
Trimethylamine N-Oxide (TMAO) Impairs Purinergic Induced Intracellular Calcium Increase and Nitric Oxide Release in Endothelial Cells


Abstract
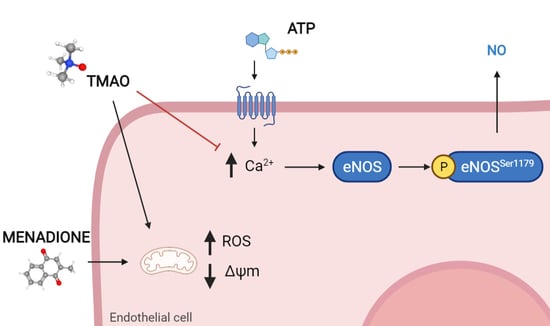
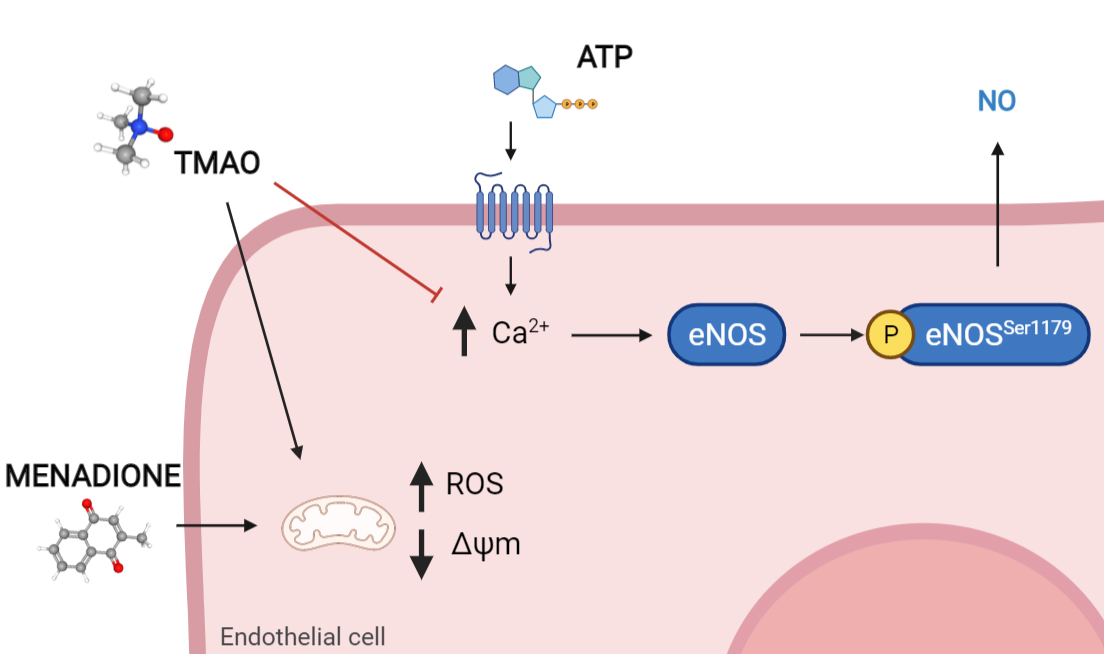
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
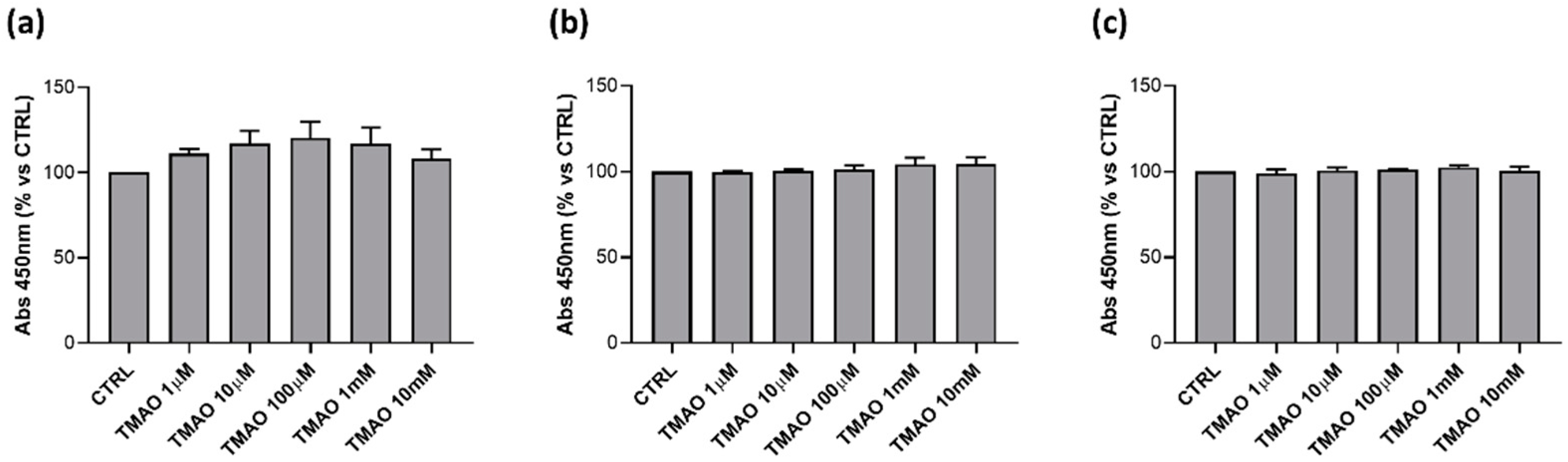
2.1. TMAO Does Not Affect Cells Viability
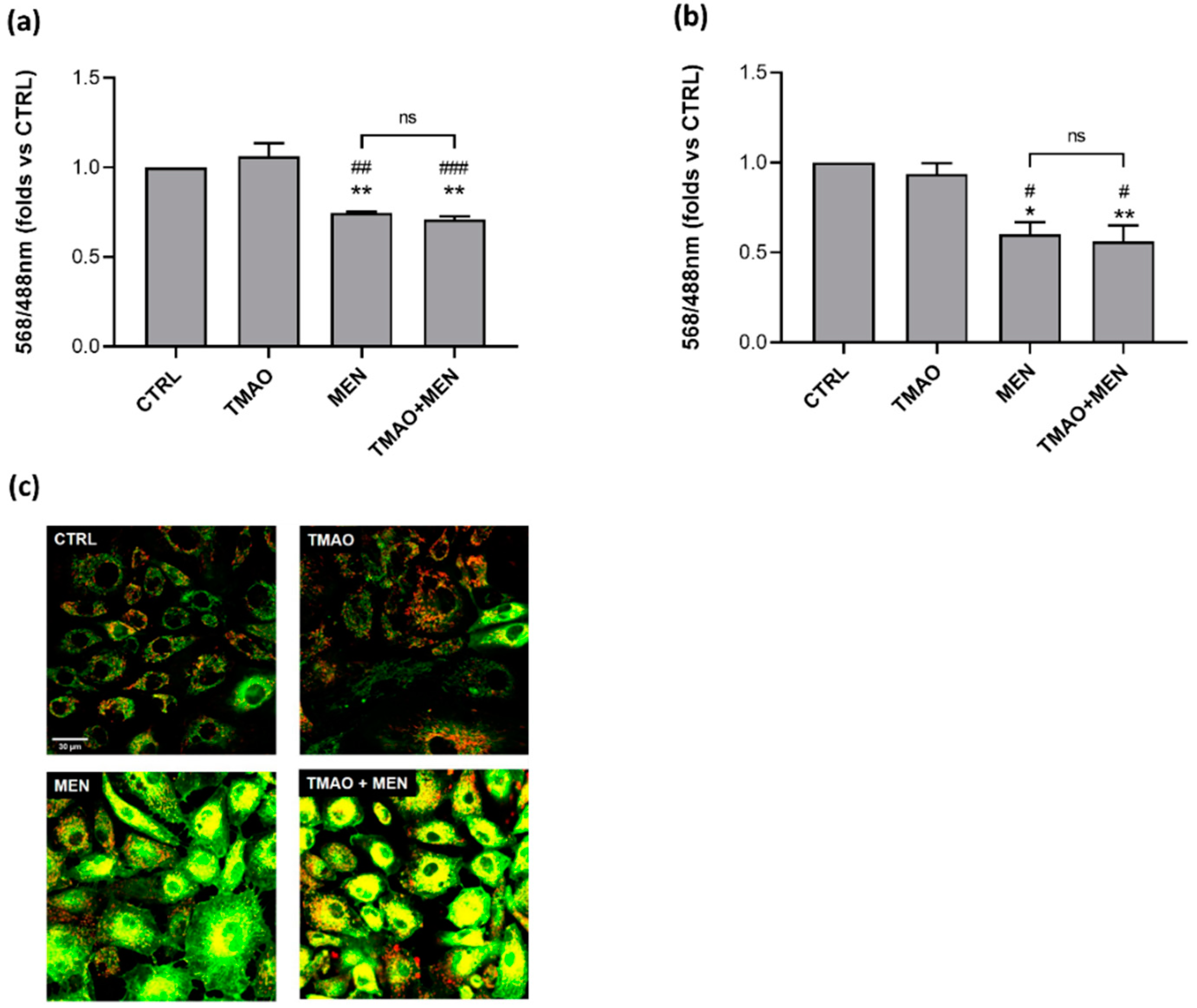
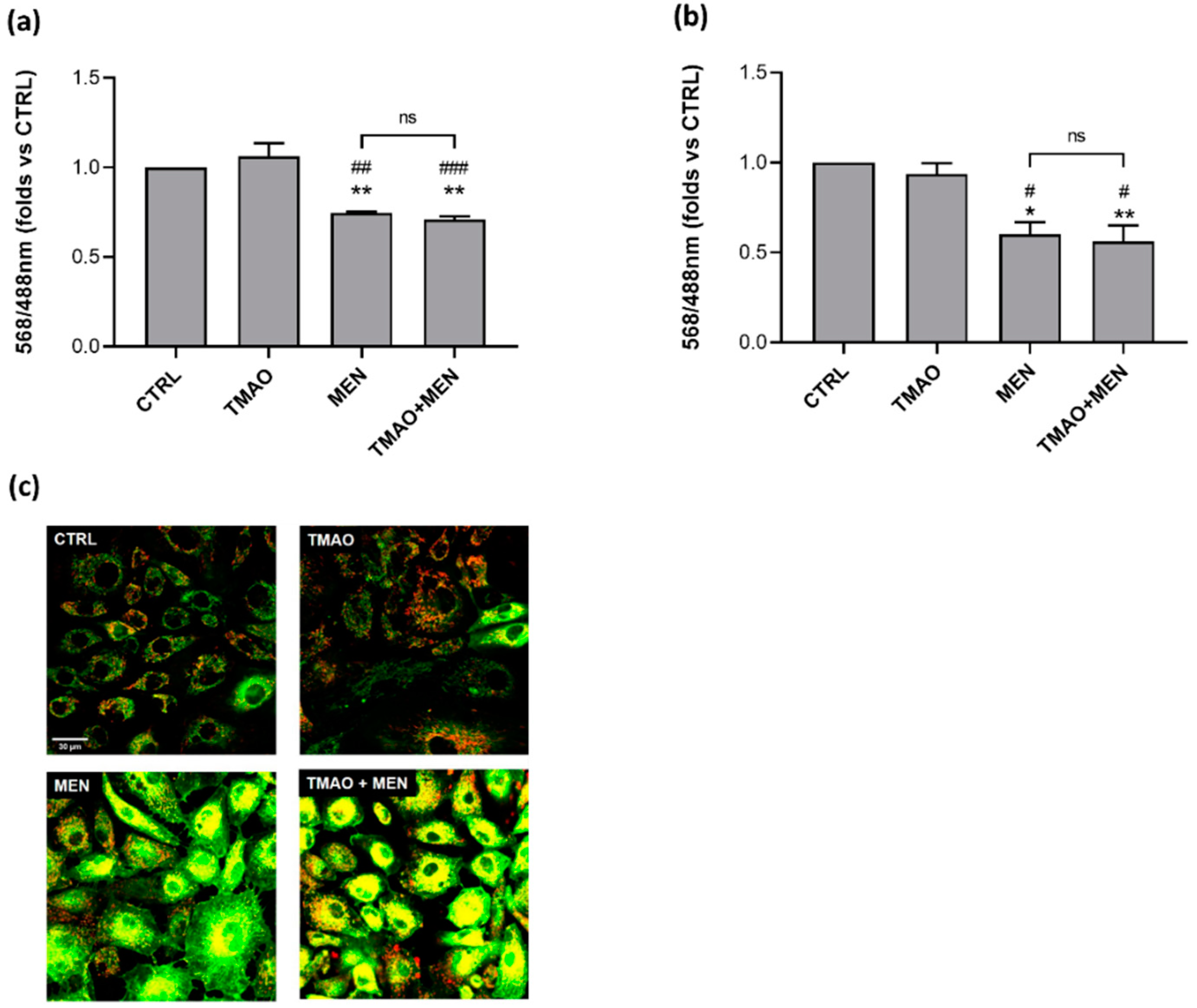
2.2. TMAO Does Not Impair Mitochondrial Membrane Potential
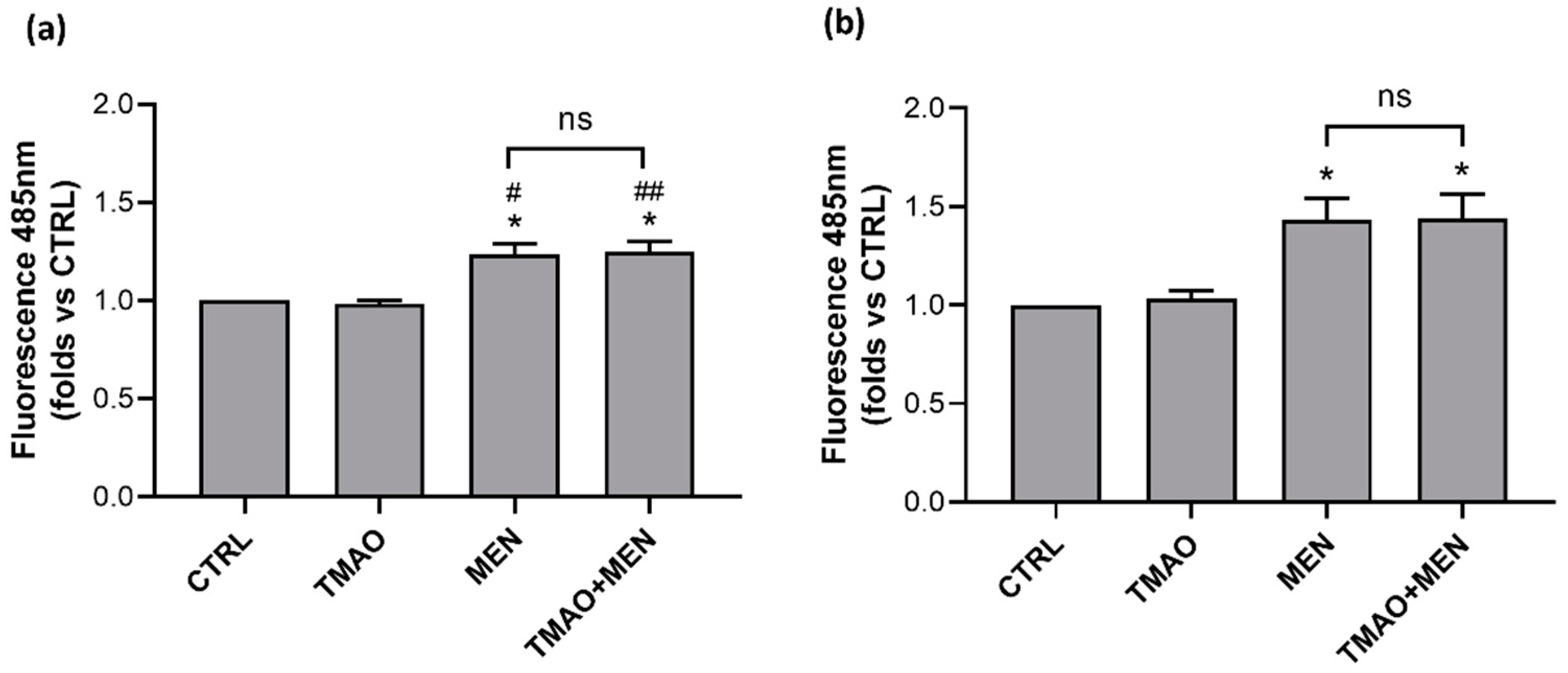
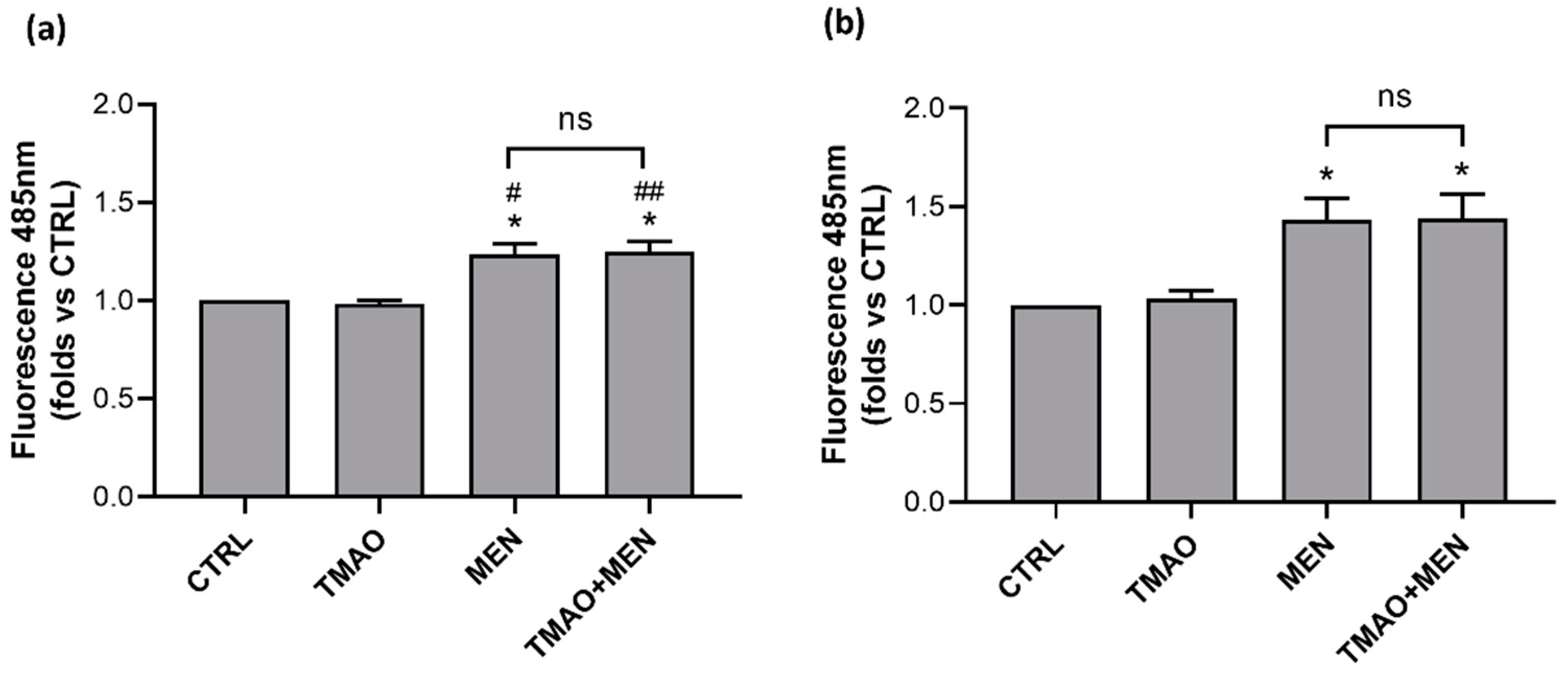
2.3. TMAO Does Not Induce the Rise of Reactive Oxygen Species
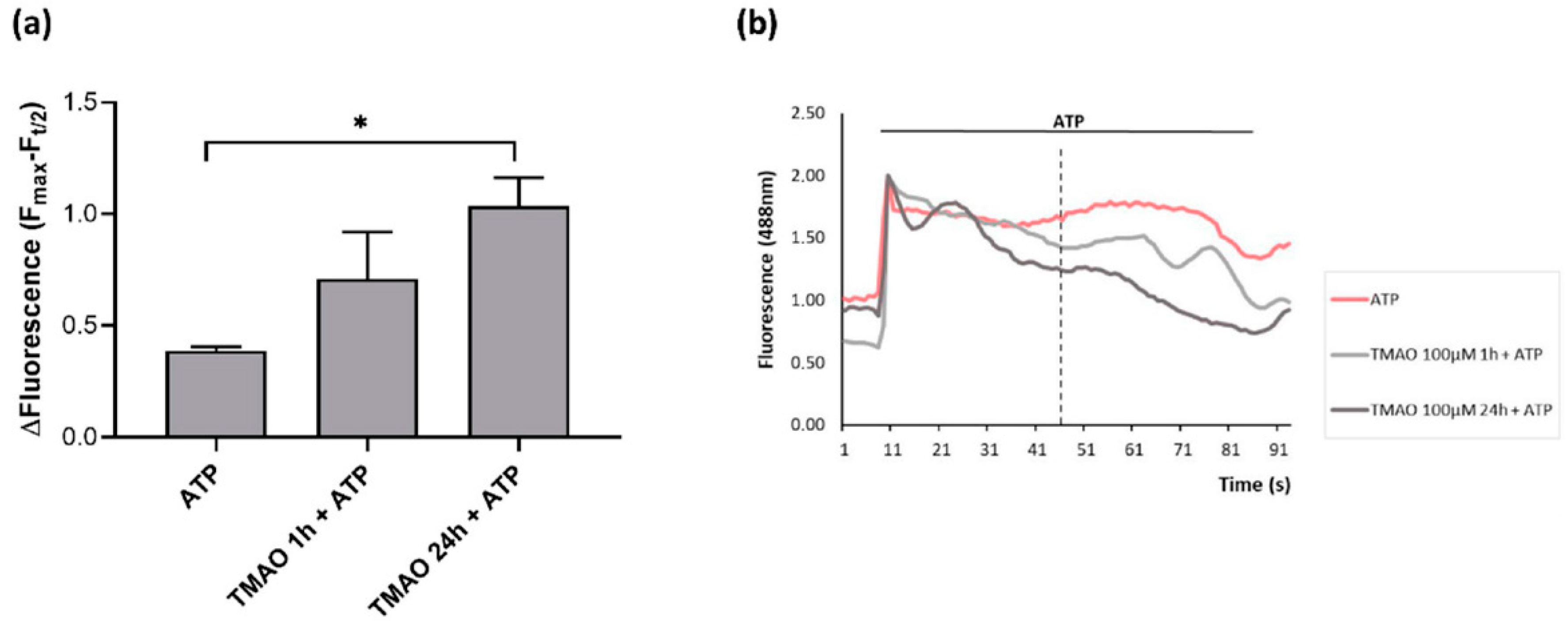
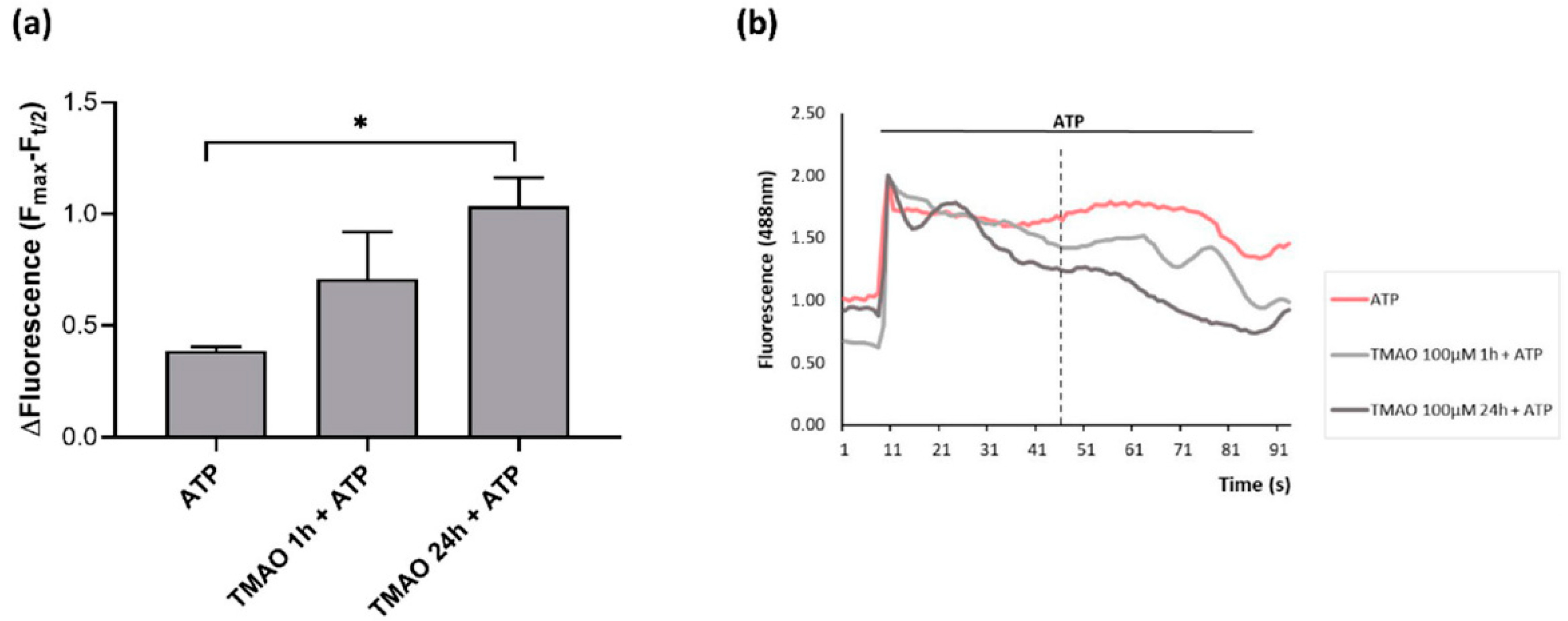
2.4. TMAO Interferes with ATP-Induced Intracellular Calcium Increase
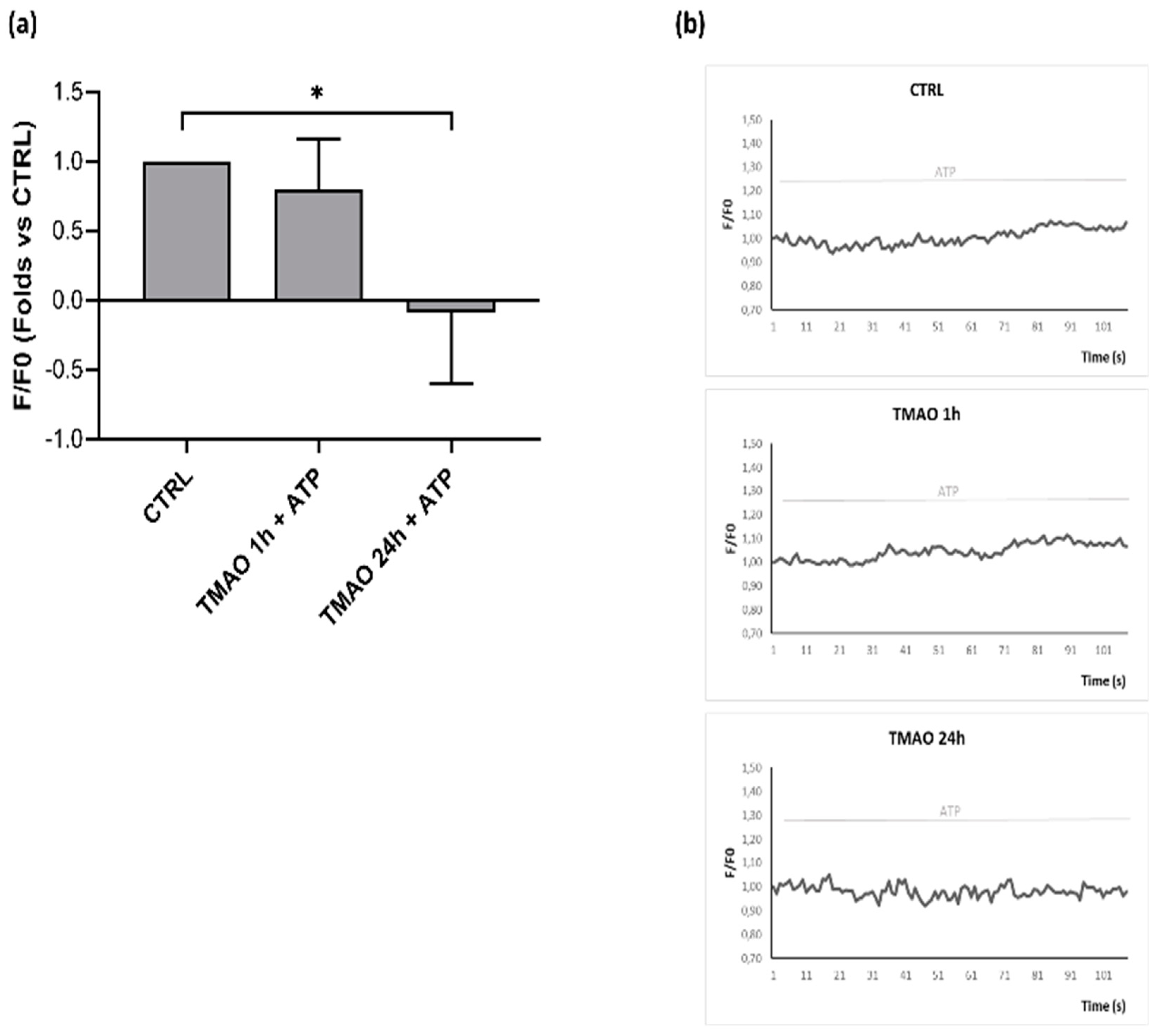
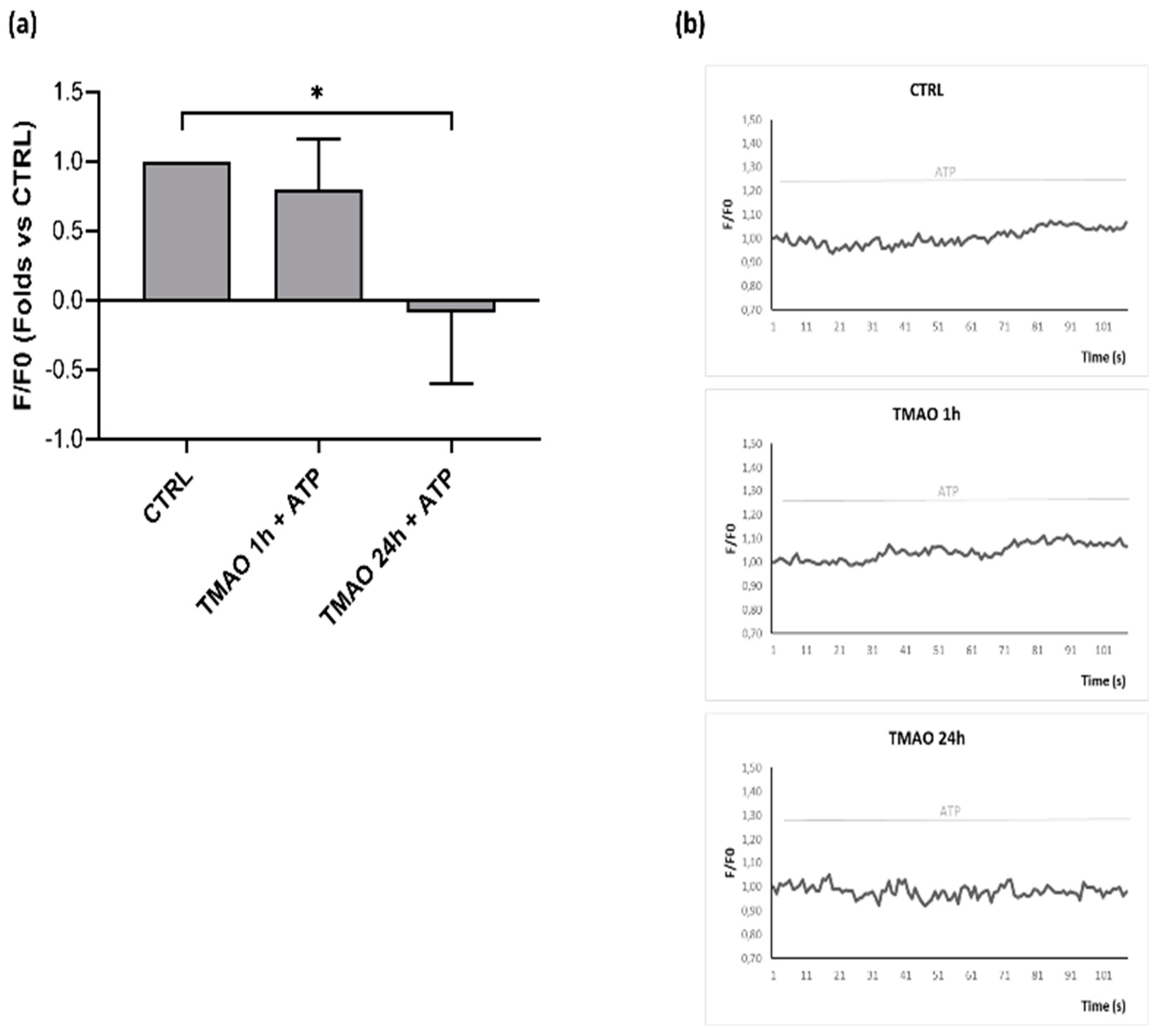
2.5. TMAO Reduces Nitric Oxide Release in Purinergic Response to ATP
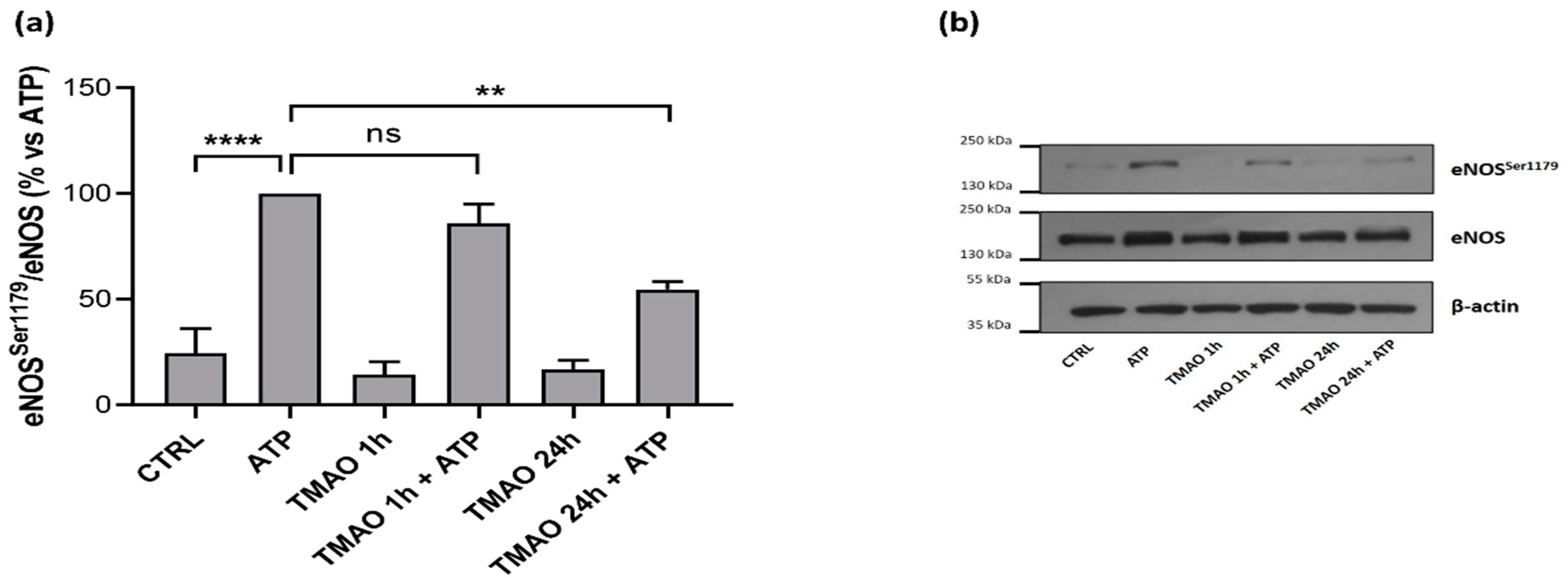
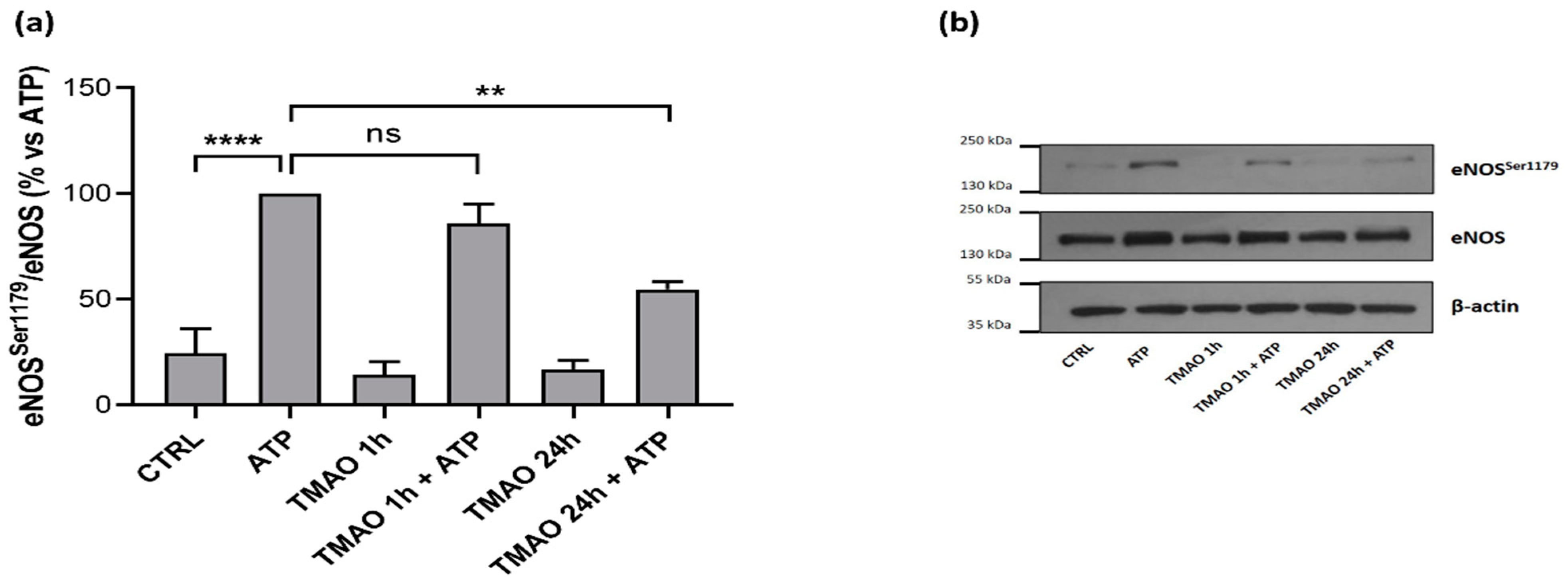
2.6. TMAO Impacts on Endothelial Nitric Oxide Synthase Phosphorylation (eNOSSer1179)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Viability
