
Aquaporins Are One of the Critical Factors in the Disruption of the Skin Barrier in Inflammatory Skin Diseases

 , , ,
, , ,  ,
,  ,
,  and
and 
Abstract
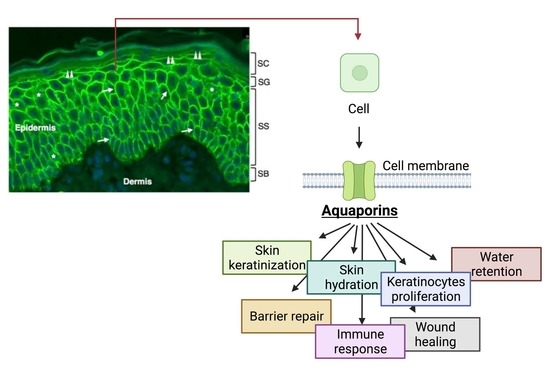
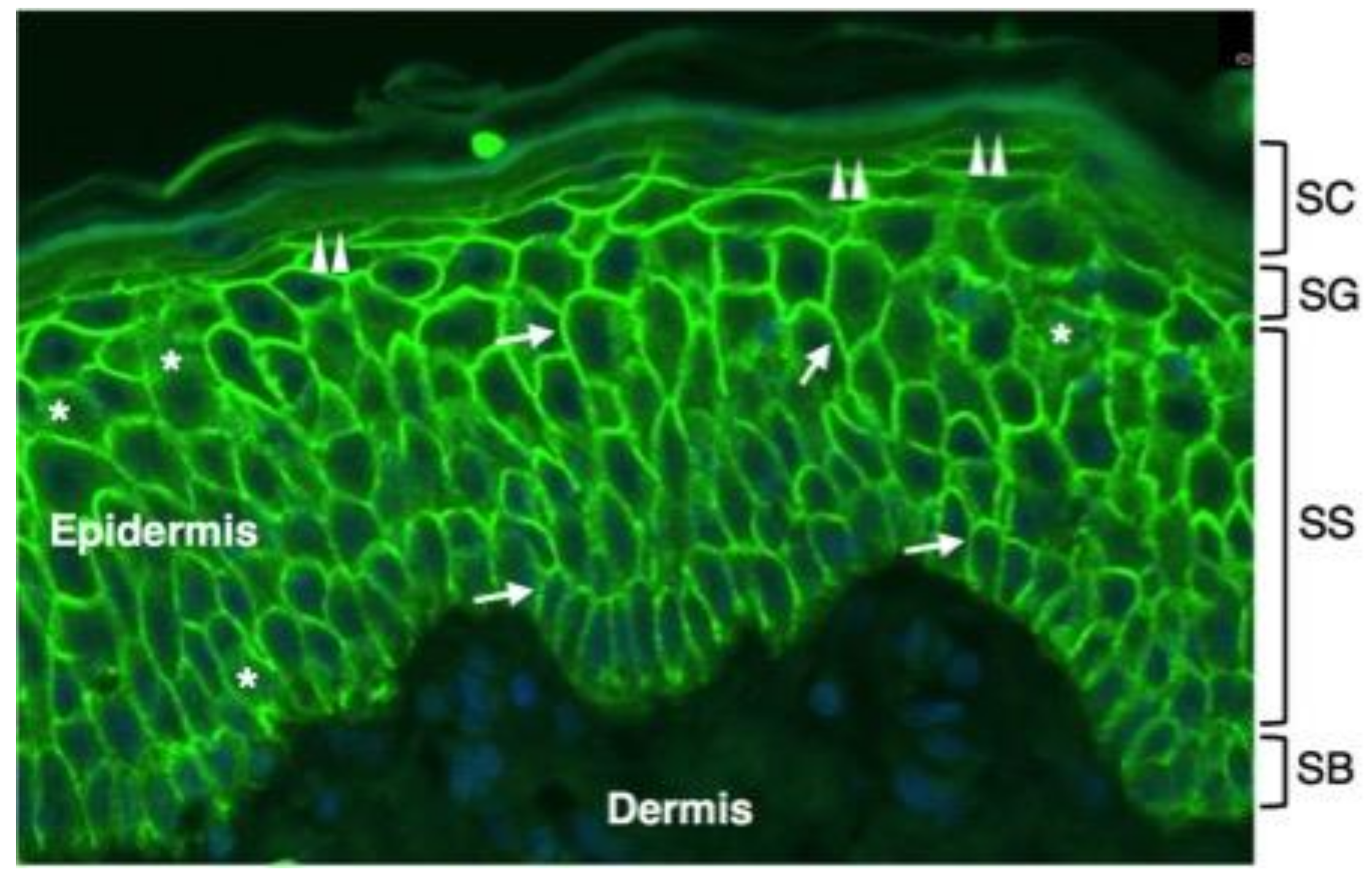
1. Introduction
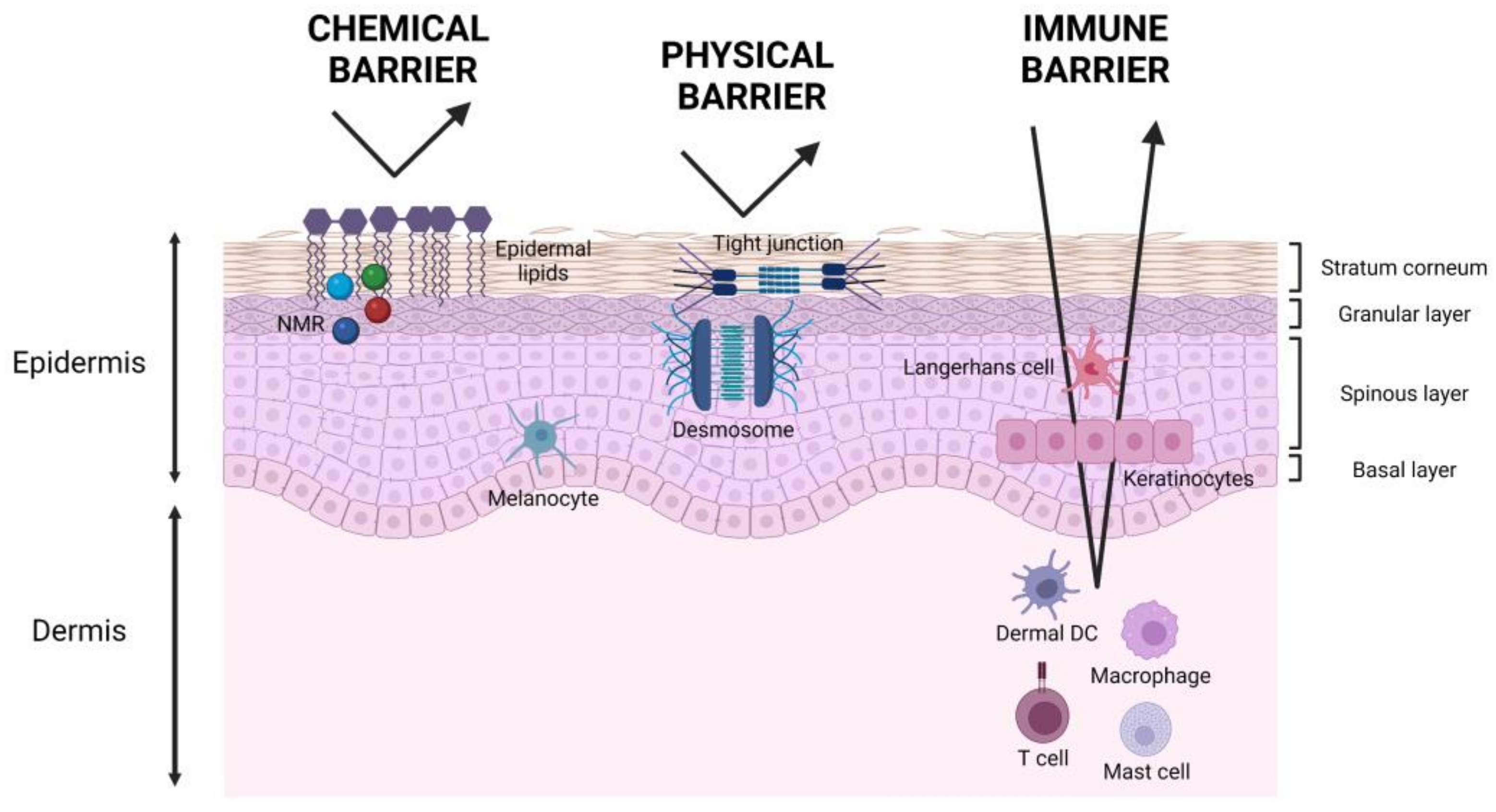
2. Skin Barrier and Water Permeability
3. Aquaporins
4. Expression, Regulation, and Physiological Relevance of Skin Aquaporins
4.1. AQP1
4.2. AQP3
4.3. AQP5
4.4. AQP7
4.5. AQP9
4.6. AQP10
5. Skin Aquaporins in Inflammatory Dermatological Diseases
5.1. Hidradenitis Suppurativa
5.2. Atopic Dermatitis
5.3. Psoriasis
6. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Baroni, A.; Buommino, E.; De Gregorio, V.; Ruocco, E.; Ruocco, V.; Wolf, R. Structure and function of the epidermis related to barrier properties. Clin. Dermatol. 2012, 30, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Proksch, E.; Brandner, J.M.; Jensen, J.M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Traidl-Hoffmann, C.; Biedermann, T. Cutaneous Barriers and Skin Immunity: Differentiating a Connected Network. Trends Immunol. 2018, 39, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Kevin Heard, L.; Chen, X.; Bollag, W.B. Aquaporins in the Skin. Adv. Exp. Med. Biol. 2017, 969, 173–191. [Google Scholar] [PubMed]
- Coates, M.; Mariottoni, P.; Corcoran, D.L.; Kirshner, H.F.; Jaleel, T.; Brown, D.A.; Brooks, S.R.; Murray, J.; Morasso, M.I.; MacLeod, A.S. The skin transcriptome in hidradenitis suppurativa uncovers an antimicrobial and sweat gland gene signature which has distinct overlap with wounded skin. PLoS ONE 2019, 14, e0216249. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, B.; Chen, L.; Xie, Y.; Liang, J.; Yang, Y.; Shao, L.; Zhang, J.; Wang, J.; Zhang, X.; et al. RNA-Seq Identifies Marked Th17 Cell Activation and Altered CFTR Expression in Different Atopic Dermatitis Subtypes in Chinese Han Populations. Front. Immunol. 2021, 12, 628512. [Google Scholar] [CrossRef]
- Ghosh, D.; Ding, L.; Sivaprasad, U.; Geh, E.; Biagini Myers, J.; Bernstein, J.A.; Khurana Hershey, G.K.; Mersha, T.B. Multiple Transcriptome Data Analysis Reveals Biologically Relevant Atopic Dermatitis Signature Genes and Pathways. PLoS ONE 2015, 10, e0144316. [Google Scholar] [CrossRef]
- Nakahigashi, K.; Kabashima, K.; Ikoma, A.; Verkman, A.S.; Miyachi, Y.; Hara-Chikuma, M. Upregulation of aquaporin-3 is involved in keratinocyte proliferation and epidermal hyperplasia. J. Invest. Dermatol. 2011, 131, 865–873. [Google Scholar] [CrossRef]
- Sun, Z.; Kong, Y.; Li, N.; Jiang, X.; Cao, T.; Jia, Y.; Zhang, Y.; Zhang, Y.; Cheng, J.; Wang, J. Effect of Yupingfeng granules on the skin barrier in atopic dermatitis mice models. J. Tradit. Chin. Med. 2018, 38, 872–878. [Google Scholar]
- Guo, L.; Chen, H.; Li, Y.; Zhou, Q.; Sui, Y. An aquaporin 3-notch1 axis in keratinocyte differentiation and inflammation. PLoS ONE 2013, 8, e80179. [Google Scholar] [CrossRef]
- Lee, Y.; Je, Y.J.; Lee, S.S.; Li, Z.J.; Choi, D.K.; Kwon, Y.B.; Sohn, K.C.; Im, M.; Seo, Y.J.; Lee, J.H. Changes in transepidermal water loss and skin hydration according to expression of aquaporin-3 in psoriasis. Ann. Dermatol. 2012, 24, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Satooka, H.; Watanabe, S.; Honda, T.; Miyachi, Y.; Watanabe, T.; Verkman, A.S. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-kappaB signalling in keratinocytes and development of psoriasis. Nat. Commun. 2015, 6, 7454. [Google Scholar] [CrossRef]
