
Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome

 ,
, 
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
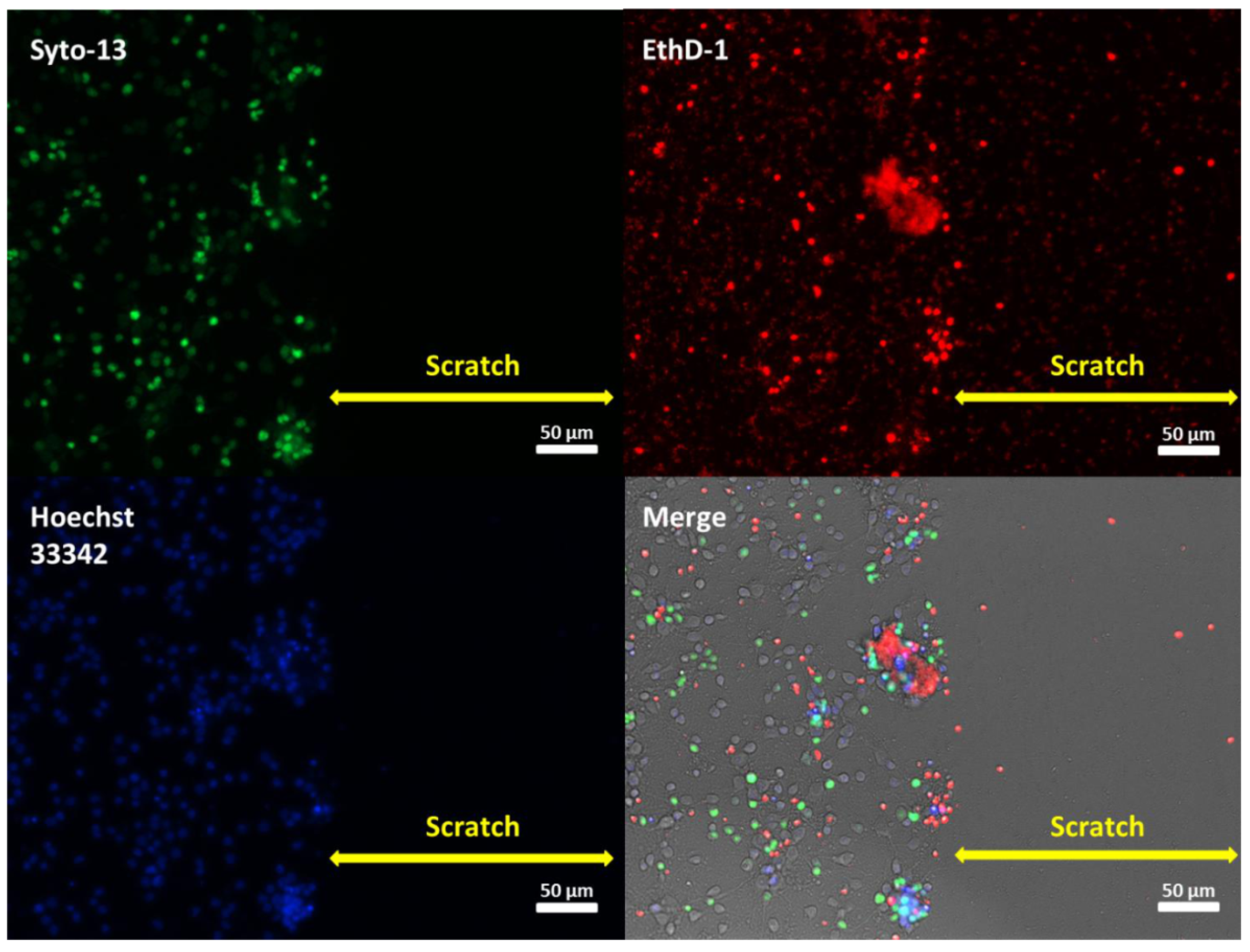
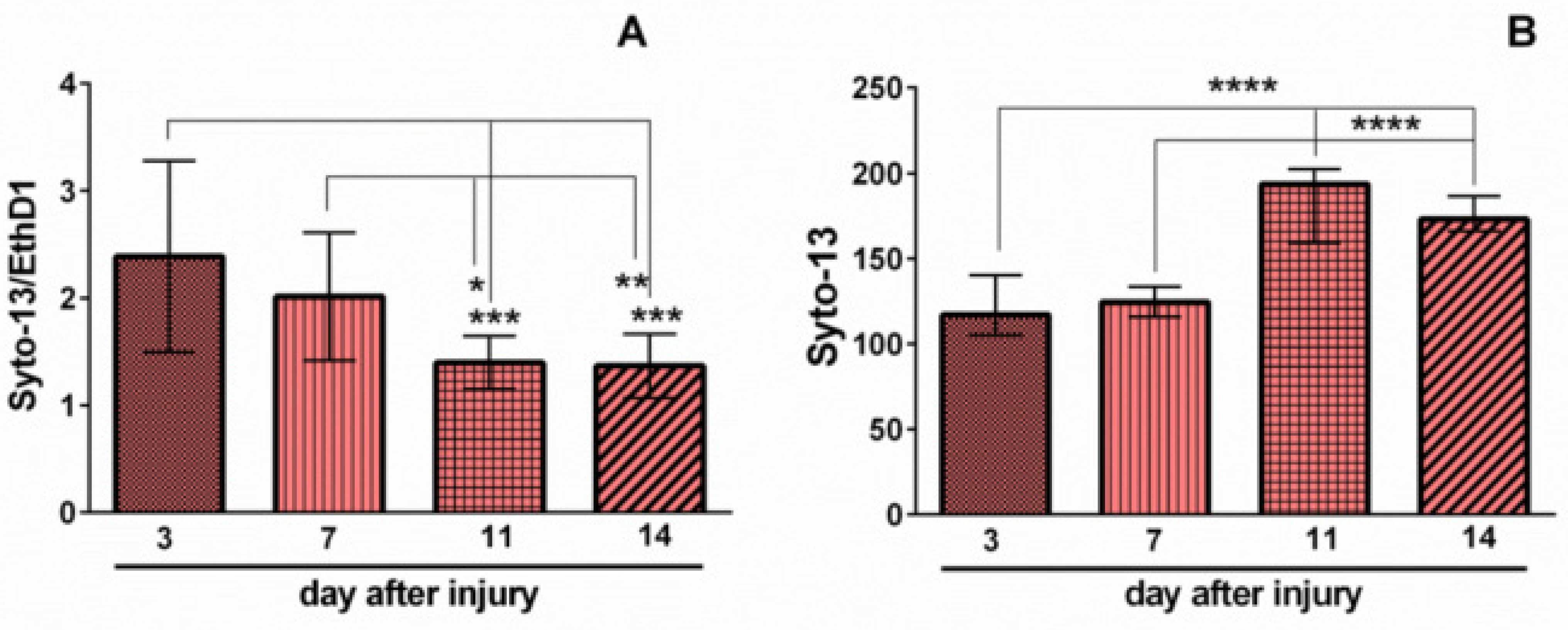
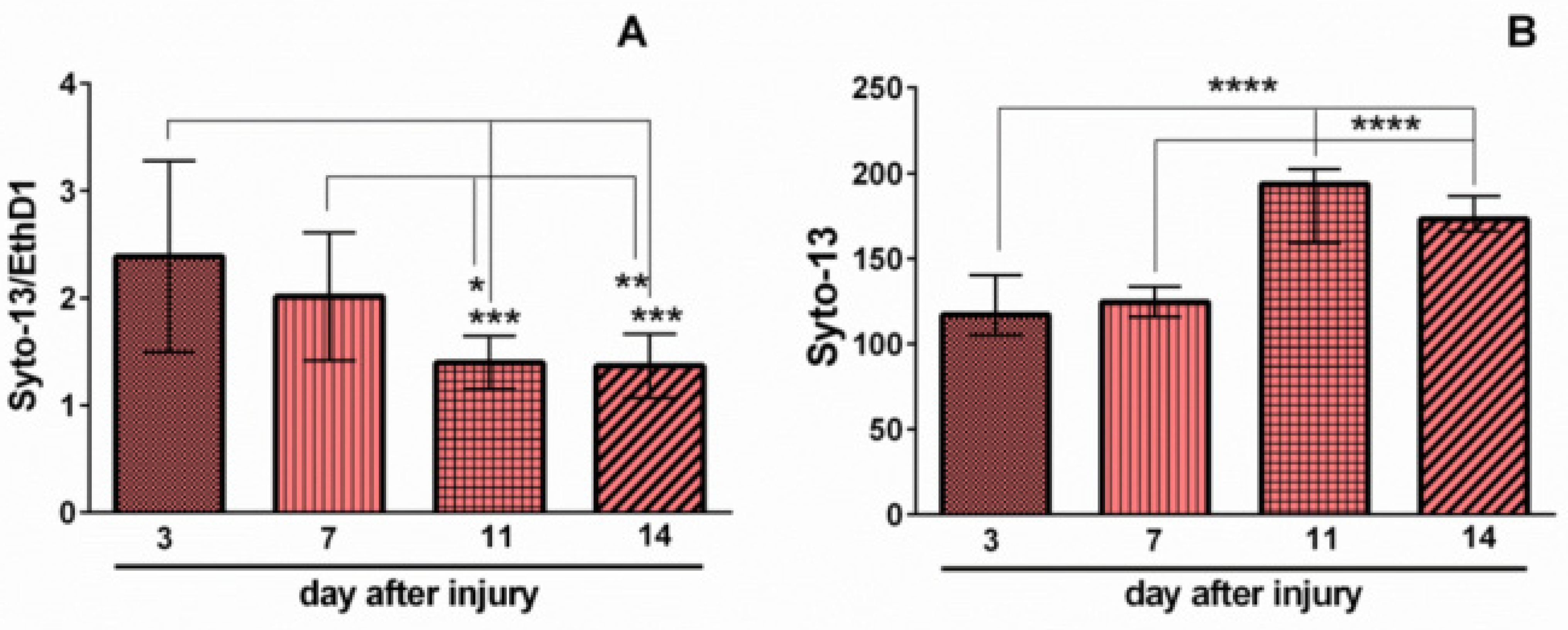
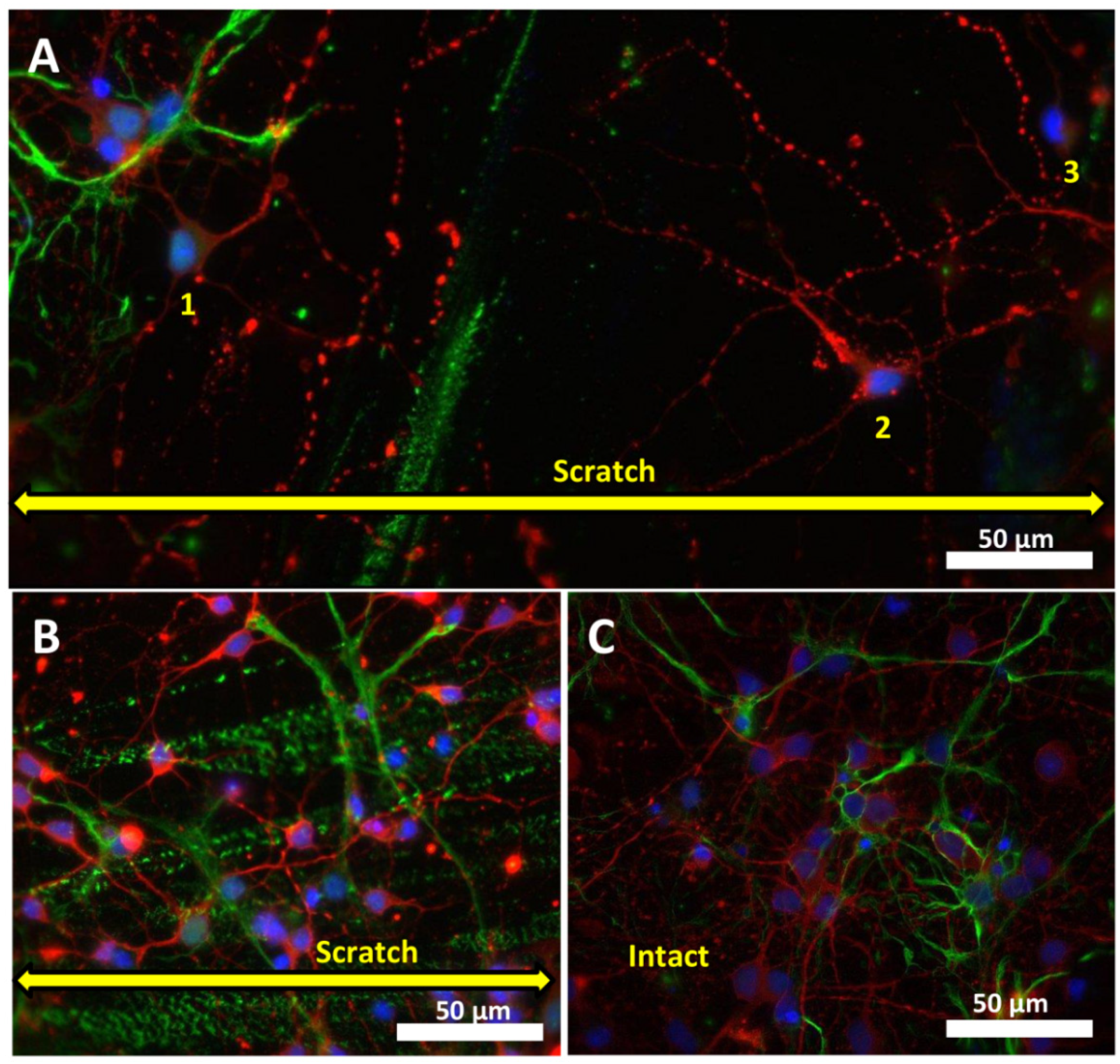
2.1. Effects of Mechanical Trauma on Cell Survival and Regeneration in Neuroglial Culture
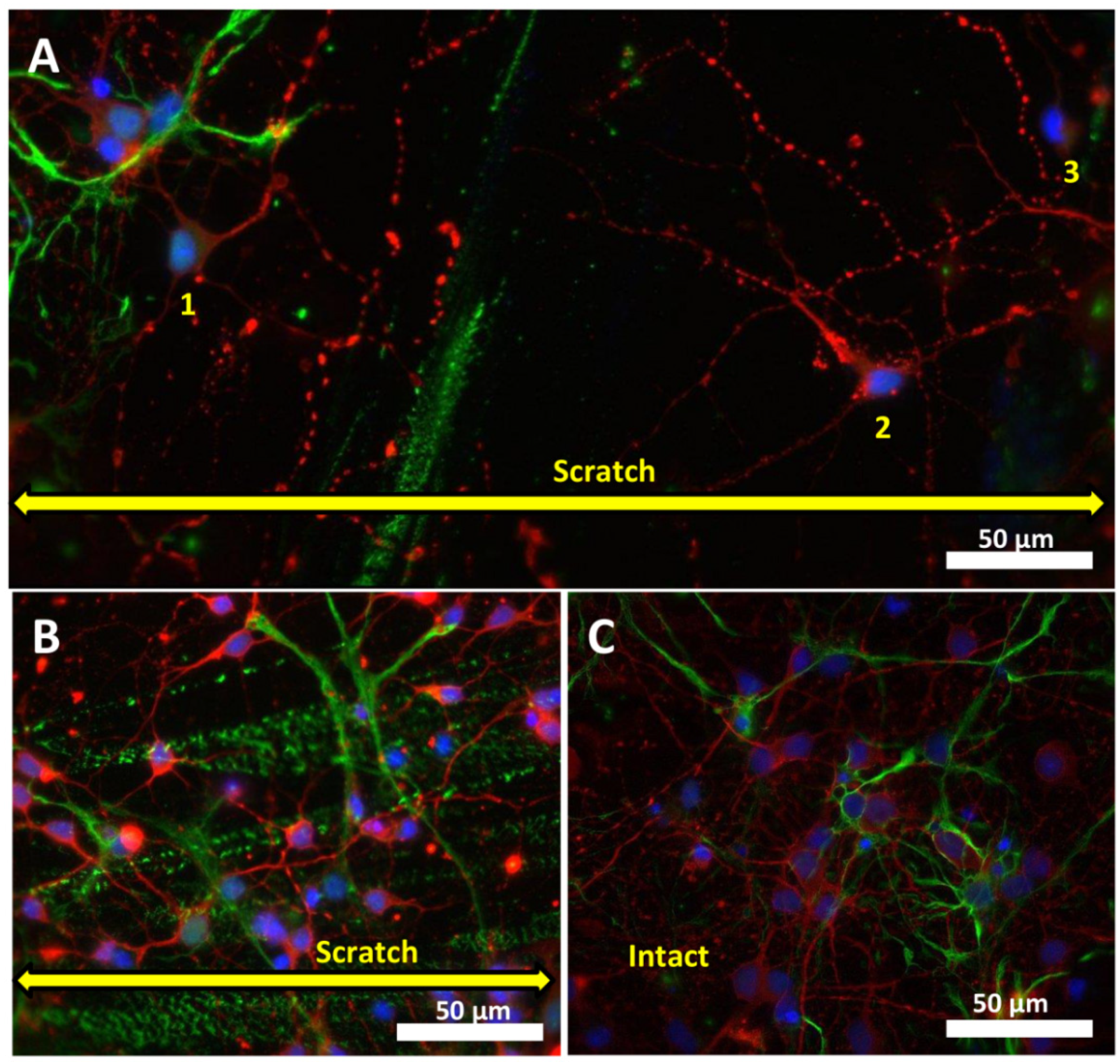
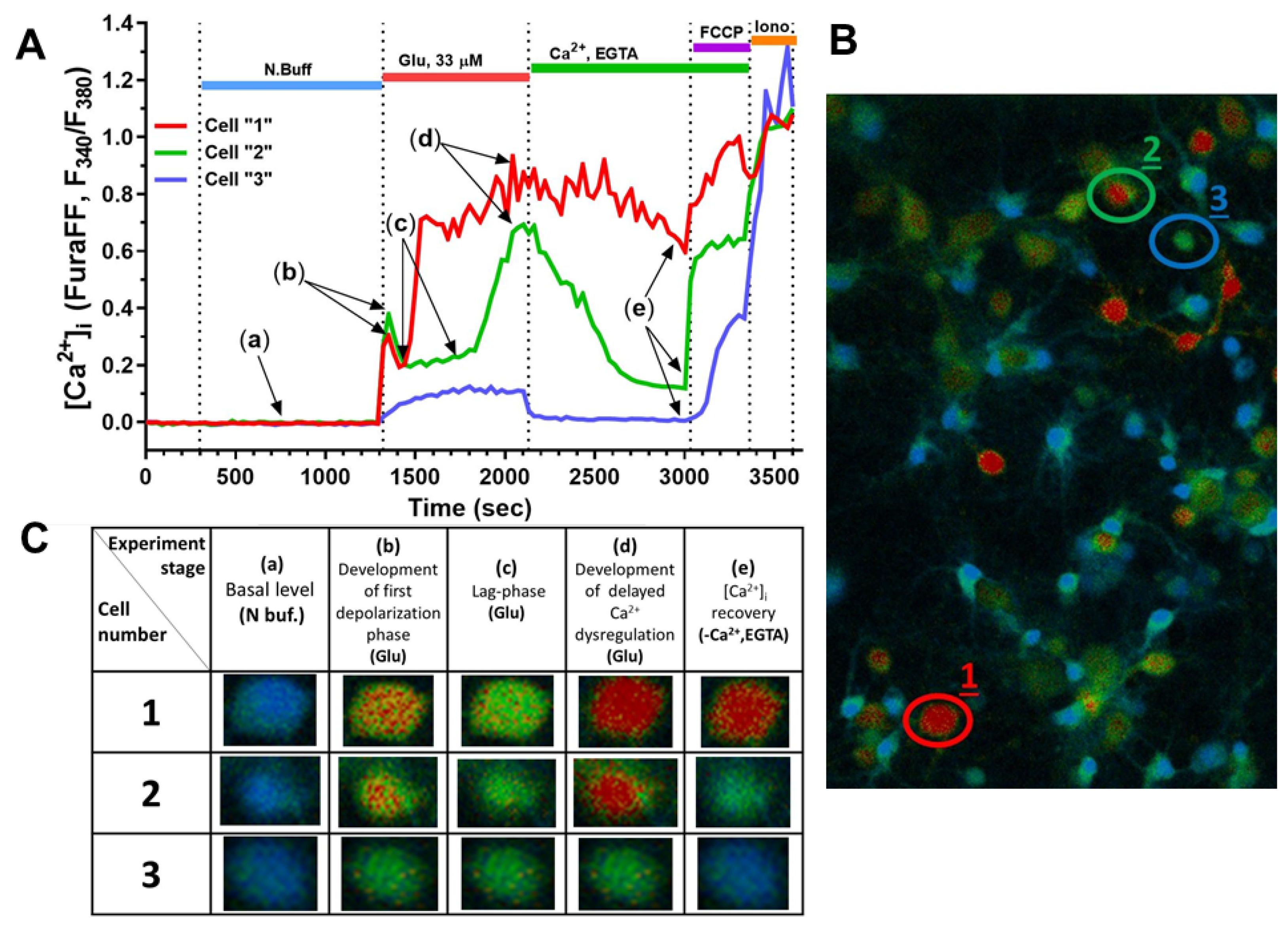
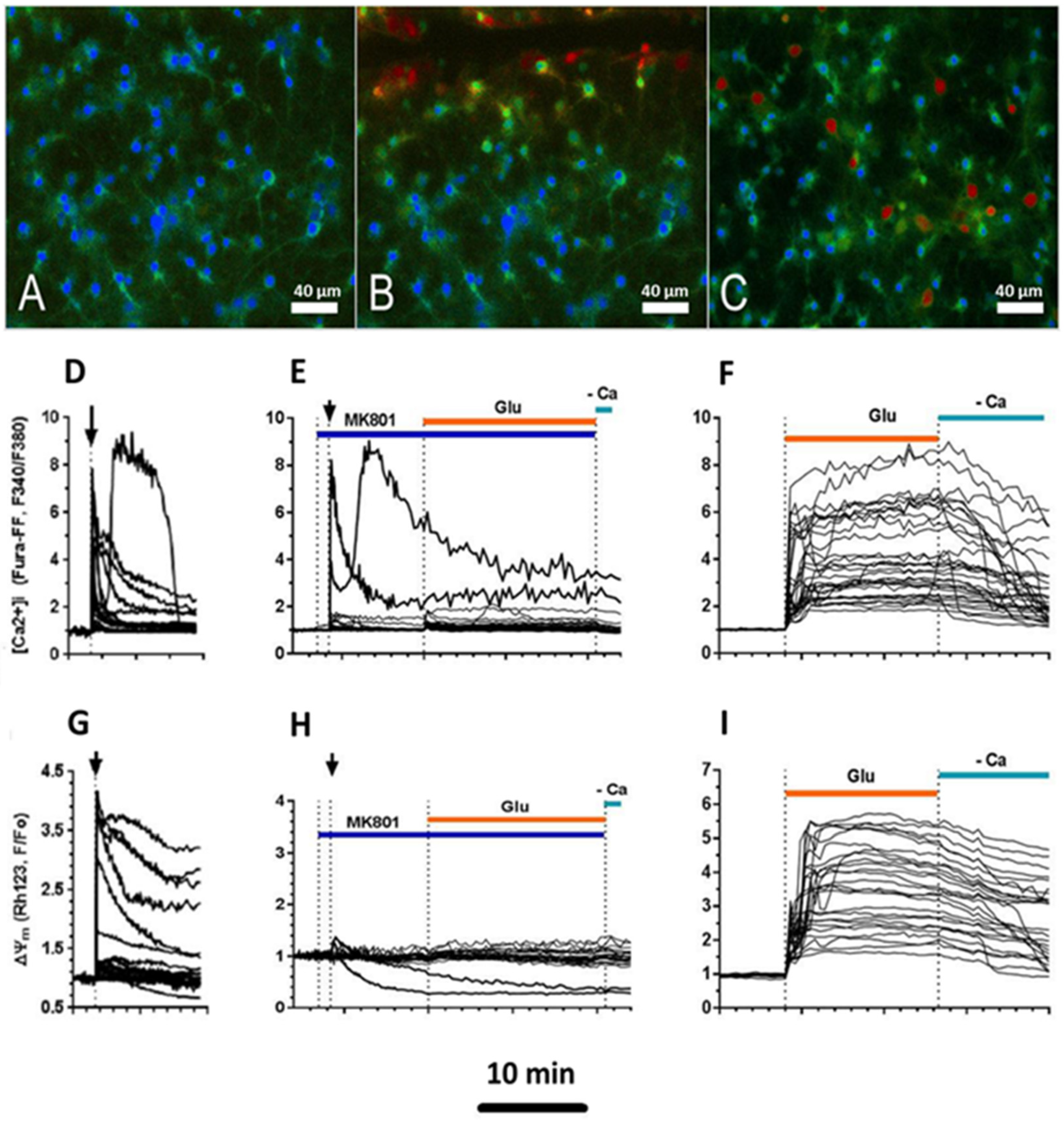
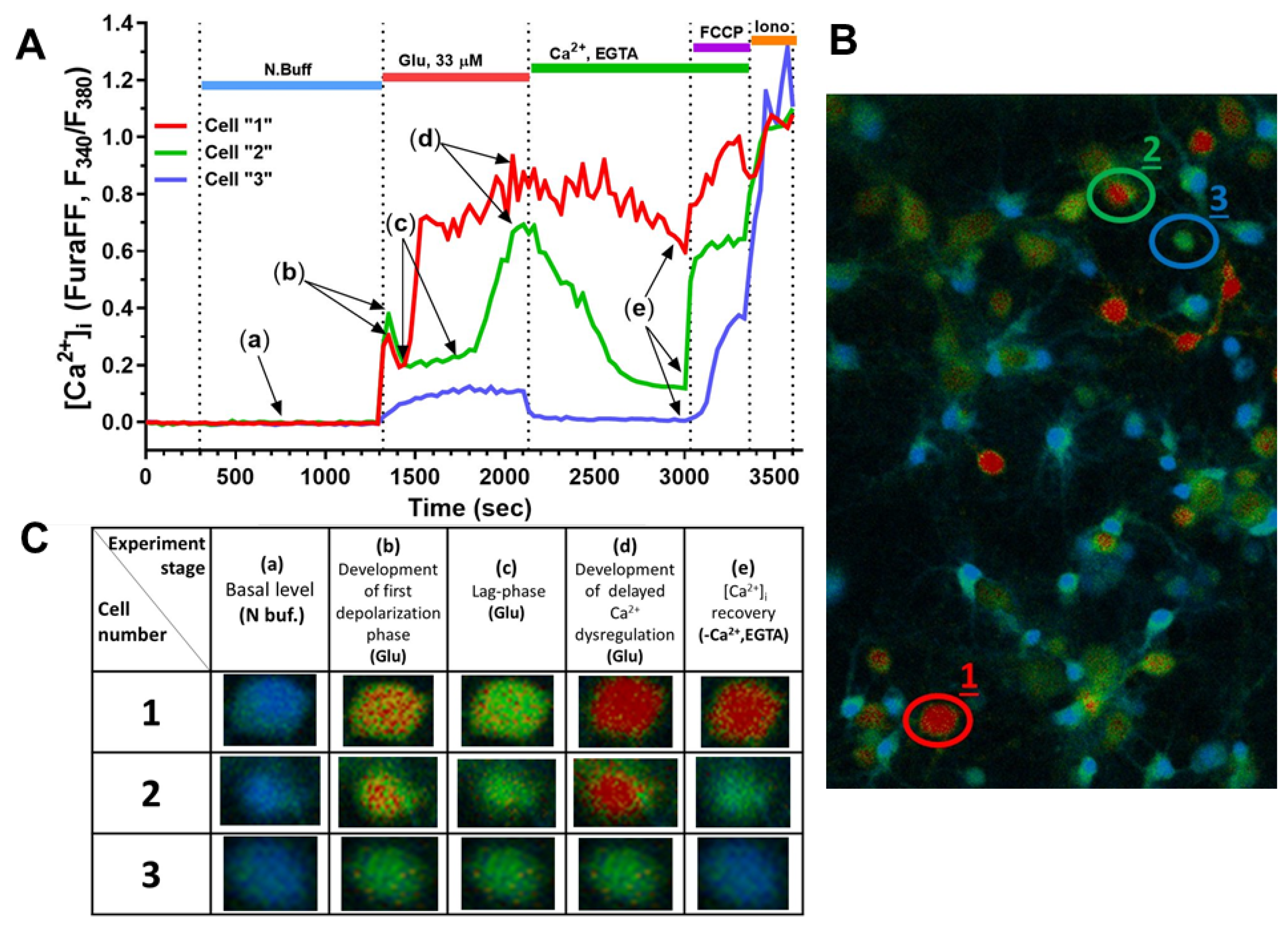
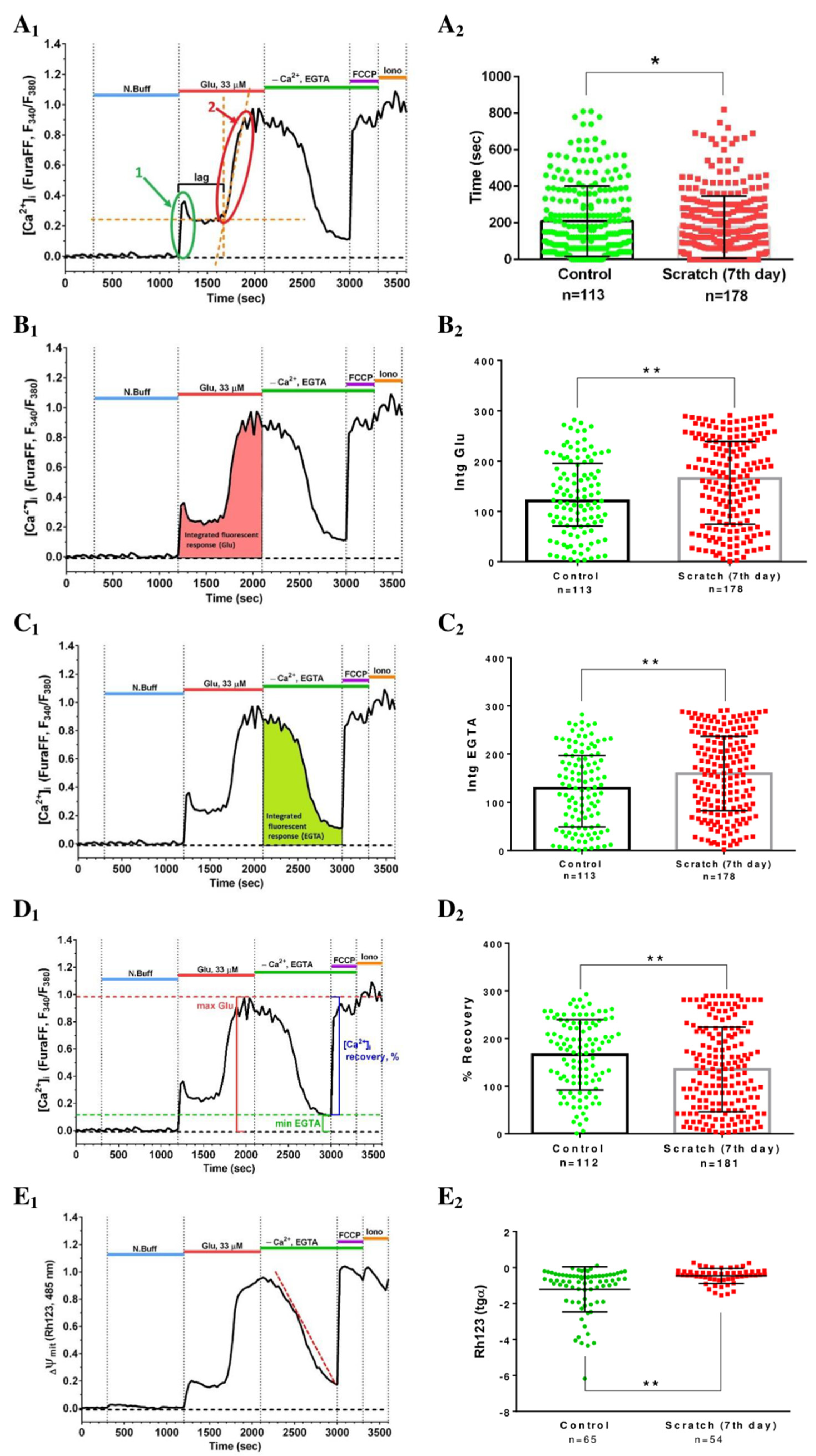
2.2. Effect of Mechanical Injury on the Parameters of Acute Changes in [Ca2+]i and ΔΨm
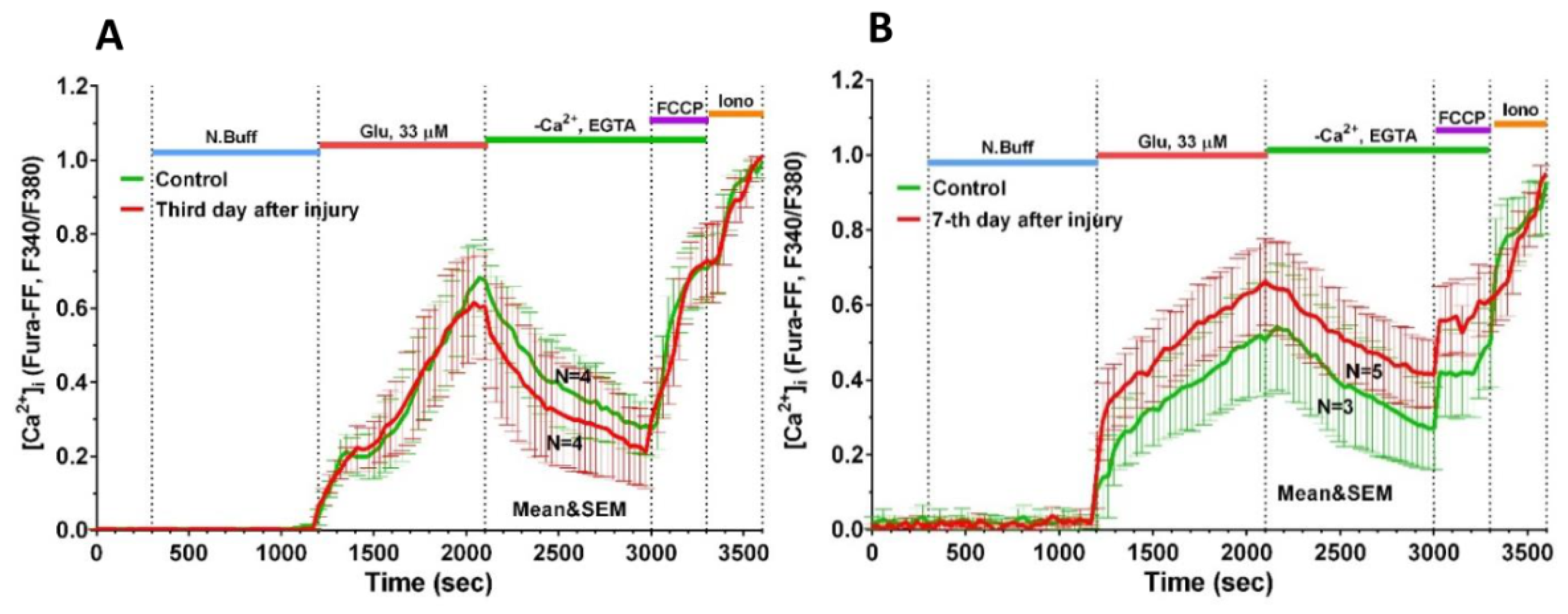
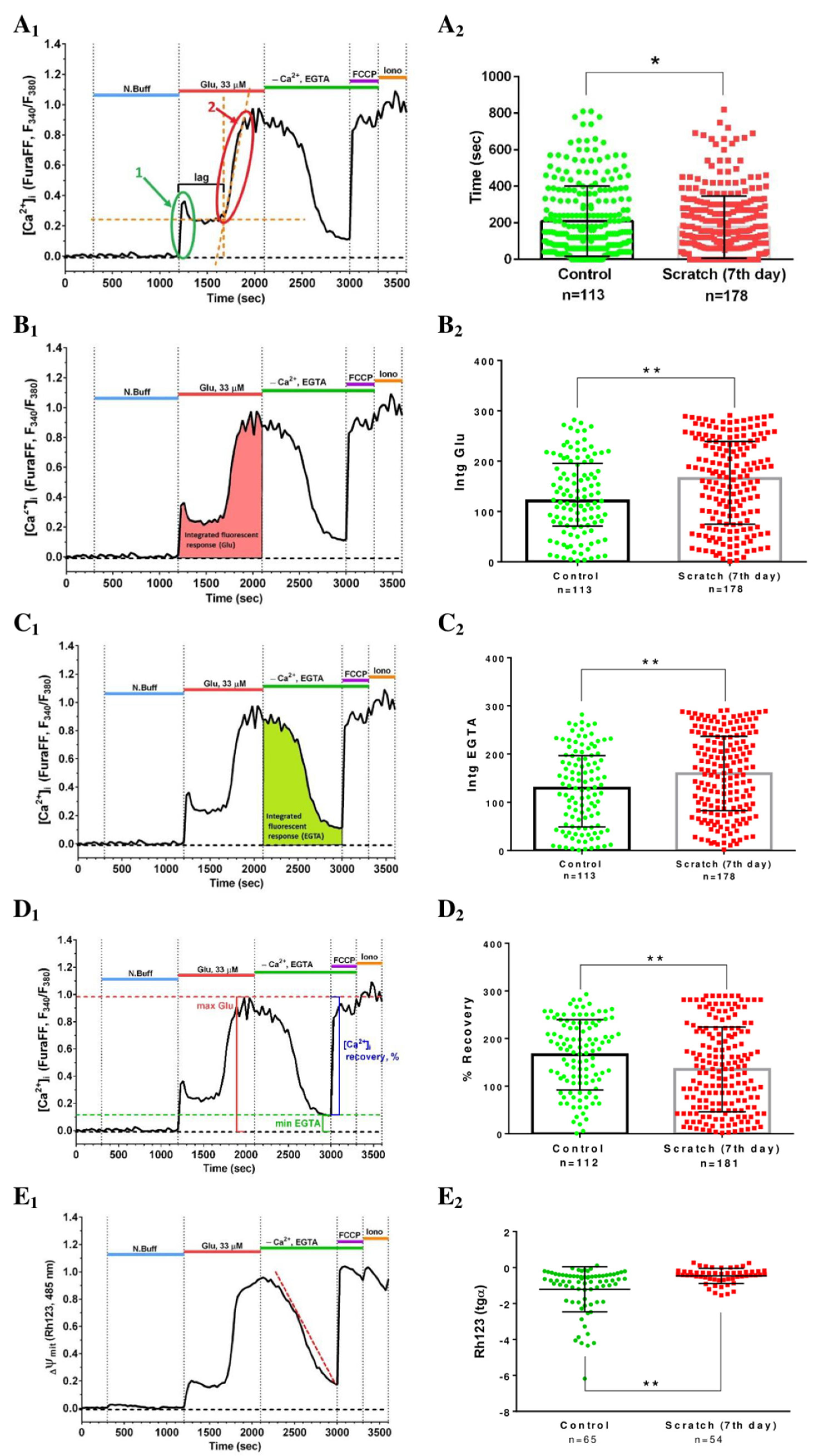
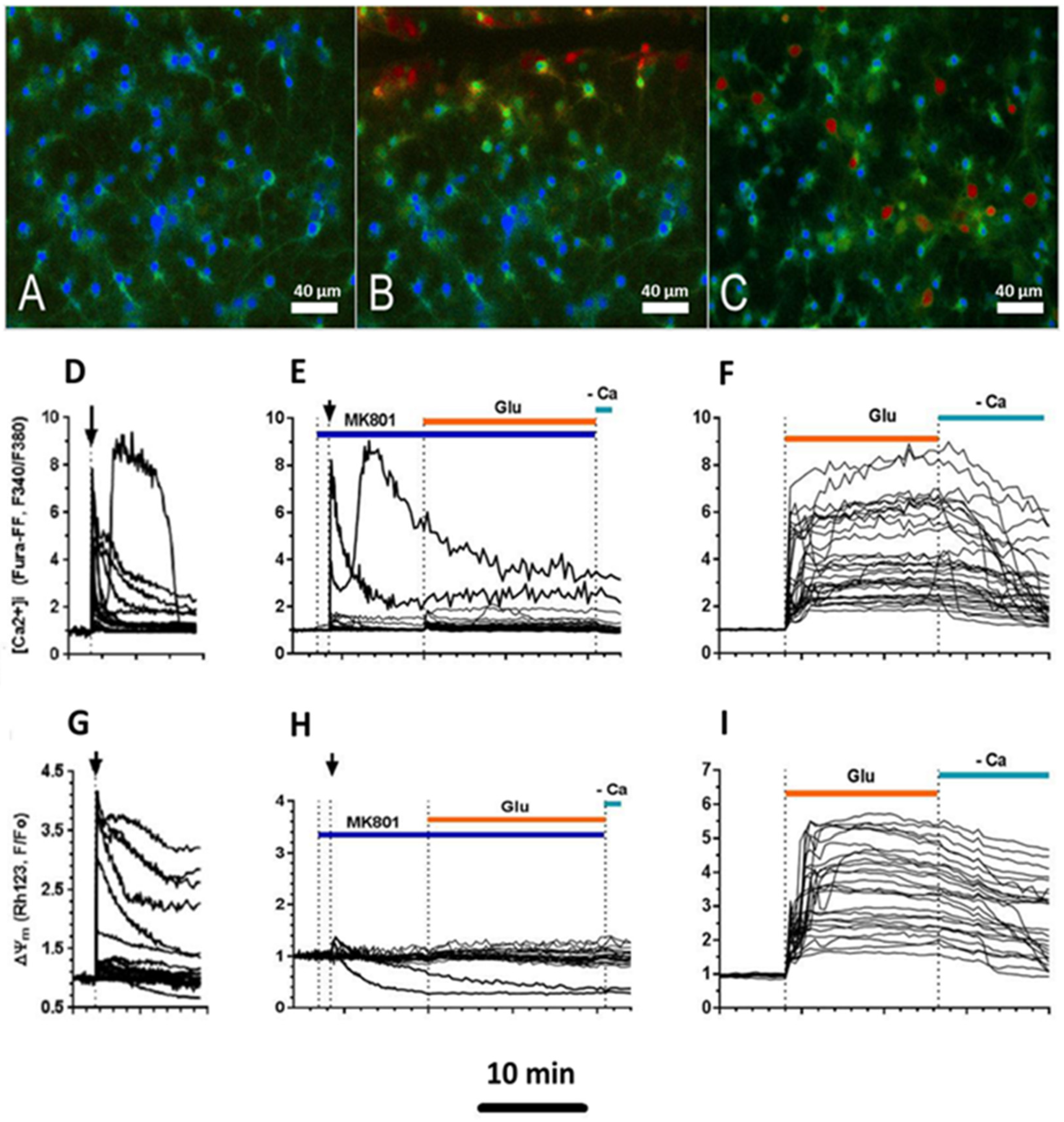
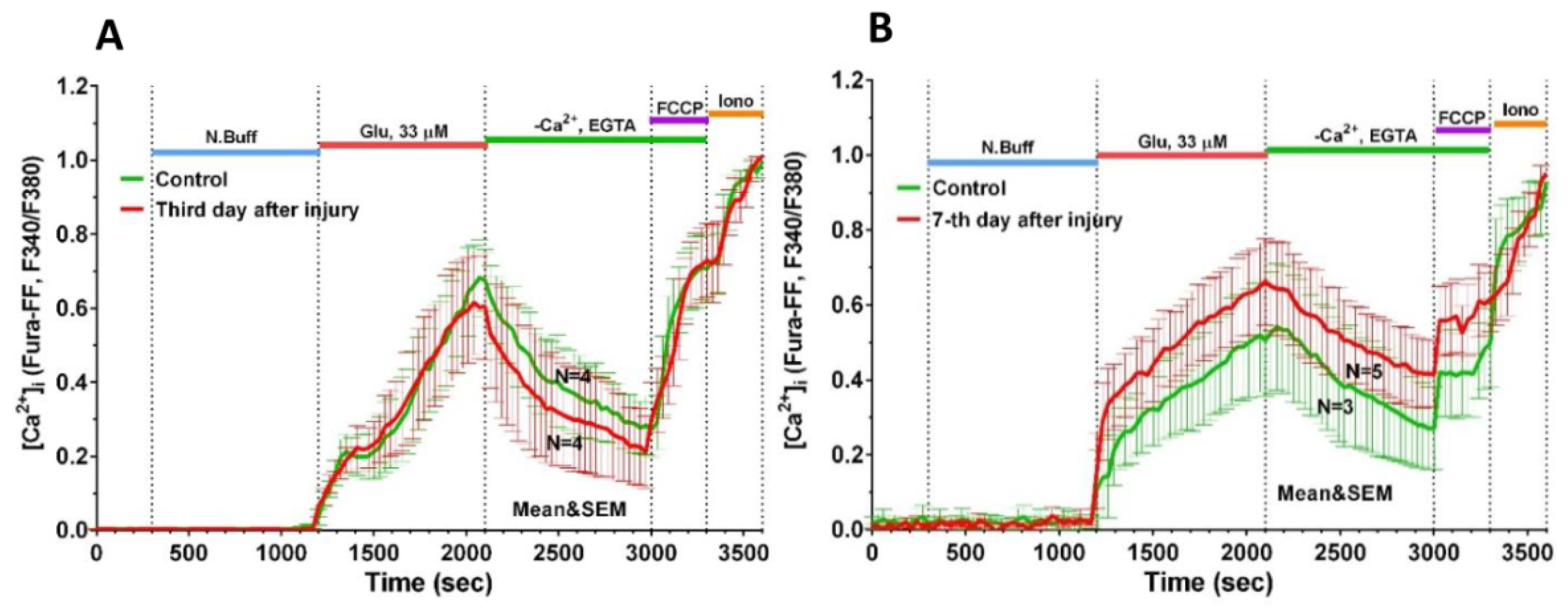
2.3. Delayed Changes in [Ca2+]i and ΔΨm in Cells of Mechanically Injured Neuroglial Cultures under Conditions of Glutamate Excitotoxicity
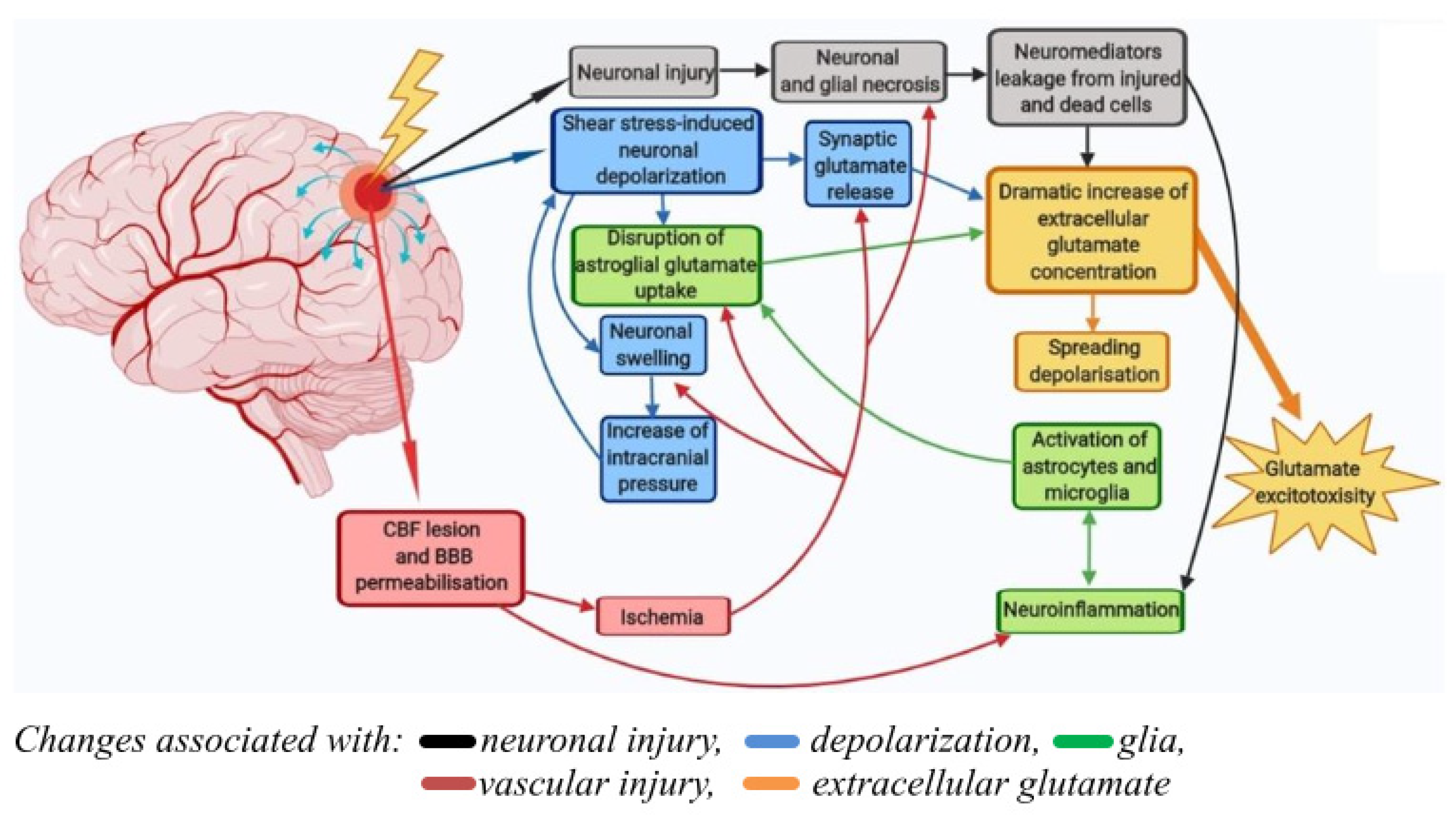
3. Discussion
4. Materials and Methods
4.1. Isolation and Cultivation of Cerebral Cortex Cells
4.2. Methods to Inflict Mechanical Injury
4.3. Evaluation of Neuronal Viability
4.4. Immunofluorescence Staining of Neuroglial Culture on Markers GFAP and Beta-3 Tubulin
4.5. Fluorescence Microscopy Measurements of [Ca2+]i and ΔΨm
4.6. Reagents