4.4. Mitochondrial Membrane Potential
4.5. Reactive Oxygen Species
4.6. Intracellular Calcium in Response to ATP Stimulation
4.7. Nitric Oxide Release after ATP Stimulation
4.8. Western Blot
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative Stress and Reactive Oxygen Species in Endothelial Dysfunction Associated with Cardiovascular and Metabolic Diseases. Vasc. Pharm. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.R.; Carr, C.S.; Al Suwaidi, J. Endothelial Dysfunction: Cardiovascular Risk Factors, Therapy, and Outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial Function and Dysfunction: Testing and Clinical Relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. ENOS Phosphorylation in Health and Disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Dudzinski, D.M.; Michel, T. Life History of ENOS: Partners and Pathways. Cardiovasc Res. 2007, 75, 247–260. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [Green Version]
- Strassheim, D.; Verin, A.; Batori, R.; Nijmeh, H.; Burns, N.; Kovacs-Kasa, A.; Umapathy, N.S.; Kotamarthi, J.; Gokhale, Y.S.; Karoor, V.; et al. P2Y Purinergic Receptors, Endothelial Dysfunction, and Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 6855. [Google Scholar] [CrossRef]
- Kramer, B.; França, L.M.; Zhang, Y.; de Paes, A.M.A.; Gerdes, A.M.; Carrillo-Sepulveda, M.A. Western Diet Triggers Toll-like Receptor 4 Signaling-Induced Endothelial Dysfunction in Female Wistar Rats. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H1735–H1747. [Google Scholar] [CrossRef]
- Elrashidy, R.A.; Zhang, J.; Liu, G. Long-Term Consumption of Western Diet Contributes to Endothelial Dysfunction and Aortic Remodeling in Rats: Implication of Rho-Kinase Signaling. Clin. Exp. Hypertens. 2019, 41, 174–180. [Google Scholar] [CrossRef]
- Simó, C.; García-Cañas, V. Dietary Bioactive Ingredients to Modulate the Gut Microbiota-Derived Metabolite TMAO. New Opportunities for Functional Food Development. Food Funct. 2020, 11, 6745–6776. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Vieira-Silva, S.; Raes, J. Microbiology Meets Big Data: The Case of Gut Microbiota-Derived Trimethylamine. Annu. Rev. Microbiol. 2015, 69, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Catucci, G.; Querio, G.; Sadeghi, S.J.; Gilardi, G.; Levi, R. Enzymatically Produced Trimethylamine N-Oxide: Conserving It or Eliminating It. Catalysts 2019, 9, 1028. [Google Scholar] [CrossRef] [Green Version]
