |
| RABV | Rabies (anxiety, hydrophobia, coma), encephalitis |
| ZIKV | Microcephaly, meningoencephalitis, Guillain-Barré syndrome, non-specific acute febrile illness |
| DENV | Fever, encephalitis, meningitis |
| MuV | Sensory motor axonopathy, meningitis |
| MV | Encephalitis, brain edema, myelitis, sclerosing panencephalitis (SSPE), measles (non-CNS) |
| NiV | Encephalitis, meningitis |
| JEV, VEEV, WEEV, EEEV | Encephalitis, meningitis |
| LACV | Encephalitis, meningitis, non-specific febrile illness |
| SARS-CoV-2 | Encephalitis, acute necrotizing encephalopathy, meningitis, acute cerebrovascular disease, confusion |
| Virus. | Viral Protein(s) | Host Target | Cell Type(s) | Reference |
|---|---|---|---|---|
| WNV | NS1, NS2A | IFN-β and NF-ⲕB signaling | BE(2)-C/m (neuroblast cell line) | [9] |
| ZIKV | NS3 | 14-3-3ε and 14-3-3η signaling | SVGA (immortalized human astrocyte cell line) | [25] |
| RABV | Capsid | TLR3 | NT2-n, SK-n-SH, Ntera-2clD/1 | [22] |
| JEV | Unknown | miR-15b targeting of RNF125 | U251 (human astrocytoma cell line), mouse brain, BV-2 (mouse microglia cell line) | [70] |
| Unknown | miR-301a targeting of IRF1 responses | HT22 (immortalized mouse hippocampal neuronal cell line) | [71] | |
| NS5 | Suppressor of cytokine signaling (SOCS3) | Mouse brain | [72] | |
| Unknown | miR-146a targeting of TRAF6, IRAK1, IRAK2, and STAT1 | Human microglial cells | [68] | |
| Unknown | miR-132 targeting of p300 co-activator of STAT1 | Mouse cortical neurons and mouse granule cell neurons | [69] | |
| Unknown | miR-432 targeting of SOCS5 | CHME3 (human microglial cells) | [73] | |
| VVEEV | sP | Macromolecular shutoff | Neuro-2a | [74] |
| Enterovirus A71 | 3C | TRIF, MAVS | SF268 (human glioblastoma) | [63] |
| Nipah virus | P protein | STAT1, IFN-β signaling | Ferret model brain | [10] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vazquez, C.; Jurado, K.A. Neurotropic RNA Virus Modulation of Immune Responses within the Central Nervous System. Int. J. Mol. Sci. 2022, 23, 4018. https://doi.org/10.3390/ijms23074018
Vazquez C, Jurado KA. Neurotropic RNA Virus Modulation of Immune Responses within the Central Nervous System. International Journal of Molecular Sciences. 2022; 23(7):4018. https://doi.org/10.3390/ijms23074018
Chicago/Turabian StyleVazquez, Christine, and Kellie A. Jurado. 2022. "Neurotropic RNA Virus Modulation of Immune Responses within the Central Nervous System" International Journal of Molecular Sciences 23, no. 7: 4018. https://doi.org/10.3390/ijms23074018
APA StyleVazquez, C., & Jurado, K. A. (2022). Neurotropic RNA Virus Modulation of Immune Responses within the Central Nervous System. International Journal of Molecular Sciences, 23(7), 4018. https://doi.org/10.3390/ijms23074018