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic Brain Injury and Mitochondrial Dysfunction. Am. J. Med. Sci. 2015, 350, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Engelhard, K. Pathophysiology of Traumatic Brain Injury. BJA Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikawa, D.G. The Role of Excitotoxic Programmed Necrosis in Acute Brain Injury. Comput. Struct. Biotechnol. J. 2015, 13, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andriessen, T.M.J.C.; Jacobs, B.; Vos, P.E. Clinical Characteristics and Pathophysiological Mechanisms of Focal and Diffuse Traumatic Brain Injury. J. Cell. Mol. Med. 2010, 14, 2381–2392. [Google Scholar] [CrossRef] [Green Version]
- Arundine, M.; Tymianski, M. Molecular Mechanisms of Glutamate-Dependent Neurodegeneration in Ischemia and Traumatic Brain Injury. Cell. Mol. Life Sci. CMLS 2004, 61, 657–668. [Google Scholar] [CrossRef]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors Affecting Excitatory Amino Acid Release Following Severe Human Head Injury. J. Neurosurg. 1998, 89, 507–518. [Google Scholar] [CrossRef]
- Yi, J.-H.; Hazell, A.S. Excitotoxic Mechanisms and the Role of Astrocytic Glutamate Transporters in Traumatic Brain Injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef]
- Dorsett, C.R.; McGuire, J.L.; Niedzielko, T.L.; DePasquale, E.A.K.; Meller, J.; Floyd, C.L.; McCullumsmith, R.E. Traumatic Brain Injury Induces Alterations in Cortical Glutamate Uptake without a Reduction in Glutamate Transporter-1 Protein Expression. J. Neurotrauma 2017, 34, 220–234. [Google Scholar] [CrossRef] [Green Version]
- Lapanantasin, S.; Chongthammakun, S.; Floyd, C.L.; Berman, R.F. Effects of 17β-estradiol on Intracellular Calcium Changes and Neuronal Survival after Mechanical Strain Injury in Neuronal–Glial Cultures. Synapse 2006, 60, 406–410. [Google Scholar] [CrossRef]