- Gessner, A.; König, J.; Fromm, M.F. Contribution of Multidrug and Toxin Extrusion Protein 1 (MATE1) to Renal Secretion of Trimethylamine-N-Oxide (TMAO). Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Samodelov, S.L.; Kullak-Ublick, G.A.; Gai, Z.; Visentin, M. Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology. Int. J. Mol. Sci. 2020, 21, 7890. [Google Scholar] [CrossRef]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and Characterization of Trimethylamine-N-Oxide Uptake and Efflux Transporters. Mol. Pharm. 2017, 14, 310–318. [Google Scholar] [CrossRef]
- Ufnal, M.; Nowiński, A. Is Increased Plasma TMAO a Compensatory Response to Hydrostatic and Osmotic Stress in Cardiovascular Diseases? Med. Hypotheses 2019, 130, 109271. [Google Scholar] [CrossRef]
- Koay, Y.C.; Chen, Y.-C.; Wali, J.A.; Luk, A.W.S.; Li, M.; Doma, H.; Reimark, R.; Zaldivia, M.T.K.; Habtom, H.T.; Franks, A.E.; et al. Plasma Levels of Trimethylamine-N-Oxide Can Be Increased with “healthy” and “Unhealthy” Diets and Do Not Correlate with the Extent of Atherosclerosis but with Plaque Instability. Cardiovasc Res. 2021, 117, 435–449. [Google Scholar] [CrossRef]
