
Sulfobutylation of Beta-Cyclodextrin Enhances the Complex Formation with Mitragynine: An NMR and Chiroptical Study

 , ,
, ,  ,
, 
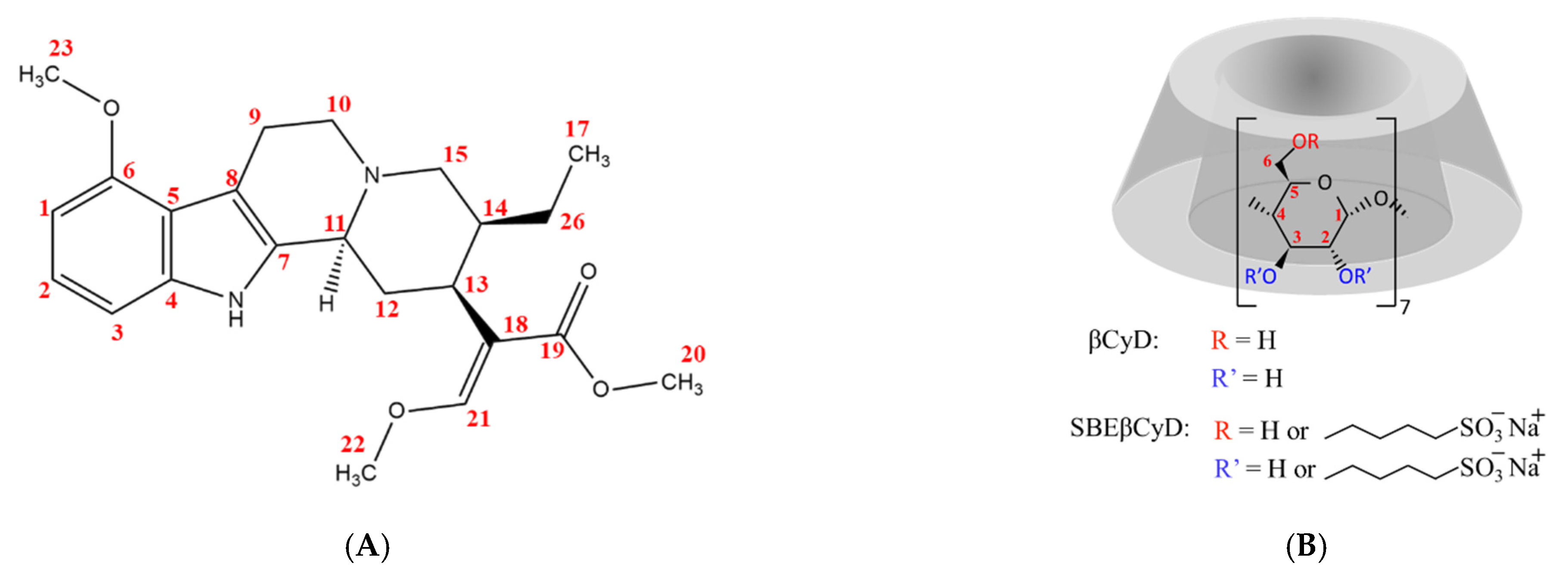{kind=link}
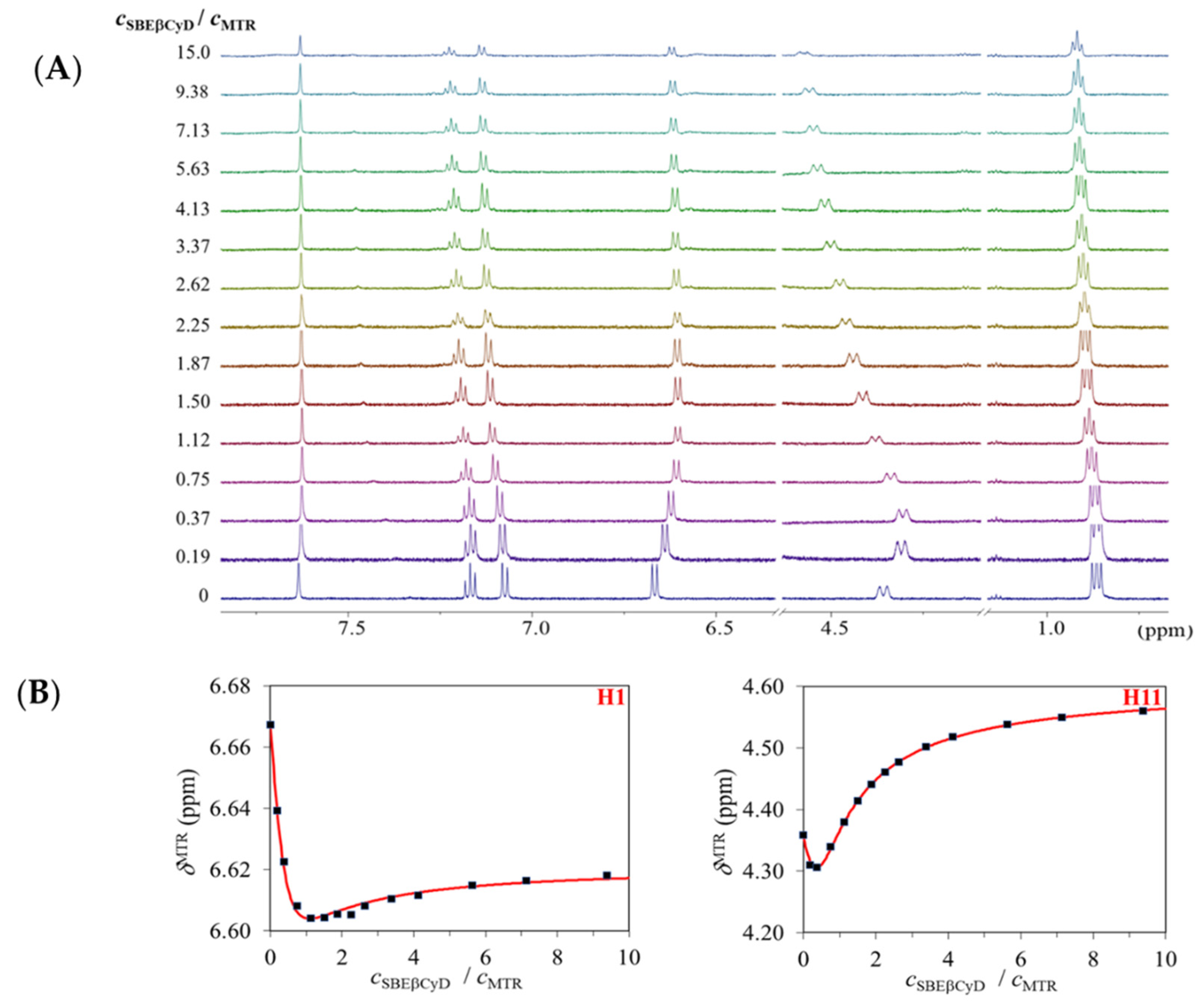{kind=link}
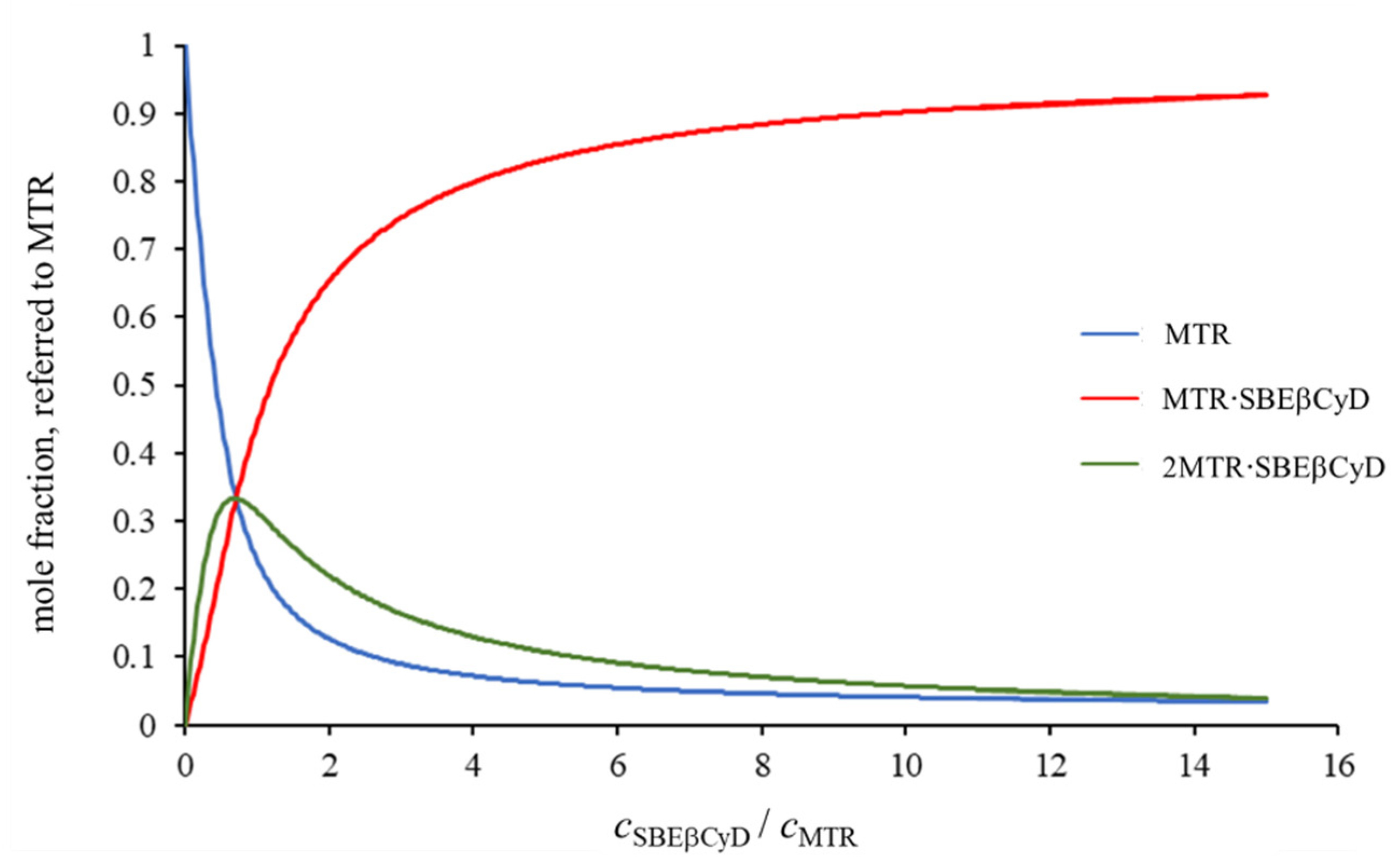{kind=link}
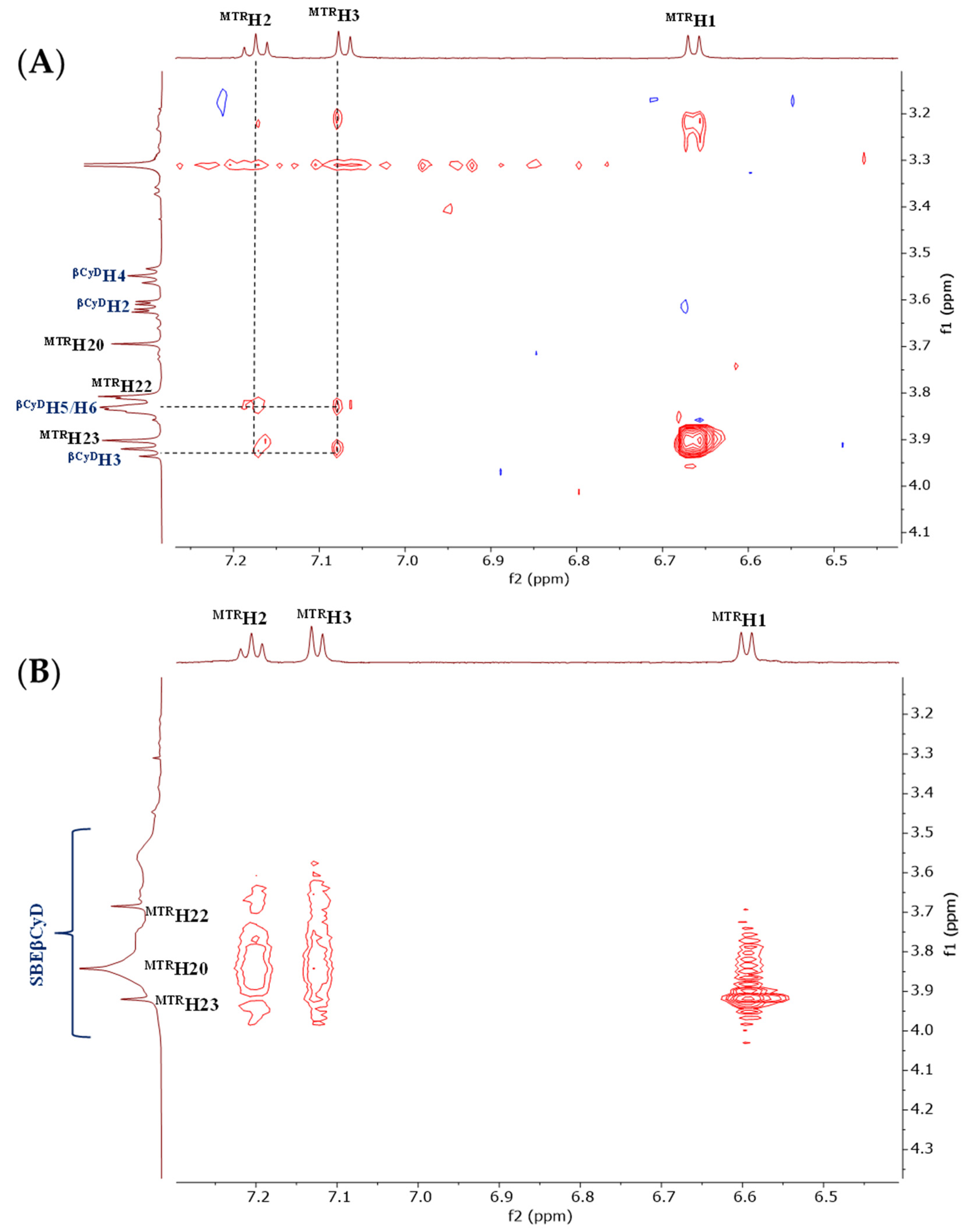{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
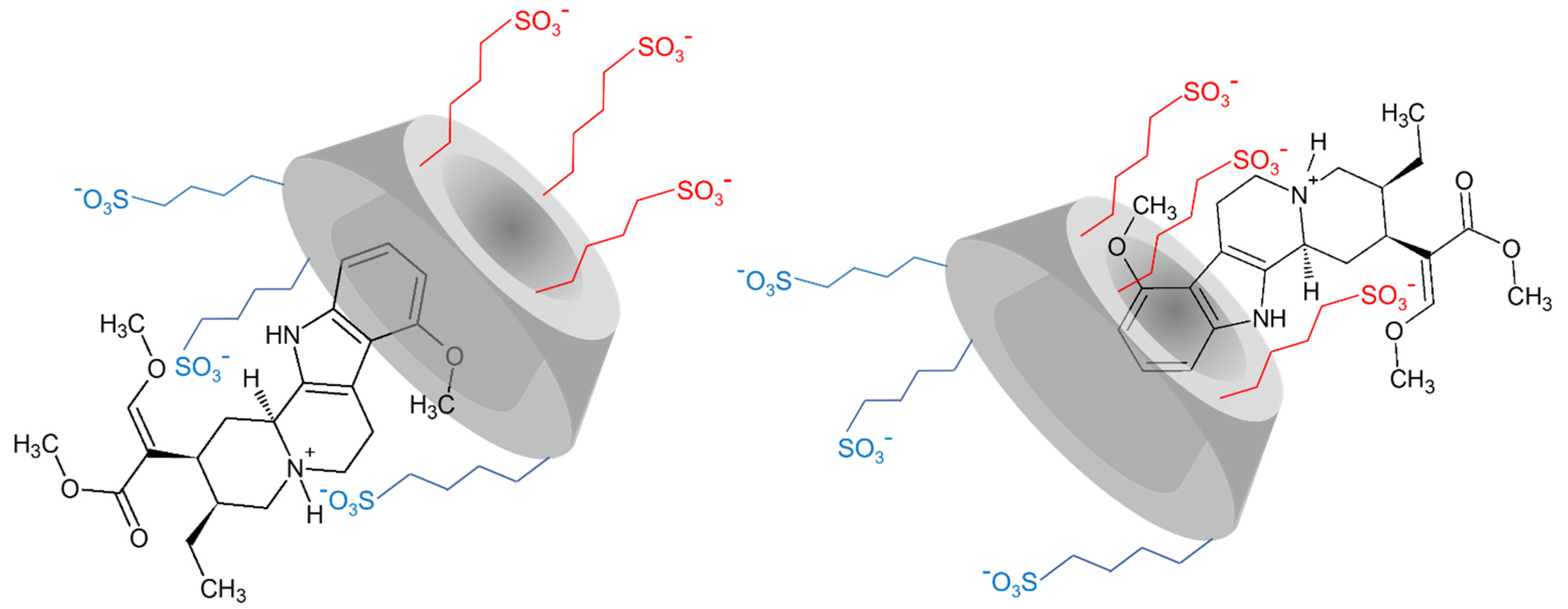
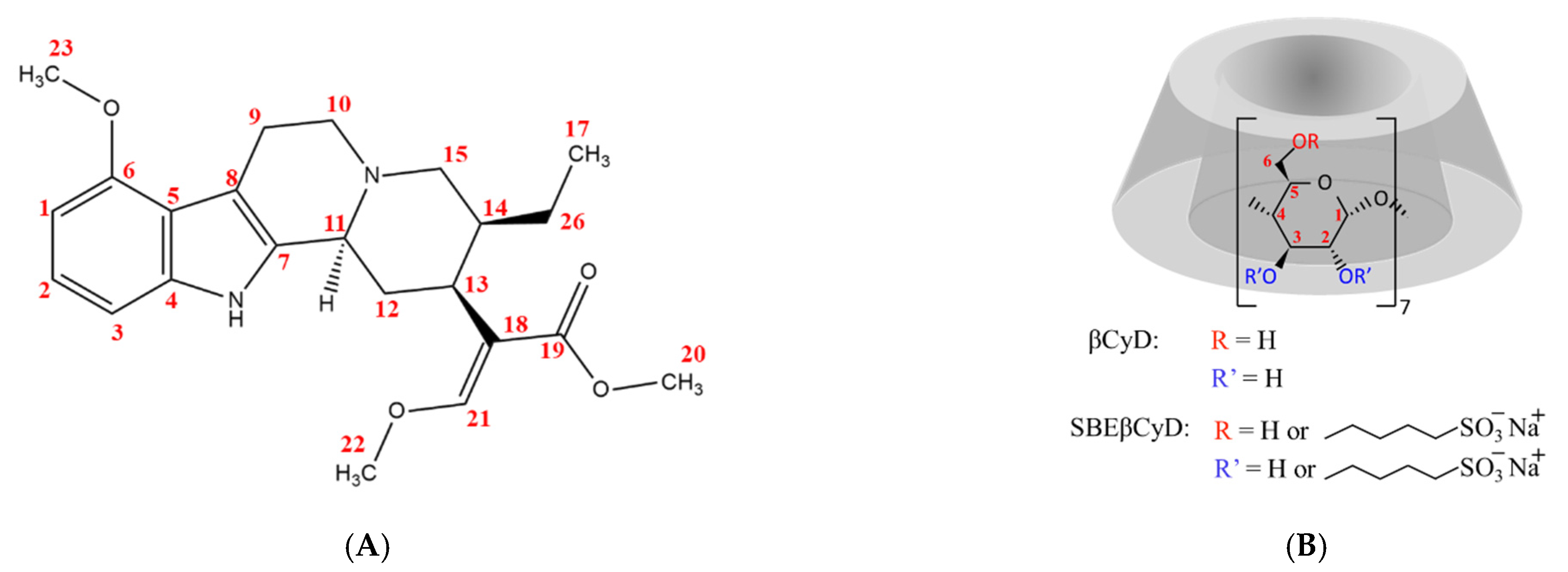
2.1. NMR Spectroscopy
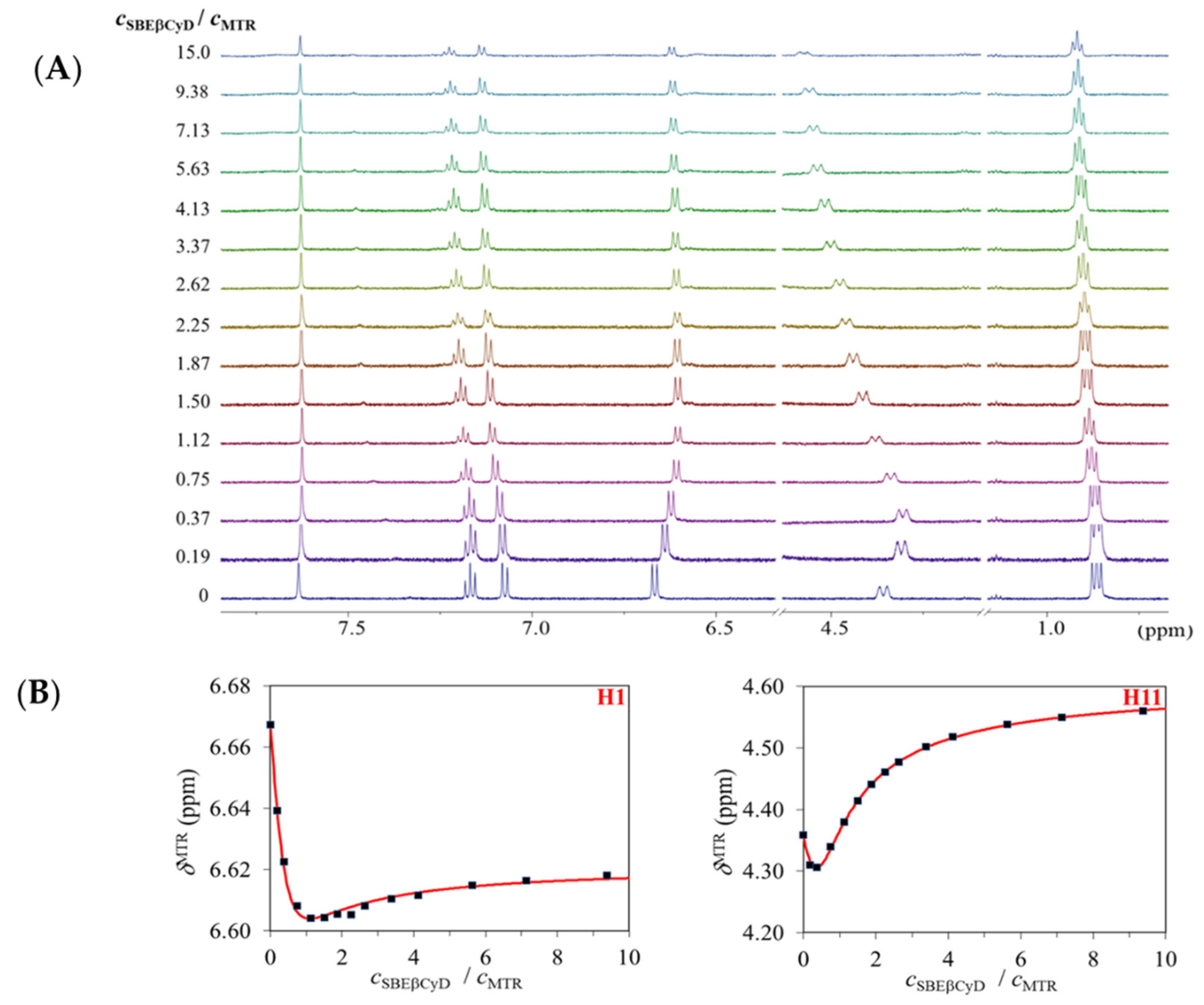
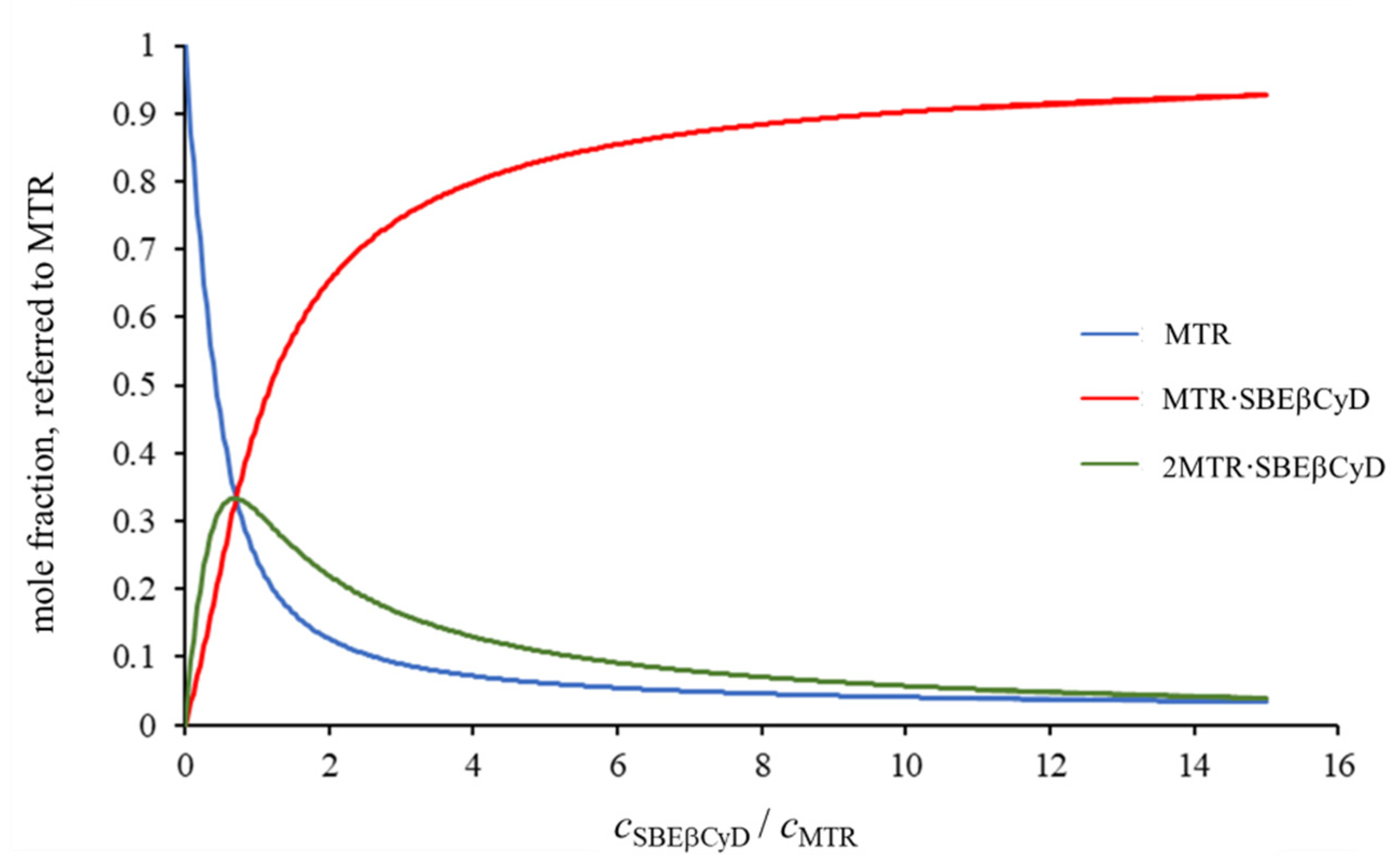
2.1.1. 1H NMR Titrations
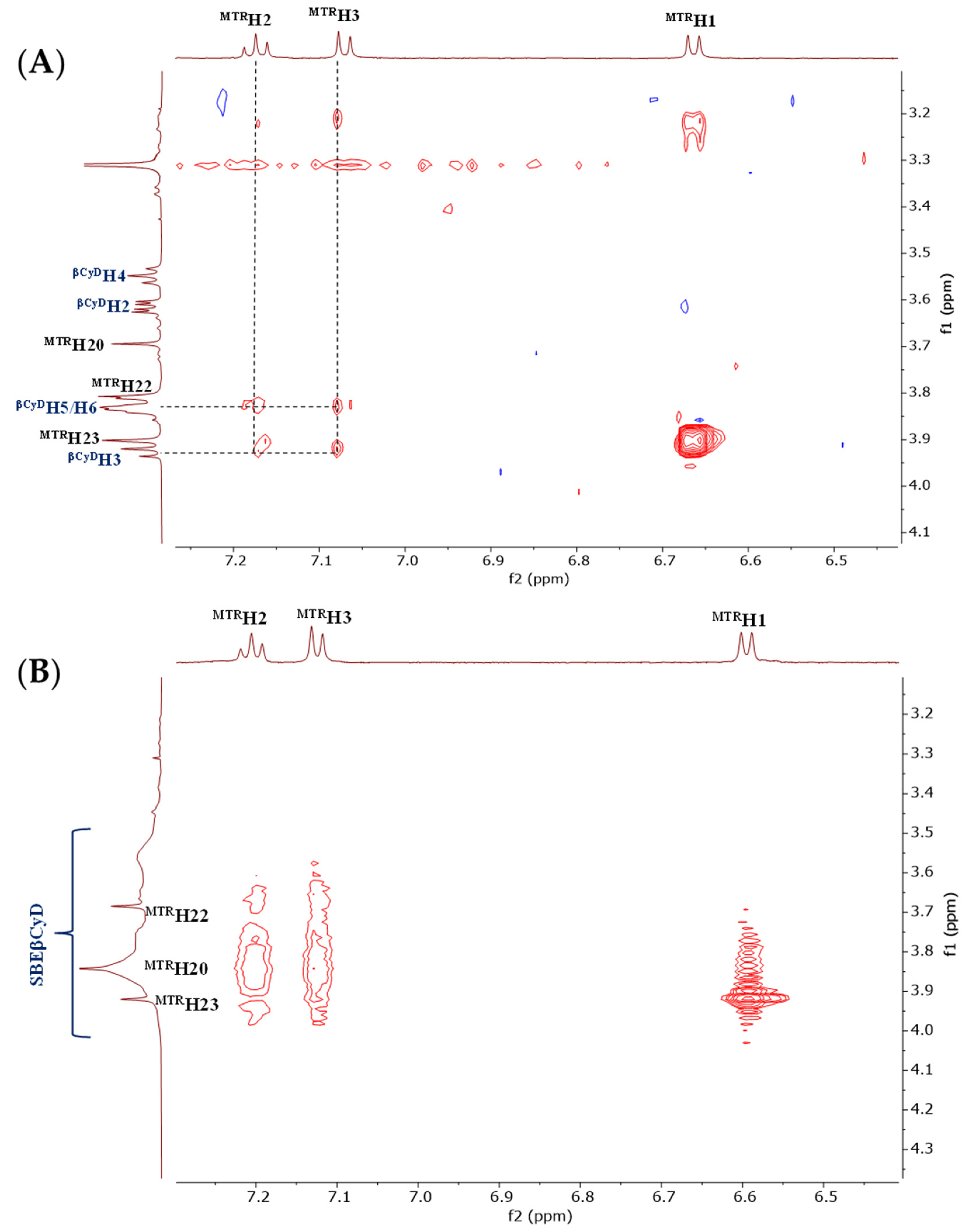
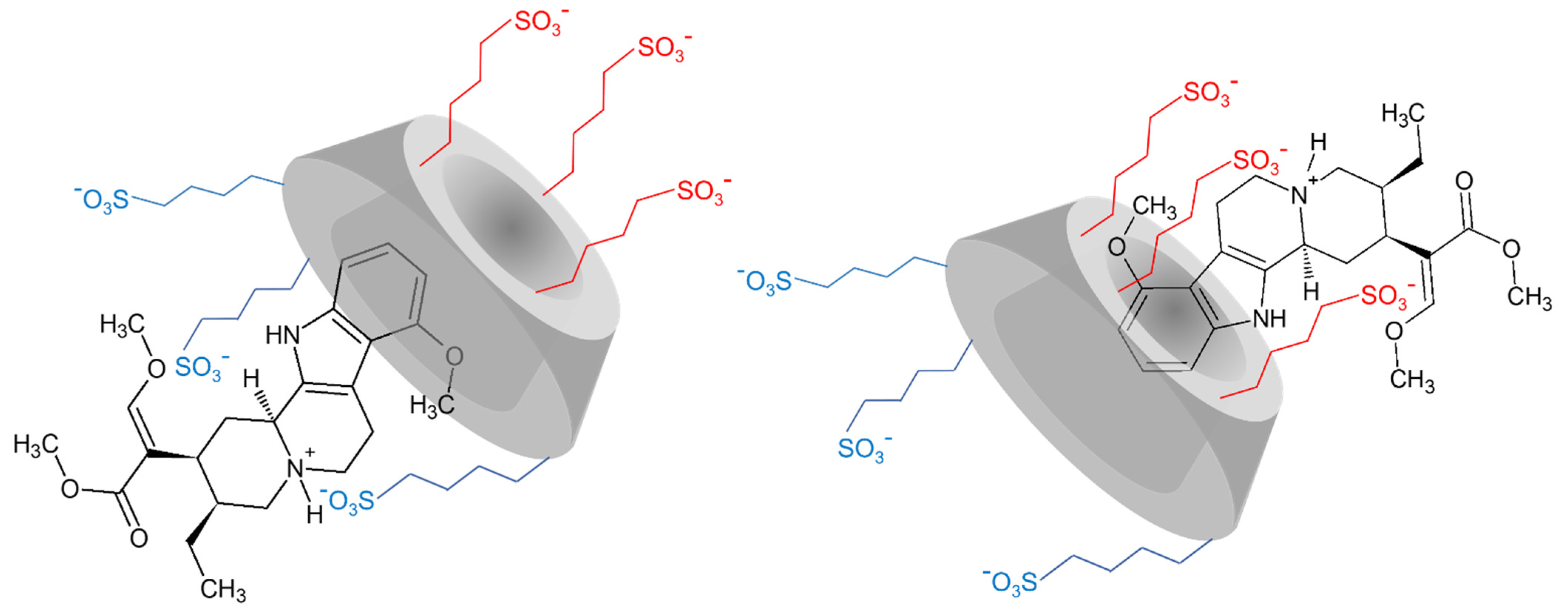
2.1.2. Structural Characterization of the Complexes by NMR