- Rungta, R.L.; Choi, H.B.; Tyson, J.R.; Malik, A.; Dissing-Olesen, L.; Lin, P.J.C.; Cain, S.M.; Cullis, P.R.; Snutch, T.P.; Macvicar, B.A. The Cellular Mechanisms of Neuronal Swelling Underlying Cytotoxic Edema. Cell 2015, 161, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.T.; Cheng, N.C.; Wu, C.Y.; Yang, Y.L. NKCC1-Mediated Traumatic Brain Injury-Induced Brain Edema and Neuron Death via Raf/MEK/MAPK Cascade. Crit. Care Med. 2008, 36, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K. Water Homeostasis in the Brain: Basic Concepts. Neuroscience 2004, 129, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Tecoma, E.S.; Monyer, H.; Goldberg, M.P.; Choi, D.W. Traumatic Neuronal Injury in Vitro Is Attenuated by NMDA Antagonists. Neuron 1989, 2, 1541–1545. [Google Scholar] [CrossRef]
- Mukhin, A.G.; Ivanova, S.A.; Knoblach, S.M.; Faden, A.I. New in Vitro Model of Traumatic Neuronal Injury: Evaluation of Secondary Injury and Glutamate Receptor-Mediated Neurotoxicity. J. Neurotrauma 1997, 14, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Mukhin, A.G.; Ivanova, S.A.; Allen, J.W.; Faden, A.I. Mechanical Injury to Neuronal/Glial Cultures in Microplates: Role of NMDA Receptors and PH in Secondary Neuronal Cell Death. J. Neurosci. Res. 1998, 51, 748–758. [Google Scholar] [CrossRef]
- Laskowski, A.; Schmidt, W.; Dinkel, K.; Martínez-Sánchez, M.; Reymann, K.G. BFGF and EGF Modulate Trauma-Induced Proliferation and Neurogenesis in Juvenile Organotypic Hippocampal Slice Cultures. Brain Res. 2005, 1037, 78–89. [Google Scholar] [CrossRef]
