
SARS-CoV-2 Morbidity in the CNS and the Aged Brain Specific Vulnerability

Abstract
:1. Introduction
2. Mechanisms of Neurotoxicity
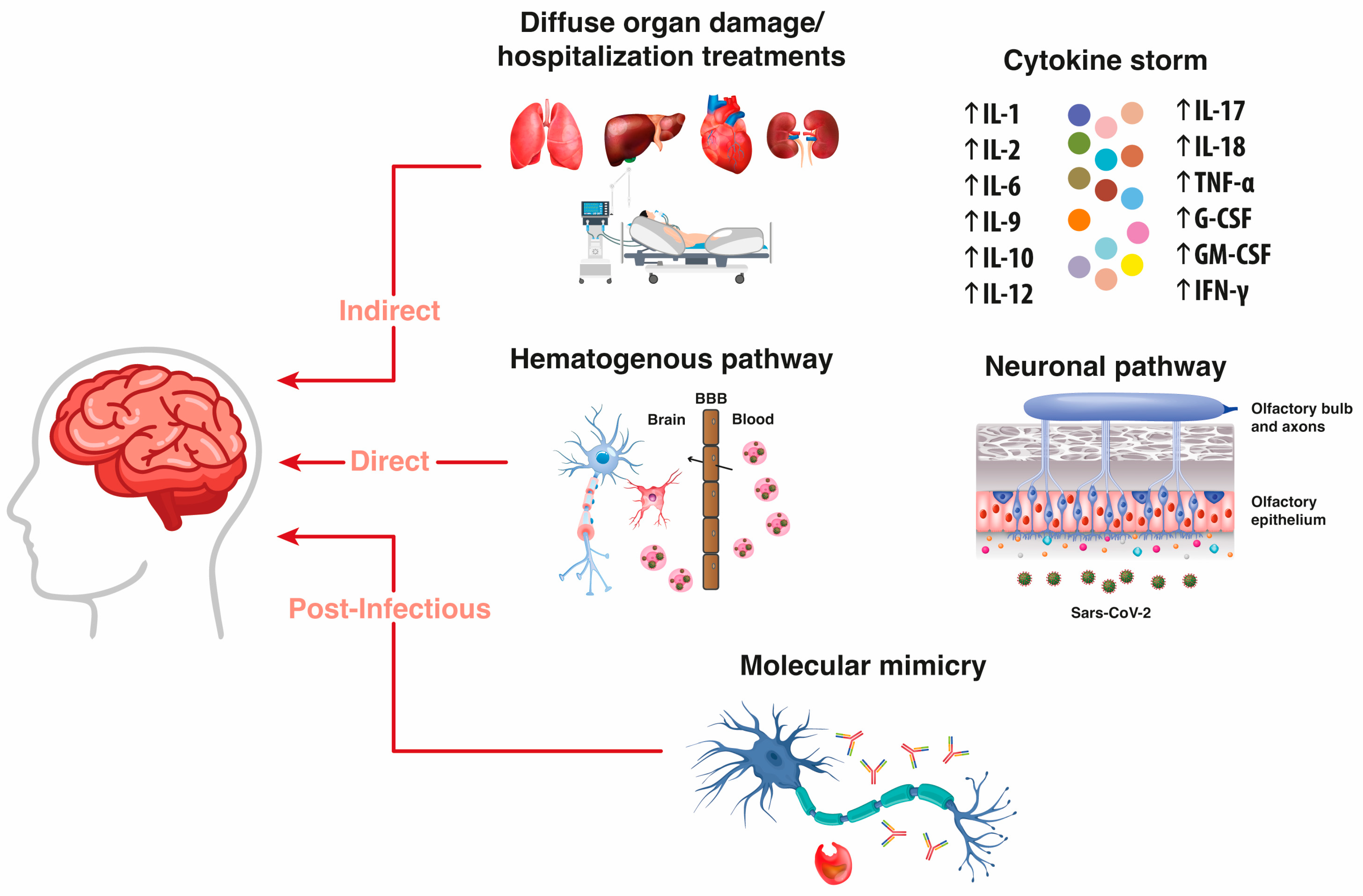
2.1. Indirect, Direct, and Postinfectious Mechanisms
2.2. Viral Infections and Neurodegeneration
2.3. APOE ε4 and COVID-19
3. Neurological Evidence of SARS-CoV-2 Damage to the CNS
3.1. CSF
3.2. Brain Autopsies
4. COVID-19 and the Aged Brain
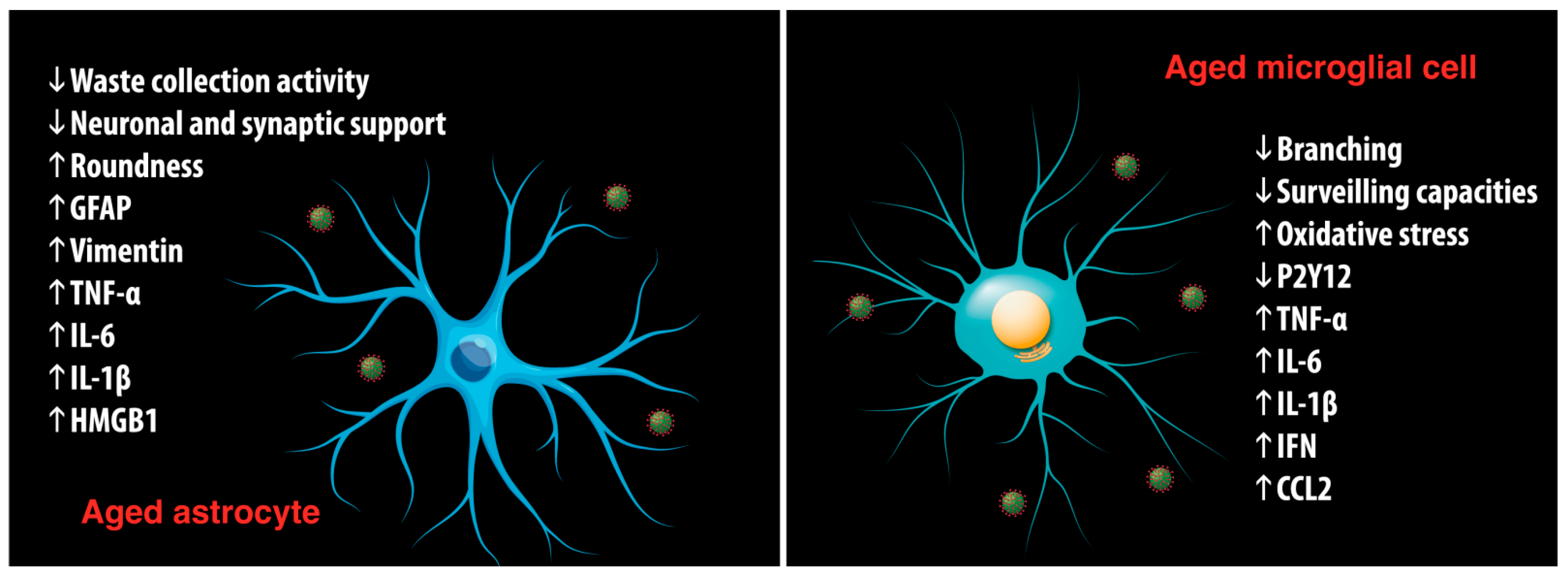
4.1. Loss of Homeostatic Control in the Elderly
4.2. Aged Brain Abnormalities Predisposing to Neurological Manifestations of COVID-19
5. Neurological Consequences, Cognitive Dysfunction and Possible Mechanisms of “Long COVID”
6. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Zhao, X.; Zai, J.; Zhao, Q.; Li, Y.; Chaillon, A. Transmission dynamics and evolutionary history of 2019-nCoV. J. Med. Virol. 2020, 92, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- European Centre for Disease Prevention and Control. Outbreak of Novel Coronavirus Disease 2019 (COVID-19): Increased Transmission Globally–Fifth Update. 2020. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/RRA-outbreak-novel-coronavirus-disease-2019-increase-transmission-globally-COVID-19.pdf (accessed on 15 May 2021).