2.2. Circular Dichroism and UV Absorption Spectroscopy
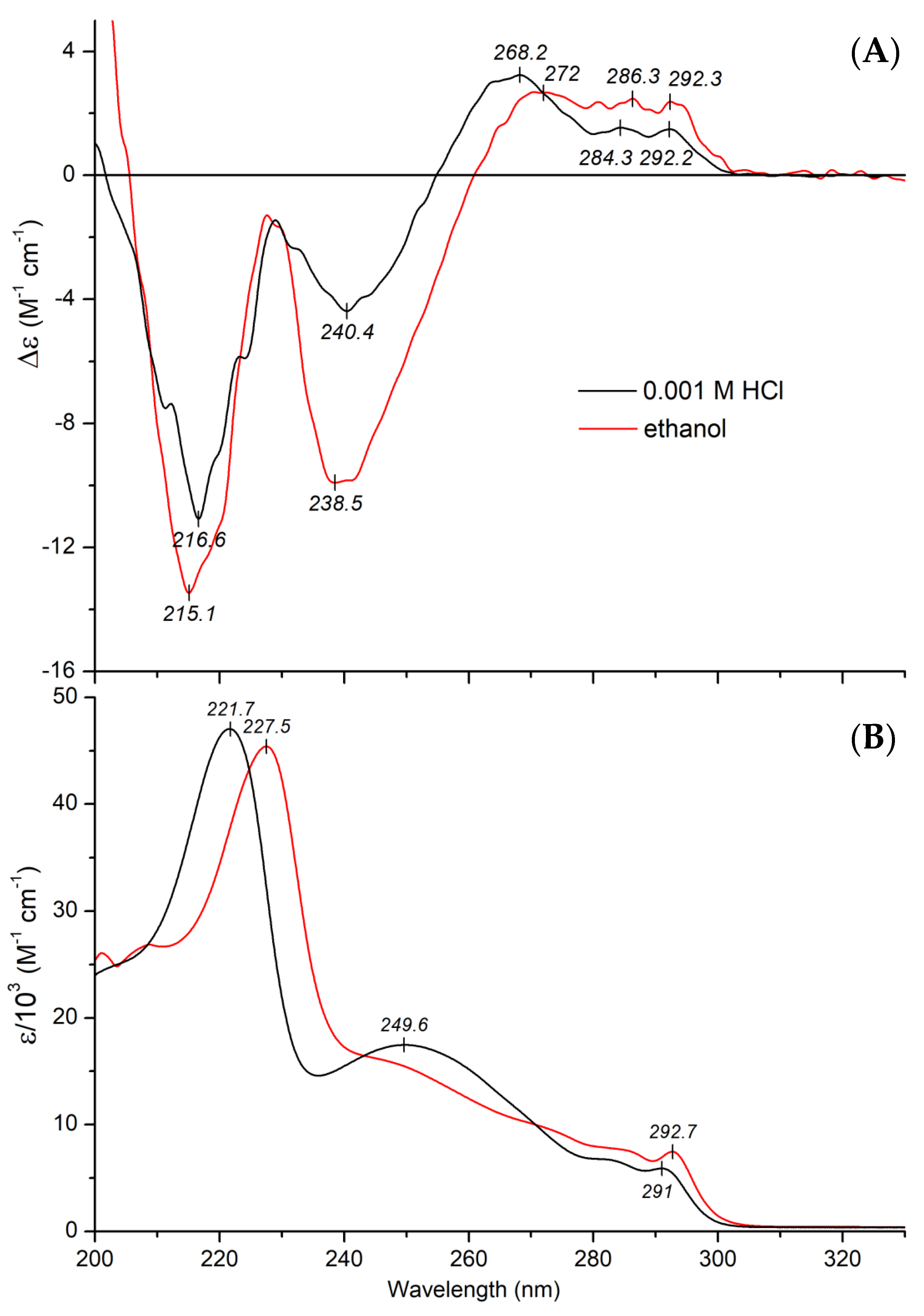
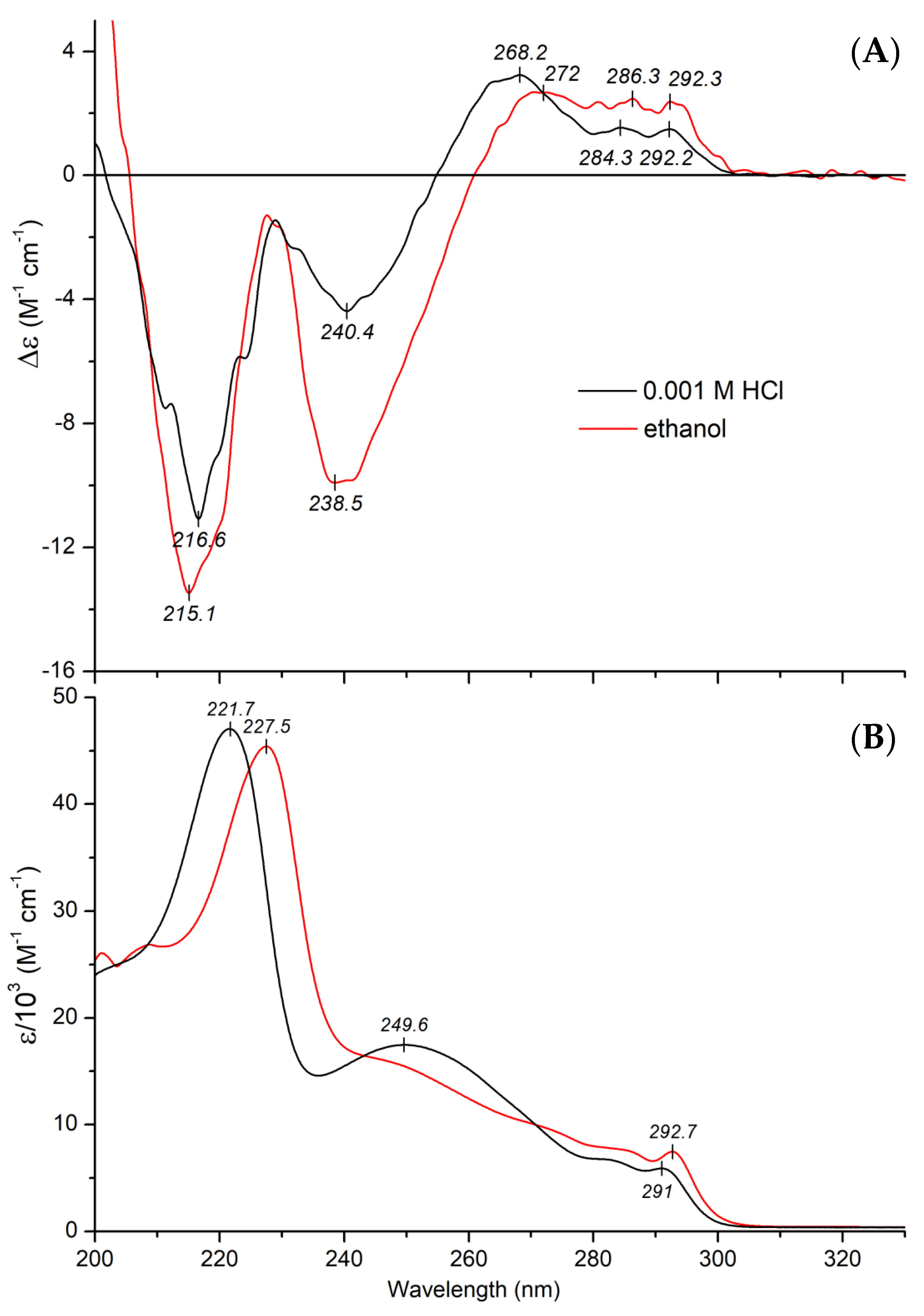
2.2.1. CD and UV Absorption Spectroscopic Properties of Mitragynine
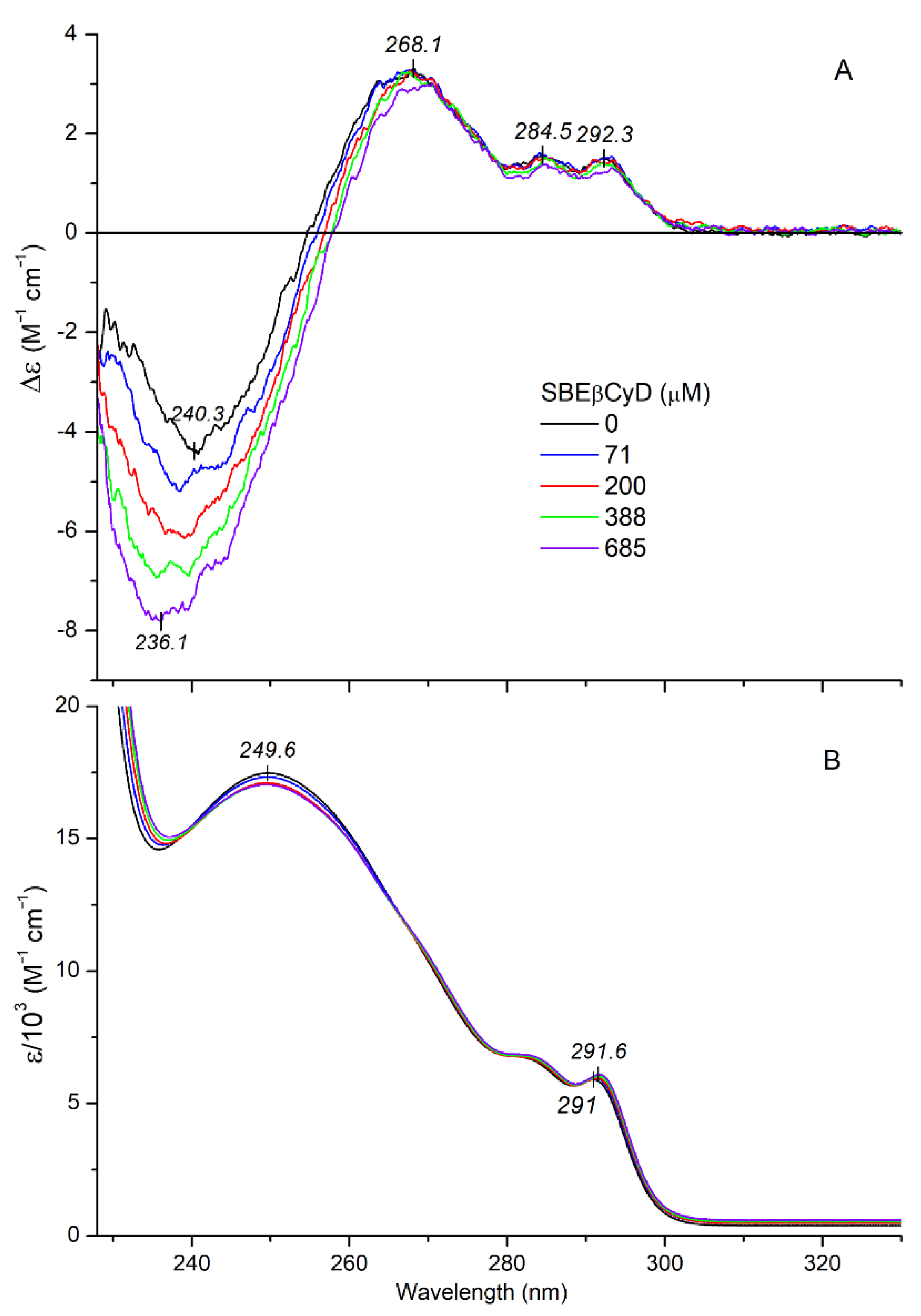
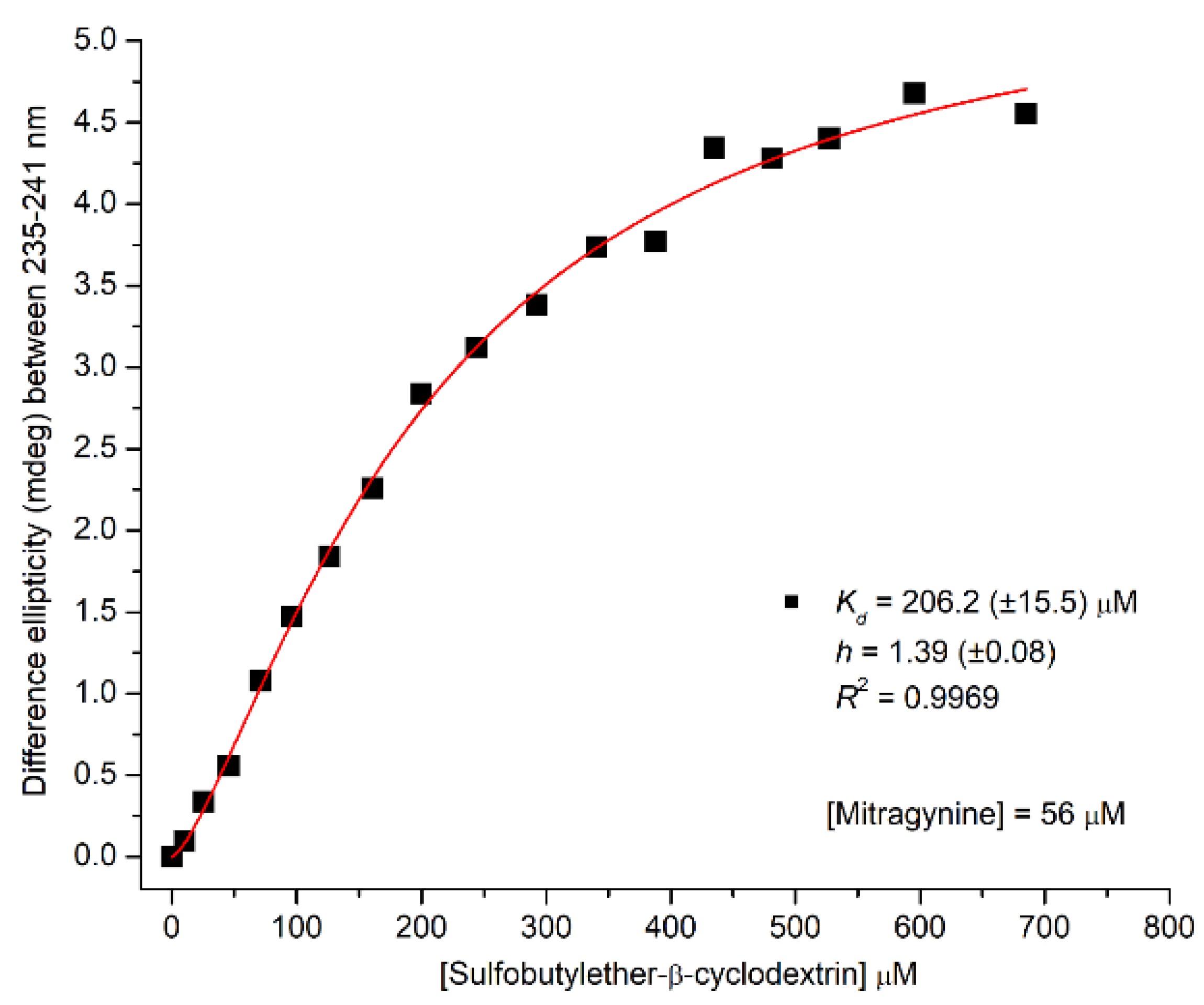
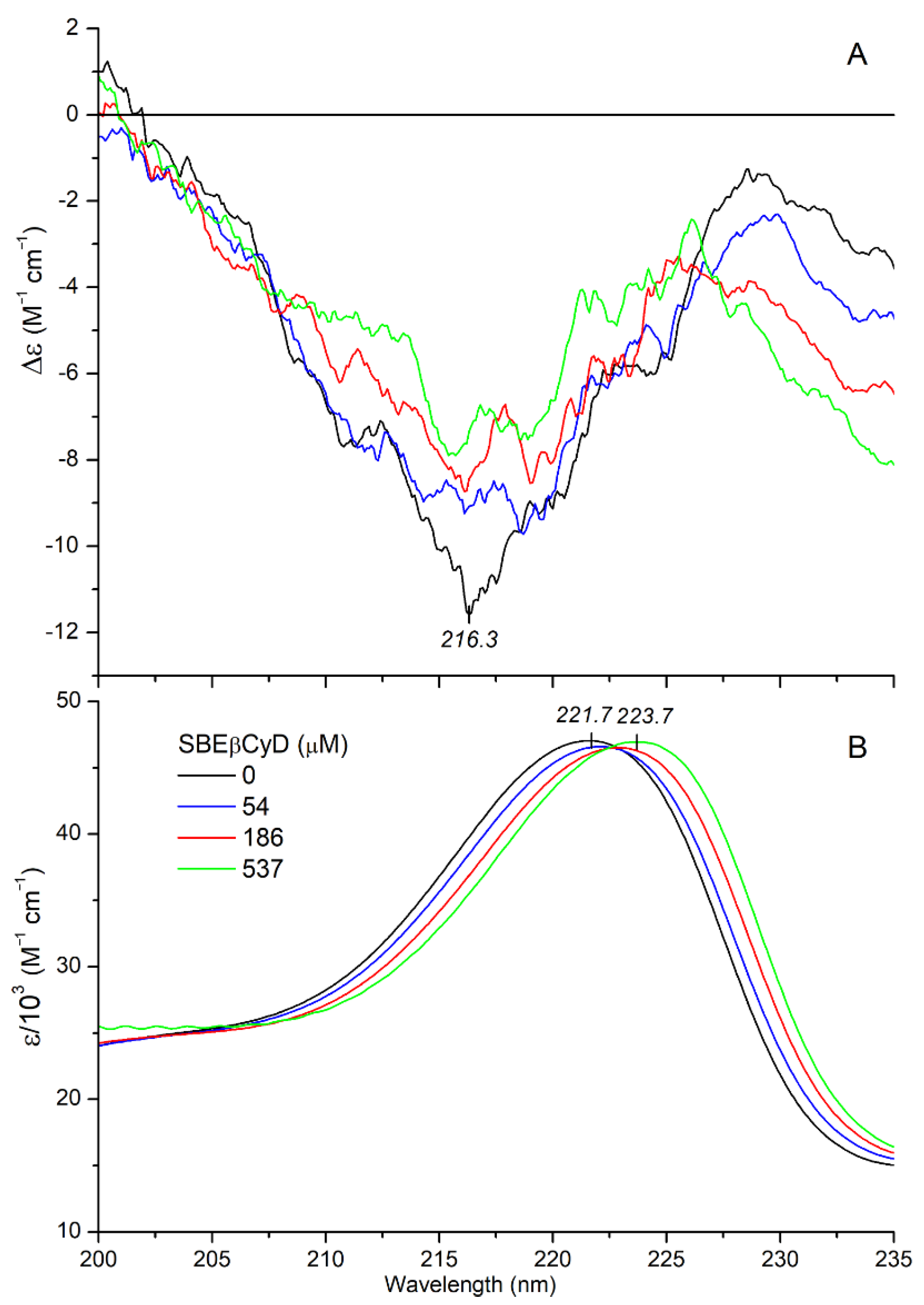
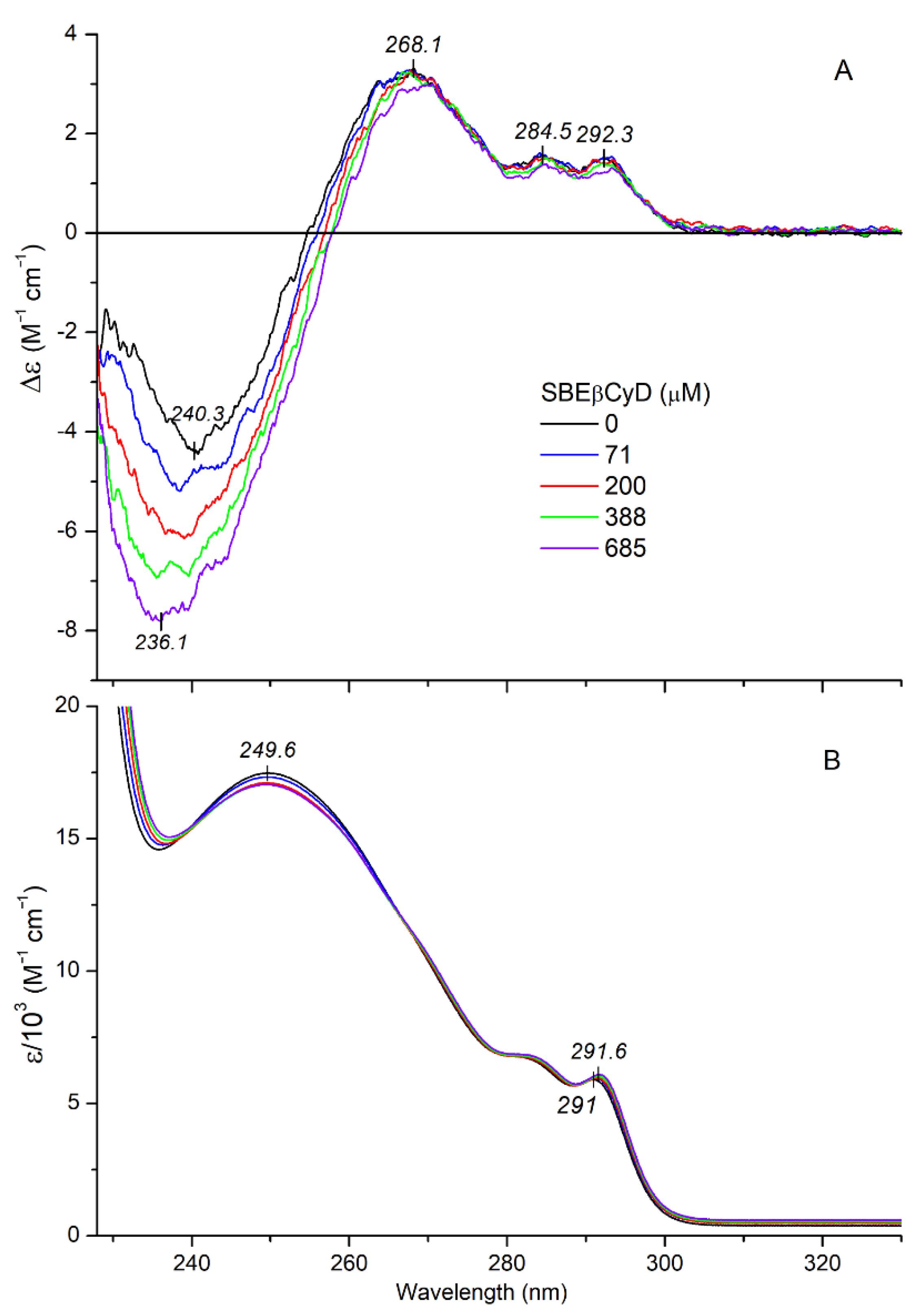
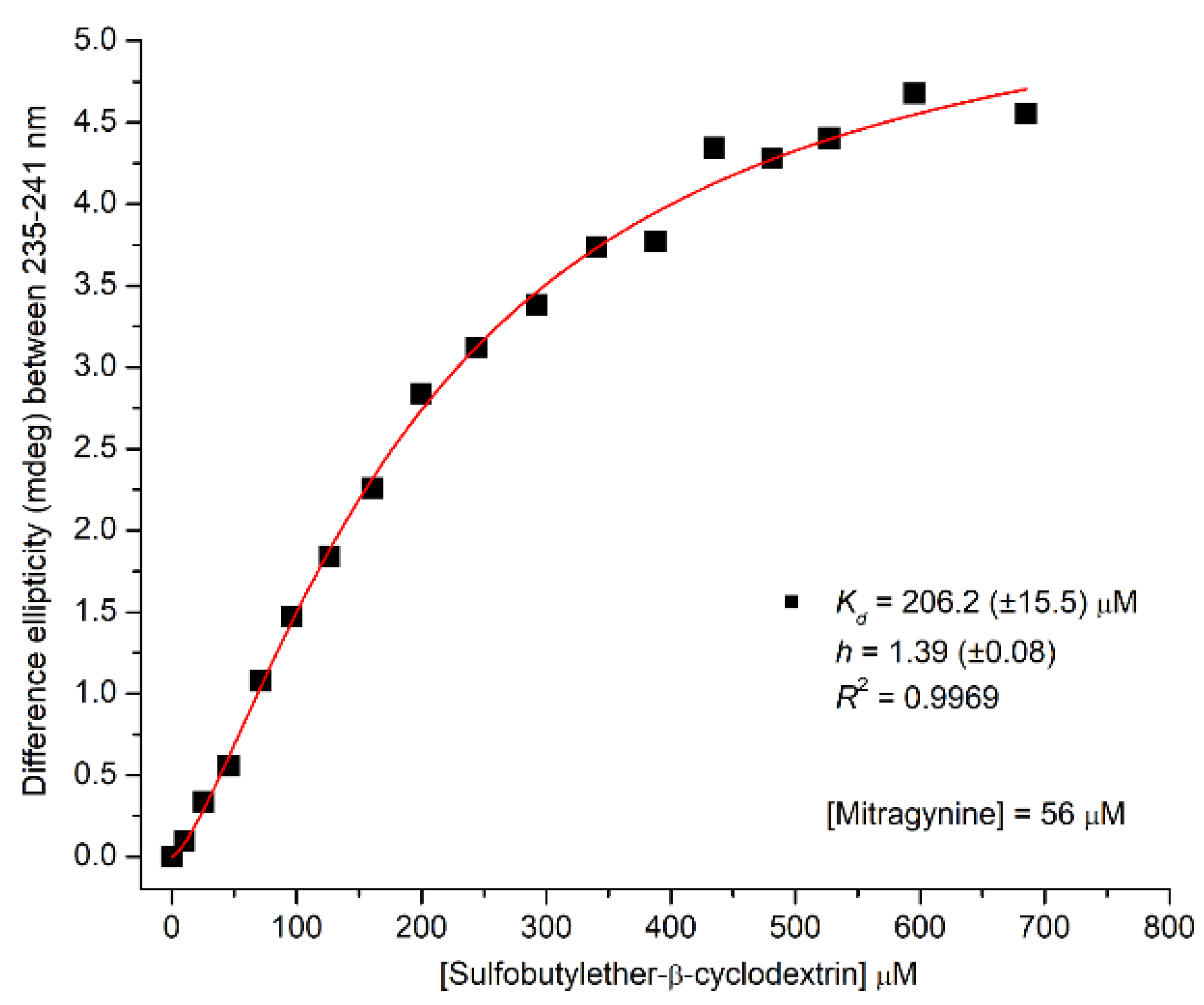
2.2.2. CD/UV Spectroscopic Evaluation of Inclusion Complexes of Mitragynine with Sulfobutylether-β-Cyclodextrin (SBEβCyD)
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. NMR Spectroscopic Measurements
3.2.1. 1H NMR Titration Experiments
3.2.2. Structural Characterization of the MTR-CyD Complexes by 2D NMR Measurements