- Allen, J.W.; Knoblach, S.M.; Faden, A.I. Combined Mechanical Trauma and Metabolic Impairment in Vitro Induces NMDA Receptor-dependent Neuronal Cell Death and Caspase-3-dependent Apoptosis. FASEB J. 1999, 13, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Bakaeva, Z.V.; Surin, A.M.; Lizunova, N.V.; Zgodova, A.E.; Krasilnikova, I.A.; Fisenko, A.P.; Frolov, D.A.; Andreeva, L.A.; Myasoedov, N.F.; Pinelis, V.G. Neuroprotective Potential of Peptides HFRWPGP (ACTH6–9PGP), KKRRPGP, and PyrRP in Cultured Cortical Neurons at Glutamate Excitotoxicity. Dokl. Biochem. Biophys. 2020, 491, 62–66. [Google Scholar] [CrossRef]
- Murphy, T.H.; Baraban, J.M. Glutamate Toxicity in Immature Cortical Neurons Precedes Development of Glutamate Receptor Currents. Dev. Brain Res. 1990, 57, 146–150. [Google Scholar] [CrossRef]
- Tabernero, A.; Bolanos, J.P.; Medina, J.M. Lipogenesis from Lactate in Rat Neurons and Astrocytes in Primary Culture. Biochem. J. 1993, 294, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Gwag, B.J.; Canzoniero, L.M.T.; Sensi, S.L.; Demaro, J.A.; Koh, J.Y.; Goldberg, M.P.; Jacquin, M.; Choi, D.W. Calcium Ionophores Can Induce Either Apoptosis or Necrosis in Cultured Cortical Neurons. Neuroscience 1999, 90, 1339–1348. [Google Scholar] [CrossRef]