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE 2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Kerkeni, M.; Gharbi, J. RAGE receptor: May be a potential inflammatory mediator for SARS-CoV-2 infection? Med. Hypotheses 2020, 144, 109950. [Google Scholar] [CrossRef]
- Cui, C.; Huang, C.; Zhou, W.; Ji, X.; Zhang, F.; Wang, L.; Zhou, Y.; Cui, Q. AGTR2, one possible novel key gene for the entry of SARS-CoV-2 into human cells. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1230–1233. [Google Scholar] [CrossRef]
- Kerslake, R.; Hall, M.; Randeva, H.S.; Spandidos, D.A.; Chatha, K.; Kyrou, I.; Karteris, E. Co-expression of peripheral olfactory receptors with SARS-CoV-2 infection mediators: Potential implications beyond loss of smell as a COVID-19 symptom. Int. J. Mol. Med. 2020, 46, 949–956. [Google Scholar] [CrossRef]
- Davies, J.; Randeva, H.S.; Chatha, K.; Hall, M.; Spandidos, D.A.; Karteris, E.; Kyrou, I. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef]
- Zubair, A.S.; McAlpine, L.S.; Gardin, T.; Farhadian, S.; Kuruvilla, D.E.; Spudich, S. Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: A review. JAMA Neurol. 2020, 77, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Michalicova, A.; Bhide, K.; Bhide, M.; Kovac, A. How viruses infiltrate the central nervous system. Acta Virol. 2017, 61, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2020, 11, 573095. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Sasannejad, C.; Ely, E.W.; Lahiri, S. Long-term cognitive impairment after acute respiratory distress syndrome: A review of clinical impact and pathophysiological mechanisms. Crit. Care 2019, 23, 352. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res. Ther. 2020, 12, 69. [Google Scholar] [CrossRef]
- Ogier, M.; Andeol, G.; Sagui, E.; Bo, G.D. How to detect and track chronic neurologic sequelae of COVID-19? Use of auditory brainstem responses and neuroimaging for long-term patient follow-up. Brain Behav. Immun. Health 2020, 5, 100081. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shen, C.; Li, J.; Yuan, J.; Wei, J.M.; Huang, F.; Wang, F.; Li, G.; Li, Y.; Xing, L.; et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J. Allergy Clin. Immunol. 2020, 146, 119–127.e4. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of Ace2 protein, the functional receptor for Sars coronavirus. A first step in understanding Sars pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus infections in the nervous system. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, P.A., 2nd; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Dahm, T.; Rudolph, H.; Schwerk, C.; Schroten, H.; Tenenbaum, T. Neuroinvasion and inflammation in viral central nervous system infections. Mediat. Inflamm. 2016, 2016, 8562805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; Bai, W.Z.; Hirano, N.; Hayashida, T.; Hashikawa, T. Coronavirus infection of rat dorsal root ganglia: Ultrastructural characterization of viral replication, transfer, and the early response of satellite cells. Virus Res. 2012, 163, 628–635. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hirano, N.; Hayashida, T.; Taniguchi, T.; Sugita, Y.; Tohyama, K.; Hashikawa, T. Neurotropic virus tracing suggests a membranous-coating-mediated mechanism for Transsynaptic communication. J. Comp. Neurol. 2013, 521, 203–212. [Google Scholar] [CrossRef]
- Bohmwald, K.; Gálvez, N.M.S.; Ríos, M.; Kalergis, A.M. Neurologic alterations due to respiratory virus infections. Front. Cell. Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef]
- Pérez, C.A. Looking ahead: The risk of neurologic complications due to COVID-19. Neurol. Clin. Pract. 2020, 10, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Chew, F.T.; Ong, S.Y.; Hew, C.L. Severe acute respiratory syndrome coronavirus and viral mimicry. Lancet 2003, 361, 2081. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wu, W.; Pan, W.; Wu, L.; Liu, K.; Zhang, H.L. Acute necrotizing encephalopathy: An underrecognized clinicoradiologic disorder. Mediat. Inflamm. 2015, 2015, 792578. [Google Scholar] [CrossRef]