3.3. Circular Dichroism and UV Absorption Spectroscopic Measurements
Analysis of CD Titration Data for Estimation the Apparent Kd Value of Inclusion Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takayama, H. Chemistry and pharmacology of analgesic indole alkaloids from the Rubiaceous plant, Mitragyna speciosa. Chem. Pharm. Bull. 2004, 52, 916–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, K.L.R.; Prast, C.J. Ethnopharmacology of kratom and the Mitragyna alkaloids. J. Ethnopharmacol. 1988, 23, 115–119. [Google Scholar] [CrossRef]
- Hassan, Z.; Muzaimi, M.; Navaratnam, V.; Yusoff, N.H.M.; Suhaimi, F.W.; Vadivelu, R.; Vicknasingam, B.K.; Amato, D.; von Hörsten, S.; Ismail, N.I.W.; et al. From Kratom to mitragynine and its derivatives: Physiological and behavioural effects related to use, abuse, and addiction. Neurosci. Biobehav. Rev. 2013, 37, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.N.; Lund, J.A.; Murch, S.J. A botanical, phytochemical and ethnomedicinal review of the genus Mitragyna korth: Implications for products sold as kratom. J. Ethnopharmacol. 2017, 202, 302–325. [Google Scholar] [CrossRef] [PubMed]
- Grewal, K.S. Observations on the pharmacology of mitragynine. J. Pharmacol. Exp. Ther. 1932, 46, 251–271. [Google Scholar]
- Babu, K.M.; McCurdy, C.R.; Boyer, E.W. Opioid receptors and legal highs: Salvia divinorum and Kratom. Clin. Toxicol. 2009, 46, 146–152. [Google Scholar] [CrossRef]