- Taguchi, R.; Nishikawa, H.; Kume, T.; Terauchi, T.; Kaneko, S.; Katsuki, H.; Yonaga, M.; Sugimoto, H.; Akaike, A. Serofendic Acid Prevents Acute Glutamate Neurotoxicity in Cultured Cortical Neurons. Eur. J. Pharmacol. 2003, 477, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Kambe, Y.; Nakamichi, N.; Georgiev, D.D.; Nakamura, N.; Taniura, H.; Yoneda, Y. Insensitivity to Glutamate Neurotoxicity Mediated by NMDA Receptors in Association with Delayed Mitochondrial Membrane Potential Disruption in Cultured Rat Cortical Neurons. J. Neurochem. 2008, 105, 1886–1900. [Google Scholar] [CrossRef]
- Grebenik, E.A.; Surin, A.M.; Bardakova, K.N.; Demina, T.S.; Minaev, N.V.; Veryasova, N.N.; Artyukhova, M.A.; Krasilnikova, I.A.; Bakaeva, Z.V.; Sorokina, E.G.; et al. Chitosan-g-Oligo(L,L-Lactide) Copolymer Hydrogel for Nervous Tissue Regeneration in Glutamate Excitotoxicity: In Vitro Feasibility Evaluation. Biomed. Mater. 2020, 15, 015011. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Maimaitili, M.; Habekost, M.; Gill, K.P.; Mermet-Joret, N.; Nabavi, S.; Febbraro, F.; Denham, M. Rapid Generation of Regionally Specified CNS Neurons by Sequential Patterning and Conversion of Human Induced Pluripotent Stem Cells. Stem Cell Res. 2020, 48, 101945. [Google Scholar] [CrossRef]
- Galiakberova, A.A.; Surin, A.M.; Bakaeva, Z.V.; Sharipov, R.R.; Zhang, D.; Dorovskoy, D.A.; Shakirova, K.M.; Fisenko, A.P.; Dashinimaev, E.B. IPSC-Derived Human Neurons with GCaMP6s Expression Allow In Vitro Study of Neurophysiological Responses to Neurochemicals. Neurochem. Res. 2021, 47, 952–966. [Google Scholar] [CrossRef]