- Pohl, D.; Alper, G.; Van Haren, K.; Kornberg, A.J.; Lucchinetti, C.F.; Tenembaum, S.; Belman, A.L. Acute disseminated encephalomyelitis: Updates on an inflammatory CNS syndrome. Neurology 2016, 87, S38–S45. [Google Scholar] [CrossRef] [PubMed]
- Burgoon, M.P.; Cohrs, R.J.; Bennett, J.L.; Anderson, S.W.; Ritchie, A.M.; Cepok, S.; Hemmer, B.; Gilden, D.; Owens, G.P. Varicella zoster virus is not a disease relevant antigen in multiple sclerosis. Ann. Neurol. 2009, 65, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Jilek, S.; Schluep, M.; Meylan, P.; Vingerhoets, F.; Guignard, L.; Monney, A.; Kleeberg, J.; Le Goff, G.; Pantaleo, G.; Du Pasquier, R.A. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain 2008, 131, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Cepok, S.; Zhou, D.; Srivastava, R.; Nessler, S.; Stei, S.; Bussow, K.; Sommer, N.; Hemmer, B. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J. Clin. Investig. 2005, 115, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [Green Version]
- Deleidi, M.; Isacson, O. Viral and inflammatory triggers of neurodegenerative diseases. Sci. Transl. Med. 2012, 4, 121ps3. [Google Scholar] [CrossRef] [Green Version]
- Piacentini, R.; Civitelli, L.; Ripoli, C.; Marcocci, M.E.; De Chiara, G.; Garaci, E.; Azzena, G.B.; Palamara, A.T.; Grassi, C. HSV-1 promotes Ca(2+)-mediated APP phosphorylation and Abeta accumulation in rat cortical neurons. Neurobiol. Aging 2011, 32, e13–e26. [Google Scholar] [CrossRef]
- Santana, S.; Recuero, M.; Bullido, M.J.; Valdivieso, F.; Aldudo, J. Herpes simplex virus type I induces the accumulation of intracellular beta-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol. Aging 2012, 33, 430.e19–430.e33. [Google Scholar] [CrossRef]
- Lerchundi, R.; Neira, R.; Valdivia, S.; Vio, K.; Concha, M.I.; Zambrano, A.; Otth, C. Tau cleavage at D421 by caspase-3 is induced in neurons and astrocytes infected with herpes simplex virus type 1. J. Alzheimers Dis. 2011, 23, 513–520. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Dubeau, P.; Bourgouin, A.; Lajoie, L.; Dubé, M.; Talbot, P.J. Human Coronaviruses and Other Respiratory Viruses: Underestimated Opportunistic Pathogens of the Central Nervous System? Viruses 2019, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Banerjea, A.C. Neurological Damage by Coronaviruses: A Catastrophe in the Queue! Front. Immunol. 2020, 11, 565521. [Google Scholar] [CrossRef] [PubMed]
- Jacomy, H.; Talbot, P.J. Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology 2003, 315, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wunderink, R.G. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology 2018, 23, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, M.; Omrani, A.S.; Baig, K.; Bahloul, A.; Elzein, F.; Matin, M.A.; Selim, M.A.; Al Mutairi, M.; Al Nakhli, D.; Al Aidaroos, A.Y.; et al. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: A single-center experience in Saudi Arabia. Int. J. Infect. Dis. 2014, 29, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K. Middle East respiratory syndrome virus pathogenesis. Semin. Respir. Crit. Care Med. 2016, 37, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Heo, J.H.; Kim, H.O.; Song, S.H.; Park, S.S.; Park, T.H.; Ahn, J.Y.; Kim, M.K.; Choi, J.P. Neurological complications during treatment of middle east respiratory syndrome. J. Clin. Neurol. 2017, 13, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Tsai, L.K.; Hsieh, S.T.; Chang, Y.C. Neurological manifestations in severe acute respiratory syndrome. Acta Neurol. Taiwan 2005, 14, 113–119. [Google Scholar] [CrossRef]