- Skagen, K.; Trøseid, M.; Ueland, T.; Holm, S.; Abbas, A.; Gregersen, I.; Kummen, M.; Bjerkeli, V.; Reier-Nilsen, F.; Russell, D.; et al. The Carnitine-Butyrobetaine-Trimethylamine-N-Oxide Pathway and Its Association with Cardiovascular Mortality in Patients with Carotid Atherosclerosis. Atherosclerosis 2016, 247, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Chou, R.-H.; Chen, C.-Y.; Chen, I.-C.; Huang, H.-L.; Lu, Y.-W.; Kuo, C.-S.; Chang, C.-C.; Huang, P.-H.; Chen, J.-W.; Lin, S.-J. Trimethylamine N-Oxide, Circulating Endothelial Progenitor Cells, and Endothelial Function in Patients with Stable Angina. Sci. Rep. 2019, 9, 4249. [Google Scholar] [CrossRef]
- Papandreou, C.; Moré, M.; Bellamine, A. Trimethylamine N-Oxide in Relation to Cardiometabolic Health-Cause or Effect? Nutrients 2020, 12, 1330. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef]
- Aldana-Hernández, P.; Leonard, K.-A.; Zhao, Y.-Y.; Curtis, J.M.; Field, C.J.; Jacobs, R.L. Dietary Choline or Trimethylamine N-Oxide Supplementation Does Not Influence Atherosclerosis Development in Ldlr-/- and Apoe-/- Male Mice. J. Nutr. 2020, 150, 249–255. [Google Scholar] [CrossRef]