- Sánchez-Alcañiz, J.A.; Haege, S.; Mueller, W.; Pla, R.; Mackay, F.; Schulz, S.; López-Bendito, G.; Stumm, R.; Marín, O. Cxcr7 Controls Neuronal Migration by Regulating Chemokine Responsiveness. Neuron 2011, 69, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Bakaeva, Z.; Lizunova, N.; Tarzhanov, I.; Boyarkin, D.; Petrichuk, S.; Pinelis, V.; Fisenko, A.; Tuzikov, A.; Sharipov, R.; Surin, A. Lipopolysaccharide From E. Coli Increases Glutamate-Induced Disturbances of Calcium Homeostasis, the Functional State of Mitochondria, and the Death of Cultured Cortical Neurons. Front. Mol. Neurosci. 2022, 14, 811171. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Kirchhoff, F. NMDA Receptors in Glia. Neuroscientist 2007, 13, 28–37. [Google Scholar] [CrossRef]
- Khodorov, B.I.; Storozhevykh, T.P.; Surin, A.M.; Yuryavichyus, A.I.; Sorokina, E.G.; Borodin, A.V.; Vinskaya, N.P.; Khaspekov, L.G.; Pinelis, V.G. The Leading Role of Mitochondrial Depolarization in the Mechanism of Glutamate-Induced Disruptions in Ca2+ Homeostasis. Neurosci. Behav. Physiol. 2002, 32, 541–547. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and Neuronal Survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodorov, B. Glutamate-Induced Deregulation of Calcium Homeostasis and Mitochondrial Dysfunction in Mammalian Central Neurones. Prog. Biophys. Mol. Biol. 2004, 86, 279–351. [Google Scholar] [CrossRef] [PubMed]