- Huang, D.Z.; Lang, Z.W.; Wen, T.; He, L.X.; Xie, L.; Zhou, Y.S. Detection of SARS coronavirus RNA in lung tissues from patients with severe acute respiratory syndrome by in situ reverse transcription polymerase chain reaction. Chin. J. Microbiol. Immunol. 2004, 10, 311–316. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. ApoE e4e4 genotype and mortality with COVID-19 in UK Biobank. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1801–1803. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, M.; Garcia, G., Jr.; Tian, E.; Cui, Q.; Chen, X.; Sun, G.; Wang, J.; Arumugaswami, V.; Shi, Y. ApoE-Isoform-Dependent SARS-CoV-2 Neurotropism and Cellular Response. Cell Stem Cell 2021, 28, 331–342.e5. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Chalmers, K.; Ince, P.; Esiri, M.; Attems, J.; Jellinger, K.; Yamada, M.; McCarron, M.; Minett, T.; Matthews, F.; et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am. J. Neurodegener. Dis. 2014, 3, 19–32. [Google Scholar] [PubMed]
- Thambisetty, M.; Beason-Held, L.; An, Y.; Kraut, M.A.; Resnick, S.M. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol. 2010, 67, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Elmashala, A.; Chopra, S.; Garg, A. The neurologic manifestations of coronavirus disease 2019. J. Neurol. Res. 2020, 10, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Tancheva, L.; Petralia, M.C.; Miteva, S.; Dragomanova, S.; Solak, A.; Kalfin, R.; Lazarova, M.; Yarkov, D.; Ciurleo, R.; Cavalli, E.; et al. Emerging neurological and psychobiological aspects of COVID-19 infection. Brain Sci. 2020, 10, 852. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan. China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Hascup, E.R.; Hascup, K.N. Does SARS-CoV-2 infection cause chronic neurological complications? Geroscience 2020, 42, 1083–1087. [Google Scholar] [CrossRef]
- Tandon, M.; Kataria, S.; Patel, J.; Mehta, T.R.; Daimee, M.; Patel, V.; Prasad, A.; Chowdhary, A.A.; Jaiswal, S.; Sriwastava, S. A comprehensive systematic review of CSF analysis that defines neurological manifestations of COVID-19. Int. J. Infect. Dis. 2021, 104, 390–397. [Google Scholar] [CrossRef]
- Johansson, A.; Mohamed, M.S.; Moulin, T.C.; Schioth, H.B. Neurological manifestations of COVID-19: A comprehensive literature review and discussion of mechanisms. J. Neuroimmunol. 2021, 358, 577658. [Google Scholar] [CrossRef]
- Li, Y.C.; Zhang, Y.; Tan, B.H. What can cerebrospinal fluid testing and brain autopsies tell us about viral neuroinvasion of SARS-CoV-2. J. Med. Virol. 2021, 93, 4247–4257. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mahadeva, U.; Green, A.; Sekhawat, V.; Barrett, N.A.; Childs, L.; Shankar-Hari, M.; Thom, M.; Jäger, H.R.; Brandner, S. Microvascular injury and hypoxic damage: Emerging neuropathological signatures in COVID-19. Acta Neuropathol. 2020, 140, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, V.P.; Foschini, M.P.; Lazzarotto, T.; Gabrielli, L.; Cenacchi, G.; Gallo, C.; Aspide, R.; Frascaroli, G.; Cortelli, P.; Riefolo, M.; et al. Brain ischemic injury in COVID-19-infected patients: A series of 10 post-mortem cases. Brain Pathol. 2021, 31, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kantonen, J.; Mahzabin, S.; Mäyränpää, M.I.; Tynninen, O.; Paetau, A.; Andersson, N.; Sajantila, A.; Vapalahti, O.; Carpén, O.; Kekäläinen, E.; et al. Neuropathologic features of four autopsied COVID-19 patients. Brain Pathol. 2020, 30, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Al-Dalahmah, O.; Thakur, K.T.; Nordvig, A.S.; Prust, M.L.; Roth, W.; Lignelli, A.; Uhlemann, A.C.; Miller, E.H.; Kunnath-Velayudhan, S.; Del Portillo, A.; et al. Neuronophagia and microglial nodules in a SARS-CoV-2 patient with cerebellar hemorrhage. Acta Neuropathol. Commun. 2020, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, M.; Kortekaas, J.; Herden, C.; Hoffmann, B.; Tappe, D.; Trebst, C.; Griffin, D.E.; Brindle, H.E.; Solomon, T.; Brown, A.S.; et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016, 131, 159–184. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, D.; Li, G. The role of microglia in viral encephalitis: A review. J. Neuroinflamm. 2019, 16, 76. [Google Scholar] [CrossRef]