- Watt, F.M. Mammalian skin cell biology: At the interface between laboratory and clinic. Science 2014, 346, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Pappas, A. Epidermal surface lipids. Dermatoendocrinology 2009, 1, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin barrier immunity and ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.D.; Baadsgaard, O.; Ellis, C.N.; Duell, E.; Voorhees, J.J. Mechanisms of cyclosporine A inhibition of antigen-presenting activity in uninvolved and lesional psoriatic epidermis. J. Invest. Dermatol. 1990, 94, 649–656. [Google Scholar] [CrossRef]
- Segre, J.A. Epidermal barrier formation and recovery in skin disorders. J. Clin. Invest. 2006, 116, 1150–1158. [Google Scholar] [CrossRef]
- Agre, P. Aquaporin water channels (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2004, 43, 4278–4290. [Google Scholar] [CrossRef]
- Gena, P.; Pellegrini-Calace, M.; Biasco, A.; Svelto, M.; Calamita, G. Aquaporin membrane channels: Biophysics, classification, functions and possible biotechnological applications. Food Biophys. 2011, 6, 241–249. [Google Scholar] [CrossRef]
- Jahn, T.P.; Moller, A.L.; Zeuthen, T.; Holm, L.M.; Klaerke, D.A.; Mohsin, B.; Kuhlbrandt, W.; Schjoerring, J.K. Aquaporin homologues in plants and mammals transport ammonia. FEBS Lett. 2004, 574, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medrano-Fernandez, I. Human aquaporin-11 guarantees efficient transport of H2O2 across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Moller, A.L.; Kristiansen, K.A.; Schulz, A.; Moller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Functional and molecular characterization of the human neutral solute channel aquaporin-9. Am. J. Physiol. 1999, 277, F685–F696. [Google Scholar] [CrossRef]
- Nakhoul, N.L.; Davis, B.A.; Romero, M.F.; Boron, W.F. Effect of expressing the water channel aquaporin-1 on the CO2 permeability of Xenopus oocytes. Am. J. Physiol. 1998, 274, C543–C548. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Hong, N.J.; Garvin, J.L. Aquaporin-1 transports NO across cell membranes. Hypertension 2006, 48, 157–164. [Google Scholar] [CrossRef]
- Wang, Y.; Tajkhorshid, E. Molecular mechanisms of conduction and selectivity in aquaporin water channels. J. Nutr. 2007, 137 (Suppl. S1), 1509S–1515S, discussion 1516S–1517S. [Google Scholar] [CrossRef]
- Calamita, G.; Perret, J.; Delporte, C. Aquaglyceroporins: Drug Targets for Metabolic Diseases? Front. Physiol. 2018, 9, 851. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Yool, A.J.; Bill, R.M. Recent breakthroughs and future directions in drugging aquaporins. Trends Pharmacol. Sci. 2022, 43, 30–42. [Google Scholar] [CrossRef]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118 Pt 15, 3225–3232. [Google Scholar] [CrossRef]
- Verkman, A.S. Knock-out models reveal new aquaporin functions. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 359–381. [Google Scholar]
- Tradtrantip, L.; Jin, B.J.; Yao, X.; Anderson, M.O.; Verkman, A.S. Aquaporin-Targeted Therapeutics: State-of-the-Field. Adv. Exp. Med. Biol. 2017, 969, 239–250. [Google Scholar] [PubMed]
- da Silva, I.V.; Silva, A.G.; Pimpao, C.; Soveral, G. Skin aquaporins as druggable targets: Promoting health by addressing the disease. Biochimie 2021, 188, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Sugiyama, Y.; Kabashima, K.; Sohara, E.; Uchida, S.; Sasaki, S.; Inoue, S.; Miyachi, Y. Involvement of aquaporin-7 in the cutaneous primary immune response through modulation of antigen uptake and migration in dendritic cells. FASEB J. 2012, 26, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Nejsum, L.N.; Kwon, T.H.; Jensen, U.B.; Fumagalli, O.; Frokiaer, J.; Krane, C.M.; Menon, A.G.; King, L.S.; Agre, P.C.; Nielsen, S. Functional requirement of aquaporin-5 in plasma membranes of sweat glands. Proc. Natl. Acad. Sci. USA 2002, 99, 511–516. [Google Scholar] [CrossRef]