- Hawley, C. Management of Minor Head Injury in Adults. Emerg. Nurse 2010, 18, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Dikmen, S.; Machamer, J.; Temkin, N. Mild Traumatic Brain Injury: Longitudinal Study of Cognition, Functional Status, and Post-Traumatic Symptoms. J. Neurotrauma 2017, 34, 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Theeler, B.; Lucas, S.; Riechers, R.G.; Ruff, R.L. Post-traumatic Headaches in Civilians and Military Personnel: A Comparative, Clinical Review. Headache J. Head Face Pain 2013, 53, 881–900. [Google Scholar] [CrossRef] [PubMed]
- Marshman, L.A.G.; Jakabek, D.; Hennessy, M.; Quirk, F.; Guazzo, E.P. Post-Traumatic Amnesia. J. Clin. Neurosci. 2013, 20, 1475–1481. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Gan, C.; Zhao, K.; Chen, J.; Song, J.; Lei, T. A Precise, Controllable in Vitro Model for Diffuse Axonal Injury through Uniaxial Stretch Injury. Front. Neurosci. 2019, 13, 1063. [Google Scholar] [CrossRef] [Green Version]
- Magou, G.C.; Pfister, B.J.; Berlin, J.R. Effect of Acute Stretch Injury on Action Potential and Network Activity of Rat Neocortical Neurons in Culture. Brain Res. 2015, 1624, 525–535. [Google Scholar] [CrossRef]
- Rosas-Hernandez, H.; Burks, S.M.; Cuevas, E.; Ali, S.F. Stretch-Induced Deformation as a Model to Study Dopaminergic Dysfunction in Traumatic Brain Injury. Neurochem. Res. 2019, 44, 2546–2555. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Waseem, T.; Mukhtarov, M.; Buldakova, S.; Medina, I.; Bregestovski, P. Genetically Encoded Cl-Sensor as a Tool for Monitoring of Cl-Dependent Processes in Small Neuronal Compartments. J. Neurosci. Methods 2010, 193, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.-Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s Disease. Neurotherapeutics 2010, 7, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.; Attwell, D. The Release and Uptake of Excitatory Amino Acids. Trends Pharmacol. Sci. 1990, 11, 462–468. [Google Scholar] [CrossRef]