- Boyer, E.W.; Babu, K.M.; Adkins, J.E.; McCurdy, C.R.; Halpern, J.H. Self-treatment of opioid withdrawal using kratom (Mitragynia speciosa korth). Addiction 2008, 103, 1048–1050. [Google Scholar] [CrossRef] [Green Version]
- Kruegel, A.C.; Gassaway, M.M.; Kapoor, A.; Váradi, A.; Majumdar, S.; Filizola, M.; Javitch, J.A.; Sames, D. Synthetic and receptor signaling explorations of the Mitragyna alkaloids: Mitragynine as an atypical molecular framework for opioid receptor modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Váradi, A.; Marrone, G.F.; Palmer, T.C.; Narayan, A.; Szabó, M.R.; Le Rouzic, V.; Grinnell, S.G.; Subrath, J.J.; Warner, E.; Kalra, S.; et al. Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit β-arrestin-2. J. Med. Chem. 2016, 59, 8381–8397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Müller, C.P.; Vicknasingam, B.K. Kratom (Mitragyna speciosa) dependence, withdrawal symptoms and craving in regular users. Drug Alcohol Depend. 2014, 139, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Swogger, M.T.; Walsh, Z. Kratom use and mental health: A systematic review. Drug Alcohol Depend. 2018, 183, 134–140. [Google Scholar] [CrossRef]
- Field, E. XCVIII—Mitragynine and mitraversine, two new alkaloids from species of mitragyne. J. Chem. Soc. Trans. 1921, 119, 887–891. [Google Scholar] [CrossRef]