- Rojek, A.; Praetorius, J.; Frokiaer, J.; Nielsen, S.; Fenton, R.A. A current view of the mammalian aquaglyceroporins. Annu. Rev. Physiol. 2008, 70, 301–327. [Google Scholar] [CrossRef]
- Leitch, V.; Agre, P.; King, L.S. Altered ubiquitination and stability of aquaporin-1 in hypertonic stress. Proc. Natl. Acad. Sci. USA 2001, 98, 2894–2898. [Google Scholar] [CrossRef]
- Boury-Jamot, M.; Sougrat, R.; Tailhardat, M.; Le Varlet, B.; Bonte, F.; Dumas, M.; Verbavatz, J.M. Expression and function of aquaporins in human skin: Is aquaporin-3 just a glycerol transporter? Biochim. Biophys. Acta 2006, 1758, 1034–1042. [Google Scholar] [CrossRef]
- Hara-Chikuma, M.; Verkman, A.S. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J. Mol. Med. (Berl) 2008, 86, 221–231. [Google Scholar] [CrossRef]
- Zheng, X.; Bollinger Bollag, W. Aquaporin 3 colocates with phospholipase d2 in caveolin-rich membrane microdomains and is downregulated upon keratinocyte differentiation. J. Invest. Dermatol. 2003, 121, 1487–1495. [Google Scholar] [CrossRef]
- Qin, H.; Zheng, X.; Zhong, X.; Shetty, A.K.; Elias, P.M.; Bollag, W.B. Aquaporin-3 in keratinocytes and skin: Its role and interaction with phospholipase D2. Arch. Biochem. Biophys. 2011, 508, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, V.; Olala, L.O.; Kagha, K.; Pan, Z.Q.; Chen, X.; Yang, R.; Cline, A.; Helwa, I.; Marshall, L.; Kaddour-Djebbar, I.; et al. Regulation of the Glycerol Transporter, Aquaporin-3, by Histone Deacetylase-3 and p53 in Keratinocytes. J. Invest. Dermatol. 2017, 137, 1935–1944. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, X. Aquaporin 3, a glycerol and water transporter, is regulated by p73 of the p53 family. FEBS Lett. 2001, 489, 4–7. [Google Scholar] [CrossRef]
- Jiang, Y.J.; Kim, P.; Lu, Y.F.; Feingold, K.R. PPARgamma activators stimulate aquaporin 3 expression in keratinocytes/epidermis. Exp. Dermatol. 2011, 20, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Chowdhury, S.; Choudhary, V.; Chen, X.; Bollag, W.B. Keratinocyte aquaporin-3 expression induced by histone deacetylase inhibitors is mediated in part by peroxisome proliferator-activated receptors (PPARs). Exp. Dermatol. 2020, 29, 380–386. [Google Scholar] [CrossRef]
- Choudhary, V.; Olala, L.O.; Qin, H.; Helwa, I.; Pan, Z.Q.; Tsai, Y.Y.; Frohman, M.A.; Kaddour-Djebbar, I.; Bollag, W.B. Aquaporin-3 re-expression induces differentiation in a phospholipase D2-dependent manner in aquaporin-3-knockout mouse keratinocytes. J. Invest. Dermatol. 2015, 135, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.A.; Ruddy, M.K.; Hosselet, C.A.; Harris, H.W. The perinatal expression of aquaporin-2 and aquaporin-3 in developing kidney. Pediatr. Res. 1998, 43, 783–790. [Google Scholar] [CrossRef]
- Zheng, X.; Ray, S.; Bollag, W.B. Modulation of phospholipase D-mediated phosphatidylglycerol formation by differentiating agents in primary mouse epidermal keratinocytes. Biochim. Biophys. Acta 2003, 1643, 25–36. [Google Scholar] [CrossRef]
- Yuspa, S.H.; Kilkenny, A.E.; Steinert, P.M.; Roop, D.R. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J. Cell Biol. 1989, 109, 1207–1217. [Google Scholar] [CrossRef]
- Hendriks, G.; Koudijs, M.; van Balkom, B.W.; Oorschot, V.; Klumperman, J.; Deen, P.M.; van der Sluijs, P. Glycosylation is important for cell surface expression of the water channel aquaporin-2 but is not essential for tetramerization in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 2975–2983. [Google Scholar] [CrossRef]