- Duchen, M.R.; Surin, A.; Jacobson, J. Imaging Mitochondrial Function in Intact Cells. Methods Enzymol. 2003, 361, 353–389. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakaeva, Z.; Goncharov, M.; Krasilnikova, I.; Zgodova, A.; Frolov, D.; Grebenik, E.; Timashev, P.; Pinelis, V.; Surin, A. Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome. Int. J. Mol. Sci. 2022, 23, 3858. https://doi.org/10.3390/ijms23073858
Bakaeva Z, Goncharov M, Krasilnikova I, Zgodova A, Frolov D, Grebenik E, Timashev P, Pinelis V, Surin A. Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome. International Journal of Molecular Sciences. 2022; 23(7):3858. https://doi.org/10.3390/ijms23073858
Chicago/Turabian StyleBakaeva, Zanda, Mikhail Goncharov, Irina Krasilnikova, Arina Zgodova, Daniil Frolov, Ekaterina Grebenik, Peter Timashev, Vsevolod Pinelis, and Alexander Surin. 2022. "Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome" International Journal of Molecular Sciences 23, no. 7: 3858. https://doi.org/10.3390/ijms23073858
APA StyleBakaeva, Z., Goncharov, M., Krasilnikova, I., Zgodova, A., Frolov, D., Grebenik, E., Timashev, P., Pinelis, V., & Surin, A. (2022). Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome. International Journal of Molecular Sciences, 23(7), 3858. https://doi.org/10.3390/ijms23073858