- Lou, J.J.; Movassaghi, M.; Gordy, D.; Olson, M.G.; Zhang, T.; Khurana, D.; Chen, Z.; Perez-Rosendahl, M.; Thammachantha, S.; Singer, E.J.; et al. Neuropathology of COVID-19 (neuro-COVID): Clinicopathological update. Free Neuropathol. 2021, 2, 2. [Google Scholar] [CrossRef]
- Maiese, A.; Manetti, A.C.; Bosetti, C.; Del Duca, F.; La Russa, R.; Frati, P.; Di Paolo, M.; Turillazzi, E.; Fineschi, V. SARS-CoV-2 and the brain: A review of the current knowledge on neuropathology in COVID-19. Brain Pathol. 2021, 31, e13013. [Google Scholar] [CrossRef]
- Hewitt, J.; Carter, B.; Vilches-Moraga, A.; Quinn, T.J.; Braude, P.; Verduri, A.; Pearce, L.; Stechman, M.; Short, R.; Price, A.; et al. COPE Study Collaborators. The effect of frailty on survival in patients with COVID-19 (COPE): A multicentre, European, observational cohort study. Lancet Public Health 2020, 5, e444–e451. [Google Scholar] [CrossRef]
- Sablerolles, R.S.G.; Lafeber, M.; van Kempen, J.A.L.; van de Loo, B.P.A.; Boersma, E.; Rietdijk, W.J.R.; Polinder-Bos, H.A.; Mooijaart, S.P.; van der Kuy, H.; Versmissen, J.; et al. Association between Clinical Frailty Scale score and hospital mortality in adult patients with COVID-19 (COMET): An international, multicentre, retrospective, observational cohort study. Lancet Healthy Longev. 2021, 2, e163–e170. [Google Scholar] [CrossRef]
- Ferrucci, L.; Levine, M.E.; Kuo, P.L.; Simonsick, E.M. Time and the Metrics of Aging. Circ. Res. 2018, 123, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera, E.; Bernardes de Jesus, B.; Foronda, M.; Flores, J.M.; Blasco, M.A. The rate of increase of short telomeres predicts longevity in mammals. Cell Rep. 2012, 2, 732–737. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.L.; Kronmal, R.A.; Gardner, J.P.; Psaty, B.M.; Jenny, N.S.; Tracy, R.P.; Walston, J.; Kimura, M.; Aviv, A. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am. J. Epidemiol. 2007, 165, 14–21. [Google Scholar] [CrossRef]
- Najarro, K.; Nguyen, H.; Chen, G.; Xu, M.; Alcorta, S.; Yao, X.; Zukley, L.; Metter, E.J.; Truong, T.; Lin, Y.; et al. Telomere length as an indicator of the robustness of B- and T-cell response to influenza in older adults. J. Infect. Dis. 2015, 212, 1261–1269. [Google Scholar] [CrossRef]
- Batsis, J.A.; Mackenzie, T.A.; Vasquez, E.; Germain, C.M.; Emeny, R.T.; Rippberger, P.; Lopez-Jimenez, F.; Bartels, S.J. Association of adiposity, telomere length and mortality: Data from the NHANES 1999–2002. Int. J. Obes. 2018, 42, 198–204. [Google Scholar] [CrossRef]
- Goglin, S.E.; Farzaneh-Far, R.; Epel, E.S.; Lin, J.; Blackburn, E.H.; Whooley, M.A. Correction: Change in leukocyte telomere length predicts mortality in patients with stable coronary heart disease from the heart and soul study. PLoS ONE 2016, 11, e0168868. [Google Scholar] [CrossRef]
- Andriani, G.A.; Almeida, V.P.; Faggioli, F.; Mauro, M.; Tsai, W.L.; Santambrogio, L.; Maslov, A.; Gadina, M.; Campisi, J.; Vijg, J.; et al. Whole chromosome instability induces senescence and promotes SASP. Sci. Rep. 2016, 6, 35218. [Google Scholar] [CrossRef]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The free radical theory of aging. Antioxid. Redox Signal. 2003, 5, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.Z.; Ding, J.; Tuke, M.A.; Wood, A.R.; Bandinelli, S.; Frayling, T.M.; Ferrucci, L. Influence of cell distribution and diabetes status on the association between mitochondrial DNA copy number and aging phenotypes in the InCHIANTI study. Aging Cell 2017, 17, e12683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genom. 2017, 18, 890. [Google Scholar] [CrossRef] [Green Version]