- Stokes, A.M.; Semmineh, N.B.; Nespodzany, A.; Bell, L.C.; Quarles, C.C. Systematic assessment of multi-echo dynamic susceptibility contrast MRI using a digital reference object. Magn. Reson. Med. 2020, 83, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.; Gondran, C.; Menon, G.; Mur, L.; Oberto, G.; Guerif, Y.; Dal Farra, C.; Domloge, N. Impact of AQP3 inducer treatment on cultured human keratinocytes, ex vivo human skin and volunteers. Int. J. Cosmet. Sci. 2011, 33, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.W.; Soon, Y.Y.; Chen, D.; Tey, J.C.S. Chemoradiotherapy versus chemotherapy for locally advanced unresectable pancreatic cancer: A systematic review and meta-analysis. Asia Pac. J. Clin. Oncol. 2018, 14, 392–401. [Google Scholar] [CrossRef]
- Cohly, H.H.; Isokpehi, R.; Rajnarayanan, R.V. Compartmentalization of aquaporins in the human intestine. Int. J. Environ. Res. Public Health 2008, 5, 115–119. [Google Scholar] [CrossRef]
- Bollag, W.B.; Xie, D.; Zheng, X.; Zhong, X. A potential role for the phospholipase D2-aquaporin-3 signaling module in early keratinocyte differentiation: Production of a phosphatidylglycerol signaling lipid. J. Invest. Dermatol. 2007, 127, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.E.; Bollag, R.J.; Fussell, N.; By, C.; Sheehan, D.J.; Bollag, W.B. Abnormal aquaporin-3 protein expression in hyperproliferative skin disorders. Arch. Dermatol. Res. 2011, 303, 591–600. [Google Scholar] [CrossRef][Green Version]
- Kim, N.H.; Lee, A.Y. Reduced aquaporin3 expression and survival of keratinocytes in the depigmented epidermis of vitiligo. J. Invest. Dermatol. 2010, 130, 2231–2239. [Google Scholar] [CrossRef]
- Hara-Chikuma, M.; Takahashi, K.; Chikuma, S.; Verkman, A.S.; Miyachi, Y. The expression of differentiation markers in aquaporin-3 deficient epidermis. Arch. Dermatol. Res. 2009, 301, 245–252. [Google Scholar] [CrossRef]
- Bollag, W.B.; Aitkens, L.; White, J.; Hyndman, K.A. Aquaporin-3 in the epidermis: More than skin deep. Am. J. Physiol. Cell Physiol. 2020, 318, C1144–C1153. [Google Scholar] [CrossRef]
- Calamita, G.; Gena, P.; Ferri, D.; Rosito, A.; Rojek, A.; Nielsen, S.; Marinelli, R.A.; Fruhbeck, G.; Svelto, M. Biophysical assessment of aquaporin-9 as principal facilitative pathway in mouse liver import of glucogenetic glycerol. Biol. Cell 2012, 104, 342–351. [Google Scholar] [CrossRef]
- Hara, M.; Ma, T.; Verkman, A.S. Selectively reduced glycerol in skin of aquaporin-3-deficient mice may account for impaired skin hydration, elasticity, and barrier recovery. J. Biol. Chem. 2002, 277, 46616–46621. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Verkman, A.S. Prevention of skin tumorigenesis and impairment of epidermal cell proliferation by targeted aquaporin-3 gene disruption. Mol. Cell. Biol. 2008, 28, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Marlar, S.; Jensen, H.H.; Login, F.H.; Nejsum, L.N. Aquaporin-3 in Cancer. Int. J. Mol. Sci. 2017, 18, 2106. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, T.; Zhou, Y.C.; Gao, F.; Zhang, Z.H.; Xu, H.; Wang, S.L.; Shen, L.Z. Aquaporin 3 promotes epithelial-mesenchymal transition in gastric cancer. J. Exp. Clin. Cancer Res. 2014, 33, 38. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef]
- Hara-Chikuma, M.; Chikuma, S.; Sugiyama, Y.; Kabashima, K.; Verkman, A.S.; Inoue, S.; Miyachi, Y. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J. Exp. Med. 2012, 209, 1743–1752. [Google Scholar] [CrossRef]
- Sugimoto, T.; Huang, L.; Minematsu, T.; Yamamoto, Y.; Asada, M.; Nakagami, G.; Akase, T.; Nagase, T.; Oe, M.; Mori, T.; et al. Impaired aquaporin 3 expression in reepithelialization of cutaneous wound healing in the diabetic rat. Biol. Res. Nurs. 2013, 15, 347–355. [Google Scholar] [CrossRef]
- Wang, P.H.; Huang, B.S.; Horng, H.C.; Yeh, C.C.; Chen, Y.J. Wound healing. J. Chin. Med. Assoc. 2018, 81, 94–101. [Google Scholar] [CrossRef]