- Brunt, V.E.; Gioscia-Ryan, R.A.; Casso, A.G.; VanDongen, N.S.; Ziemba, B.P.; Sapinsley, Z.J.; Richey, J.J.; Zigler, M.C.; Neilson, A.P.; Davy, K.P.; et al. Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension 2020, 76, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-Oxide in Atherogenesis: Impairing Endothelial Self-Repair Capacity and Enhancing Monocyte Adhesion. Biosci. Rep. 2017, 37, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-Oxide Induces Inflammation and Endothelial Dysfunction in Human Umbilical Vein Endothelial Cells via Activating ROS-TXNIP-NLRP3 Inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Missailidis, C.; Hällqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [Green Version]
- Gatarek, P.; Kaluzna-Czaplinska, J. Trimethylamine N-Oxide (TMAO) in Human Health. Excli. J. 2021, 20, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Bae, O.N.; Chung, S.M.; Lee, M.Y.; Chung, J.H. Menadione Induces Endothelial Dysfunction Mediated by Oxidative Stress and Arylation. Chem. Biol. Interact 2001, 137, 169–183. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, M.Y.; Chung, S.M.; Chung, J.H. Menadione-Induced Vascular Endothelial Dysfunction and Its Possible Significance. Toxicol. Appl. Pharm. 1999, 161, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Querio, G.; Antoniotti, S.; Levi, R.; Gallo, M.P. Trimethylamine N-Oxide Does Not Impact Viability, ROS Production, and Mitochondrial Membrane Potential of Adult Rat Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Xiong, S. TMAO-Protein Preferential Interaction Profile Determines TMAO’s Conditional In Vivo Compatibility. Biophys. J. 2016, 111, 1866–1875. [Google Scholar] [CrossRef] [Green Version]
- Senthong, V.; Li, X.S.; Hudec, T.; Coughlin, J.; Wu, Y.; Levison, B.; Wang, Z.; Hazen, S.L.; Tang, W.H.W. Plasma Trimethylamine N-Oxide, a Gut Microbe-Generated Phosphatidylcholine Metabolite, Is Associated With Atherosclerotic Burden. J. Am. Coll. Cardiol. 2016, 67, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhu, X.; Ran, L.; Lang, H.; Yi, L.; Mi, M. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart. Assoc. 2017, 6, 9. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Querio, G.; Antoniotti, S.; Geddo, F.; Levi, R.; Gallo, M.P. Trimethylamine N-Oxide (TMAO) Impairs Purinergic Induced Intracellular Calcium Increase and Nitric Oxide Release in Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 3982. https://doi.org/10.3390/ijms23073982
Querio G, Antoniotti S, Geddo F, Levi R, Gallo MP. Trimethylamine N-Oxide (TMAO) Impairs Purinergic Induced Intracellular Calcium Increase and Nitric Oxide Release in Endothelial Cells. International Journal of Molecular Sciences. 2022; 23(7):3982. https://doi.org/10.3390/ijms23073982
Chicago/Turabian StyleQuerio, Giulia, Susanna Antoniotti, Federica Geddo, Renzo Levi, and Maria Pia Gallo. 2022. "Trimethylamine N-Oxide (TMAO) Impairs Purinergic Induced Intracellular Calcium Increase and Nitric Oxide Release in Endothelial Cells" International Journal of Molecular Sciences 23, no. 7: 3982. https://doi.org/10.3390/ijms23073982
APA StyleQuerio, G., Antoniotti, S., Geddo, F., Levi, R., & Gallo, M. P. (2022). Trimethylamine N-Oxide (TMAO) Impairs Purinergic Induced Intracellular Calcium Increase and Nitric Oxide Release in Endothelial Cells. International Journal of Molecular Sciences, 23(7), 3982. https://doi.org/10.3390/ijms23073982