- Casoli, T.; Spazzafumo, L.; Di Stefano, G.; Conti, F. Role of diffuse low-level heteroplasmy of mitochondrial DNA in Alzheimer’s disease neurodegeneration. Front. Aging Neurosci. 2015, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, N.; Appleby, J.C.; Austad, S.N.; Cuervo, A.M.; Kaeberlein, M.; Gonzalez-Billault, C.; Lederman, S.; Stambler, I.; Sierra, F. Geroscience in the Age of COVID-19. Aging Dis. 2020, 11, 725–729. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Graham, S.H.; Liu, H. Life and death in the trash heap: The ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral ischemia. Ageing Res. Rev. 2017, 34, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Thibault, O.; Hadley, R.; Landfield, P.W. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: Relationship to impaired synaptic plasticity. J. Neurosci. 2001, 21, 9744–9756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toescu, E.C.; Verkhratsky, A.; Landfield, P.W. Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci. 2004, 27, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of brain Aging: Adaptive and Pathological Modifications by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Mattson, M.P. Recruiting adaptive cellular stress responses for successful brain ageing. Nat. Rev. Neurosci. 2012, 13, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Nichols, N.R.; Day, J.R.; Laping, N.J.; Johnson, S.A.; Finch, C.E. GFAP mRNA increases with age in rat and human brain. Neurobiol. Aging 1993, 14, 421–429. [Google Scholar] [CrossRef]
- Rozovsky, I.; Finch, C.E.; Morgan, T.E. Age-related activation of microglia and astrocytes: In vitro studies show persistent phenotypes of aging, increased proliferation and resistance to down-regulation. Neurobiol. Aging 1998, 19, 97–103. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S. Life and Death of Microglia. J. Neuroimmune Pharmacol. 2009, 4, 371–379. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, J.K.; Shin, O.S. Aging and the immune system: The impact of immunosenescence on viral infection, immunity and vaccine immunogenicity. Immune Netw. 2019, 19, e37. [Google Scholar] [CrossRef]
- McElhaney, J.E.; Verschoor, C.P.; Andrew, M.K.; Haynes, L.; Kuchel, G.A.; Pawelec, G. The immune response to influenza in older humans: Beyond immune senescence. Immun. Ageing 2020, 17, 10. [Google Scholar] [CrossRef]
- Flanary, B.E.; Streit, W.J. Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 2004, 45, 75–88. [Google Scholar] [CrossRef]
- Flanary, B.E.; Streit, W.J. Telomeres shorten with age in rat cerebellum and cortex in vivo. J. Anti-Aging Med. 2003, 6, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are astrocytes important? Neurochem. Res. 2015, 40, 389–401. [Google Scholar] [CrossRef]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.S. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 2007, 35, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Stefanou, M.I.; Palaiodimou, L.; Bakola, E.; Smyrnis, N.; Papadopoulou, M.; Paraskevas, G.P.; Rizos, E.; Boutati, E.; Grigoriadis, N.; Krogias, C. Neurological manifestations of long-COVID syndrome: A narrative review. Ther. Adv. Chronic Dis. 2022, 13, 20406223221076890. [Google Scholar] [CrossRef]
- Doykov, I.; Hällqvist, J.; Gilmour, K.C.; Grandjean, L.; Mills, K.; Heywood, W.E. ‘The long tail of COVID-19’—The detection of a prolonged inflammatory response after a SARS-CoV-2 infection in asymptomatic and mildly affected patients. F1000Research 2020, 9, 1349. [Google Scholar] [CrossRef]
- Yao, X.H.; He, Z.C.; Li, T.Y.; Zhang, H.R.; Wang, Y.; Mou, H.; Guo, Q.; Yu, S.C.; Ding, Y.; Liu, X.; et al. Pathological evidence for residual SARS-CoV-2 in pulmonary tissues of a ready-for-discharge patient. Cell Res. 2020, 30, 541–543. [Google Scholar] [CrossRef]