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules 2021, 11, 1095. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, J.S.; Lee, J.W.; Choi, K.Y.; Yang, J.D.; Cho, B.C.; Oh, E.J.; Kim, T.J.; Ko, U.H.; Shin, J.H.; et al. Effect of Keratinocytes on Myofibroblasts in Hypertrophic Scars. Aesthetic Plast. Surg. 2019, 43, 1371–1380. [Google Scholar] [CrossRef]
- Ma, T.; Hara, M.; Sougrat, R.; Verbavatz, J.M.; Verkman, A.S. Impaired stratum corneum hydration in mice lacking epidermal water channel aquaporin-3. J. Biol. Chem. 2002, 277, 17147–17153. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Verkman, A.S. Glycerol replacement corrects defective skin hydration, elasticity, and barrier function in aquaporin-3-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 7360–7365. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Itcho, K.; Hamamura, K.; Ikeda, E.; Ikeyama, H.; Furuichi, Y.; Watanabe, M.; Koyanagi, S.; Ohdo, S. 24-hour rhythm of aquaporin-3 function in the epidermis is regulated by molecular clocks. J. Invest. Dermatol. 2014, 134, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Fluhr, J.W.; Mao-Qiang, M.; Brown, B.E.; Wertz, P.W.; Crumrine, D.; Sundberg, J.P.; Feingold, K.R.; Elias, P.M. Glycerol regulates stratum corneum hydration in sebaceous gland deficient (asebia) mice. J. Invest. Dermatol. 2003, 120, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, N.; Kon, R.; Kaneko, M.; Mizukami, N.; Kusunoki, Y.; Sugiyama, K. Relationship between Aging-Related Skin Dryness and Aquaporins. Int. J. Mol. Sci. 2017, 18, 1559. [Google Scholar] [CrossRef]
- Inoue, N.; Iida, H.; Yuan, Z.; Ishikawa, Y.; Ishida, H. Age-related decreases in the response of aquaporin-5 to acetylcholine in rat parotid glands. J. Dent. Res. 2003, 82, 476–480. [Google Scholar] [CrossRef]
- Zhang, M.; Zeng, S.; Zhang, L.; Li, H.; Chen, L.; Zhang, X.; Li, X.; Lin, C.; Shu, S.; Xie, S.; et al. Localization of Na(+)-K(+)-ATPase alpha/beta, Na(+)-K(+)-2Cl-cotransporter 1 and aquaporin-5 in human eccrine sweat glands. Acta Histochem. 2014, 116, 1374–1381. [Google Scholar] [CrossRef]
- Sato, K.; Cavallin, S.; Sato, K.T.; Sato, F. Secretion of ions and pharmacological responsiveness in the mouse paw sweat gland. Clin. Sci. (Lond.) 1994, 86, 133–139. [Google Scholar] [CrossRef]
- Song, Y.; Sonawane, N.; Verkman, A.S. Localization of aquaporin-5 in sweat glands and functional analysis using knockout mice. J. Physiol. 2002, 541 Pt 2, 561–568. [Google Scholar] [CrossRef]
- Zhou, J.; Dong, Y.; Liu, J.; Ren, J.; Wu, J.; Zhu, N. AQP5 regulates the proliferation and differentiation of epidermal stem cells in skin aging. Braz. J. Med. Biol. Res. 2020, 53, e10009. [Google Scholar] [CrossRef]
- Du, Q.; Lin, M.; Yang, J.H.; Chen, J.F.; Tu, Y.R. Overexpression of AQP5 Was Detected in Axillary Sweat Glands of Primary Focal Hyperhidrosis Patients. Dermatology 2016, 232, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Huang, Y.G.; Deng, Y.C.; Tian, J.Y.; Rao, Z.R.; Che, H.L.; Zhang, H.F.; Zhao, G. Topiramate reduced sweat secretion and aquaporin-5 expression in sweat glands of mice. Life Sci. 2007, 80, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Nakahigashi, K.; Nomura, T.; Miyachi, Y.; Kabashima, K. Normal immunostaining pattern for aquaporin-5 in the lesions of palmoplantar hyperhidrosis. Case Rep. Dermatol. 2013, 5, 61–63. [Google Scholar] [CrossRef]
- Takata, K.; Matsuzaki, T.; Tajika, Y. Aquaporins: Water channel proteins of the cell membrane. Prog. Histochem. Cytochem. 2004, 39, 1–83. [Google Scholar] [CrossRef] [PubMed]
- Rojek, A.M.; Skowronski, M.T.; Fuchtbauer, E.M.; Fuchtbauer, A.C.; Fenton, R.A.; Agre, P.; Frokiaer, J.; Nielsen, S. Defective glycerol metabolism in aquaporin 9 (AQP9) knockout mice. Proc. Natl. Acad. Sci. USA 2007, 104, 3609–3614. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Ota, Y.; Hara, M.; Inoue, S. Osmotic stress up-regulates aquaporin-3 gene expression in cultured human keratinocytes. Biochim. Biophys. Acta 2001, 1522, 82–88. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Yamazaki, K.; Kusaka-Kikushima, A.; Nakahigashi, K.; Hagiwara, H.; Miyachi, Y. Analysis of aquaporin 9 expression in human epidermis and cultured keratinocytes. FEBS Open Bio 2014, 4, 611–616. [Google Scholar] [CrossRef]
- Moniaga, C.S.; Watanabe, S.; Honda, T.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9-expressing neutrophils are required for the establishment of contact hypersensitivity. Sci. Rep. 2015, 5, 15319. [Google Scholar] [CrossRef]
- Jungersted, J.M.; Bomholt, J.; Bajraktari, N.; Hansen, J.S.; Klaerke, D.A.; Pedersen, P.A.; Hedfalk, K.; Nielsen, K.H.; Agner, T.; Helix-Nielsen, C. In vivo studies of aquaporins 3 and 10 in human stratum corneum. Arch. Dermatol. Res. 2013, 305, 699–704. [Google Scholar] [CrossRef]
- Soler, D.C.; Bai, X.; Ortega, L.; Pethukova, T.; Nedorost, S.T.; Popkin, D.L.; Cooper, K.D.; McCormick, T.S. The key role of aquaporin 3 and aquaporin 10 in the pathogenesis of pompholyx. Med. Hypotheses 2015, 84, 498–503. [Google Scholar] [CrossRef]
- Pasparakis, M.; Haase, I.; Nestle, F.O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014, 14, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Mariajoseph-Antony, L.F.; Kannan, A.; Panneerselvam, A.; Loganathan, C.; Shankar, E.M.; Anbarasu, K.; Prahalathan, C. Role of Aquaporins in Inflammation—A Scientific Curation. Inflammation 2020, 43, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Alhusayen, R.; Lansang, P.; Shear, N.; Yeung, J. What is hidradenitis suppurativa? Can. Fam. Physician 2017, 63, 114–120. [Google Scholar] [PubMed]
- Wolk, K.; Join-Lambert, O.; Sabat, R. Aetiology and pathogenesis of hidradenitis suppurativa. Br. J. Dermatol. 2020, 183, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Ovadja, Z.N.; Schuit, M.M.; van der Horst, C.; Lapid, O. Inter- and intrarater reliability of Hurley staging for hidradenitis suppurativa. Br. J. Dermatol. 2019, 181, 344–349. [Google Scholar] [CrossRef]
- Schell, S.L.; Schneider, A.M.; Nelson, A.M. Yin and Yang: A disrupted skin microbiome and an aberrant host immune response in hidradenitis suppurativa. Exp. Dermatol. 2021, 30, 1453–1470. [Google Scholar] [CrossRef]
- Bonamonte, D.; De Marco, A.; Giuffrida, R.; Conforti, C.; Barlusconi, C.; Foti, C.; Romita, P. Topical antibiotics in the Dermatol.ogical clinical practice: Indications, efficacy, and adverse effects. Dermatol. Ther. 2020, 33, e13824. [Google Scholar] [CrossRef]
- Liu, T.; Li, S.; Ying, S.; Tang, S.; Ding, Y.; Li, Y.; Qiao, J.; Fang, H. The IL-23/IL-17 Pathway in Inflammatory Skin Diseases: From Bench to Bedside. Front. Immunol. 2020, 11, 594735. [Google Scholar] [CrossRef]
- Nikolakis, G.; Kokolakis, G.; Kaleta, K.; Wolk, K.; Hunger, R.; Sabat, R.; Zouboulis, C.C. Pathogenesis of hidradenitis suppurativa/acne inversa. Hautarzt 2021, 72, 658–665. [Google Scholar] [CrossRef]
- Jemec, G.B. Clinical practice. Hidradenitis suppurativa. N. Engl. J. Med. 2012, 366, 158–164. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Benhadou, F.; Byrd, A.S.; Chandran, N.S.; Giamarellos-Bourboulis, E.J.; Fabbrocini, G.; Frew, J.W.; Fujita, H.; Gonzalez-Lopez, M.A.; Guillem, P.; et al. What causes hidradenitis suppurativa?-15 years after. Exp. Dermatol. 2020, 29, 1154–1170. [Google Scholar] [CrossRef] [PubMed]