- Varatharaj, A.; Thomas, N.; Ellul, M.A.; Davies, N.W.S.; Pollak, T.A.; Tenorio, E.L.; Sultan, M.; Easton, A.; Breen, G.; Zandi, M.; et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: A UK-wide surveillance study. Lancet Psychiatry 2020, 7, 875–882. [Google Scholar] [CrossRef]
- Kumar, S.; Veldhuis, A.; Malhotra, T. Neuropsychiatric and cognitive sequelae of COVID-19. Front. Psychol. 2021, 12, 577529. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, X.; Geng, D.; Mei, N.; Wu, P.Y.; Huang, C.C.; Jia, T.; Zhao, Y.; Wang, D.; Xiao, A. Cerebral micro-structural changes in COVID-19 patients–An MRI-based 3-month follow-up study. EClinicalMedicine 2020, 25, 100484. [Google Scholar] [CrossRef]
- Duong, D. Even mild COVID-19 may have long-term brain impacts. CMAJ 2021, 193, E1360–E1361. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Ptacek, R.; Ptackova, H.; Martin, A.; Kream, R.M. Selective neuronal mitochondrial targeting in SARS-CoV-2 infection affects cognitive processes to induce ‘brain fog’ and results in behavioral changes that favor viral survival. Med. Sci. Monit. 2021, 27, e930886. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Das, N.; Mukherjee, P. Picking up a fight: Fine tuning mitochondrial innate immune defenses against RNA viruses. Front. Microbiol. 2020, 11, 1990. [Google Scholar] [CrossRef]
- Harris, H.M.B.; Hill, C. A place for viruses on the tree of life. Front. Microbiol. 2020, 11, 604048. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, S.; Chen, J.; Wei, N.; Wang, D.; Lyu, H.; Shi, C.; Hu, S. The landscape of cognitive function in recovered COVID-19 patients. J. Psychiatr. Res. 2020, 129, 98–102. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Virus Strain | Effect of Infection Triggering Neurodegeneration | References |
|---|---|---|
| Epstein–Barr | Brain tissue damage initiated by specific response of CD8+ T cell to infection | [33,34] |
| Autoimmunity by molecular mimicry with myelin antigens | [35] | |
| Hemagglutinin type 5 and neuraminidase type 1 | Increased levels of interleukin-18, interleukin-6, granulocyte colony-stimulating factor, and monocyte chemoattractant protein-1 | [36] |
| Microglial activation and dopaminergic neuronal loss in the substantia nigra | [37] | |
| Herpes simplex 1 | Neurotoxic amyloid-β accumulation | [38,39] |
| Tau phosphorylation | [40] |
| Age-Associated Changes in the Brain | References |
|---|---|
| General | |
| Genomic instability | [74] |
| Telomere shortening | [75,76,77,78,79] |
| Cellular senescence | [80,81] |
| Epigenetic changes | [82,83,84] |
| Mitochondrial impairment | [85,86,87,88] |
| Specific/neurons | |
| Impaired lysosomal and proteasomal degradation | [90,91] |
| Altered calcium regulation | [92,93,94] |
| Distorted adaptive stress response | [95,96] |
| Specific/glia | |
| Increased activation of microglia and astrocytes | [97,98,99,100] |
| Immunosenescence | [101,102,103,104,105,106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casoli, T. SARS-CoV-2 Morbidity in the CNS and the Aged Brain Specific Vulnerability. Int. J. Mol. Sci. 2022, 23, 3782. https://doi.org/10.3390/ijms23073782
Casoli T. SARS-CoV-2 Morbidity in the CNS and the Aged Brain Specific Vulnerability. International Journal of Molecular Sciences. 2022; 23(7):3782. https://doi.org/10.3390/ijms23073782
Chicago/Turabian StyleCasoli, Tiziana. 2022. "SARS-CoV-2 Morbidity in the CNS and the Aged Brain Specific Vulnerability" International Journal of Molecular Sciences 23, no. 7: 3782. https://doi.org/10.3390/ijms23073782
APA StyleCasoli, T. (2022). SARS-CoV-2 Morbidity in the CNS and the Aged Brain Specific Vulnerability. International Journal of Molecular Sciences, 23(7), 3782. https://doi.org/10.3390/ijms23073782