- Gratton, R.; Tricarico, P.M.; Moltrasio, C.; Lima Estevao de Oliveira, A.S.; Brandao, L.; Marzano, A.V.; Zupin, L.; Crovella, S. Pleiotropic Role of Notch Signaling in Human Skin Diseases. Int. J. Mol. Sci. 2020, 21, 4214. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Singdia, H.; Nijhawan, S.; Mathur, D.K.; Bhargava, R.K. A study of biophysical profile of inguinal skin: An implication for health and disease. Indian J. Sex. Transm. Dis. AIDS 2021, 42, 7–13. [Google Scholar] [PubMed]
- Avena-Woods, C. Overview of atopic dermatitis. Am. J. Manag. Care 2017, 23 (Suppl. 8), S115–S123. [Google Scholar]
- Sahni, V.N.; Balogh, E.A.; Strowd, L.C.; Feldman, S.R. The evolving atopic dermatitis management landscape. Expert Opin. Pharm. 2022, 23, 517–526. [Google Scholar] [CrossRef]
- Sroka-Tomaszewska, J.; Trzeciak, M. Molecular Mechanisms of Atopic Dermatitis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4130. [Google Scholar] [CrossRef]
- Proksch, E.; Brasch, J. Abnormal epidermal barrier in the pathogenesis of contact dermatitis. Clin. Dermatol. 2012, 30, 335–344. [Google Scholar] [CrossRef]
- Irvine, A.D.; McLean, W.H.; Leung, D.Y. Filaggrin mutations associated with skin and allergic diseases. N. Engl. J. Med. 2011, 365, 1315–1327. [Google Scholar] [CrossRef]
- Ferrucci, S.; Romagnuolo, M.; Maronese, C.A.; Germiniasi, F.; Tavecchio, S.; Angileri, L.; Casazza, G.; Marzano, A.V.; Genovese, G. Skin barrier status during dupilumab treatment in patients with severe atopic dermatitis. Ther. Adv. Chronic Dis. 2021, 12, 20406223211058332. [Google Scholar] [CrossRef]
- Yoshida, T.; Beck, L.A.; De Benedetto, A. Skin barrier defects in atopic dermatitis: From old idea to new opportunity. Allergol. Int. 2022, 71, 3–13. [Google Scholar] [CrossRef]
- Dumortier, A.; Durham, A.D.; Di Piazza, M.; Vauclair, S.; Koch, U.; Ferrand, G.; Ferrero, I.; Demehri, S.; Song, L.L.; Farr, A.G.; et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PLoS ONE 2010, 5, e9258. [Google Scholar] [CrossRef] [PubMed]
- Rendon, A.; Schakel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Barker, J.N. Pathogenesis and clinical features of psoriasis. Lancet 2007, 370, 263–271. [Google Scholar] [CrossRef]
- Orsmond, A.; Bereza-Malcolm, L.; Lynch, T.; March, L.; Xue, M. Skin Barrier Dysregulation in Psoriasis. Int. J. Mol. Sci. 2021, 22, 10841. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Lai, Y. Keratinocyte: A trigger or an executor of psoriasis? J. Leukoc. Biol. 2020, 108, 485–491. [Google Scholar] [CrossRef]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef]
- Montero-Vilchez, T.; Segura-Fernandez-Nogueras, M.V.; Perez-Rodriguez, I.; Soler-Gongora, M.; Martinez-Lopez, A.; Fernandez-Gonzalez, A.; Molina-Leyva, A.; Arias-Santiago, S. Skin Barrier Function in Psoriasis and Atopic Dermatitis: Transepidermal Water Loss and Temperature as Useful Tools to Assess Disease Severity. J. Clin. Med. 2021, 10, 359. [Google Scholar] [CrossRef]
- Nakamura-Pereira, M.; Knobel, R.; Menezes, M.O.; Andreucci, C.B.; Takemoto, M.L.S. The impact of the COVID-19 pandemic on maternal mortality in Brazil: 523 maternal deaths by acute respiratory distress syndrome potentially associated with SARS-CoV-2. Int. J. Gynaecol. Obstet. 2021, 153, 360–362. [Google Scholar] [CrossRef]
- Tesse, A.; Grossini, E.; Tamma, G.; Brenner, C.; Portincasa, P.; Marinelli, R.A.; Calamita, G. Aquaporins as Targets of Dietary Bioactive Phytocompounds. Front. Mol. Biosci. 2018, 5, 30. [Google Scholar] [CrossRef]
- Tamma, G.; Valenti, G.; Grossini, E.; Donnini, S.; Marino, A.; Marinelli, R.A.; Calamita, G. Aquaporin Membrane Channels in Oxidative Stress, Cell Signaling, and Aging: Recent Advances and Research Trends. Oxid. Med. Cell. Longev. 2018, 2018, 1501847. [Google Scholar] [CrossRef]
- Sonntag, Y.; Gena, P.; Maggio, A.; Singh, T.; Artner, I.; Oklinski, M.K.; Johanson, U.; Kjellbom, P.; Nieland, J.D.; Nielsen, S.; et al. Identification and characterization of potent and selective aquaporin-3 and aquaporin-7 inhibitors. J. Biol. Chem. 2019, 294, 7377–7387. [Google Scholar] [CrossRef] [PubMed]
- Pimpao, C.; Wragg, D.; da Silva, I.V.; Casini, A.; Soveral, G. Aquaglyceroporin Modulators as Emergent Pharmacological Molecules for Human Diseases. Front. Mol. Biosci. 2022, 9, 845237. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AQP | Permeability | Skin Layer, Cell Type and Subcellular Location | Agents/Conditions Regulating the Expression/Trafficking of the AQP | Suggested Roles in the Skin |
|---|---|---|---|---|
| AQP1 | Water, hydrogen peroxide | Keratinocytes (ED) | Undefined | Creation of hydraulic pressure for cell migration |
| Melanocytes (ED; SB) | Hypertonic stress ↑ (?) | Growth of melanocyte dendrites (?); melanosome transfer to keratinocytes (?) | ||
| Fibroblast (D) | Hypertonic stress ↑ | Cellular response to hypertonic stress (?) | ||
| Vascular endothelial cells (D, HD) | Undefined | Water exchange blood-dermis (skin hydration) | ||
| AQP3 | Water, glycerol, urea, hydrogen peroxide, ammonia | Keratinocytes (ED; SB: PM and IC; SS: PM and IC; SG: PM) | HDAC3 ↓; p73 ↑; PPARγ ↑; LXR, RAR, RXR ↑; VD3 ↓; * high levels of extracellular Ca2+ ↓↑; osmotic stress (translocation to PM↑); circadian rhythm | Keratinocyte early differentiation **; keratinocyte proliferation and migration during wound healing; skin hydration (circadian rhythm); maintenance of epidermal water permeability barrier |
| Dermal-resident T cells (PM) | Hapten-induced skin contact | Chemokine-dependent T cell migration in skin hypersensitivity to haptens | ||
| AQP5 | Water, hydrogen peroxide | Sweat glands: secretory cells (APM, BPM); excretory duct cells (APM) | Muscarinic agonists | Sweat secretion |
| AQP7 | Water, glycerol | Langherans cells (ED); Dermal dendritic cells (D) | Undefined | Cell migration, antigen uptake, immune surveillance |
| AQP9 | Water, glycerol, hydrogen peroxide, ammonia | Keratinocytes (ED; upper SG) | VD3 ↑ | Keratinocyte late differentiation; transcellular route for glycerol and urea movement (?) |
| AQP10 | Water, glycerol | Keratinocytes (ED; SC) | Undefined | Barrier function |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tricarico, P.M.; Mentino, D.; De Marco, A.; Del Vecchio, C.; Garra, S.; Cazzato, G.; Foti, C.; Crovella, S.; Calamita, G. Aquaporins Are One of the Critical Factors in the Disruption of the Skin Barrier in Inflammatory Skin Diseases. Int. J. Mol. Sci. 2022, 23, 4020. https://doi.org/10.3390/ijms23074020
Tricarico PM, Mentino D, De Marco A, Del Vecchio C, Garra S, Cazzato G, Foti C, Crovella S, Calamita G. Aquaporins Are One of the Critical Factors in the Disruption of the Skin Barrier in Inflammatory Skin Diseases. International Journal of Molecular Sciences. 2022; 23(7):4020. https://doi.org/10.3390/ijms23074020
Chicago/Turabian StyleTricarico, Paola Maura, Donatella Mentino, Aurora De Marco, Cecilia Del Vecchio, Sabino Garra, Gerardo Cazzato, Caterina Foti, Sergio Crovella, and Giuseppe Calamita. 2022. "Aquaporins Are One of the Critical Factors in the Disruption of the Skin Barrier in Inflammatory Skin Diseases" International Journal of Molecular Sciences 23, no. 7: 4020. https://doi.org/10.3390/ijms23074020
APA StyleTricarico, P. M., Mentino, D., De Marco, A., Del Vecchio, C., Garra, S., Cazzato, G., Foti, C., Crovella, S., & Calamita, G. (2022). Aquaporins Are One of the Critical Factors in the Disruption of the Skin Barrier in Inflammatory Skin Diseases. International Journal of Molecular Sciences, 23(7), 4020. https://doi.org/10.3390/ijms23074020