- Saenger, W. Cyclodextrin inclusion compounds in research and industry. Angew. Chem. Int. Ed. Engl. 1980, 19, 344–362. [Google Scholar] [CrossRef]
- Fenyvesi, E. Cyclodextrin polymers in the pharmaceutical industry. J. Incl. Phenom. 1988, 6, 537–545. [Google Scholar] [CrossRef]
- Szente, L.; Puskás, I.; Sohajda, T.; Varga, E.; Vass, P.; Nagy, Z.K.; Farkas, A.; Várnai, B.; Béni, S.; Hazai, E. Sulfobutylether-beta-cyclodextrin-enabled antiviral remdesivir: Characterization of electrospun- and lyophilized formulations. Carbohydr. Polym. 2021, 264, 118011. [Google Scholar] [CrossRef]
- Li, S. Liquid Chromatographic Separation of Enantiomers and Structurally-Related Compounds on β-Cyclodextrin Stationary Phases. Ph.D. Thesis, McGill University, Montreal, QC, Canada, 1992. [Google Scholar]
- Szabó, Z.I.; Ludmerczki, R.; Fiser, B.; Noszál, B.; Tóth, G. Chiral separation of rasagiline using sulfobutylether-β-cyclodextrin: Capillary electrophoresis, NMR and molecular modeling study. Electrophoresis 2019, 40, 1897–1903. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Kusukawa, T.; Fujita, M. Wacker Oxidation in an aqueous phase through the reverse phase-transfer catalysis of a self-assembled nanocage. Chem. Lett. 2004, 29, 598–599. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Inclusion complex of Beta Cyclodextrin and Mitragynine. Available online: https://www.researchgate.net/publication/265088570_Inclusion_complex_of_Beta_Cyclodextrin_and_Mitragynine (accessed on 23 March 2022).
- Job, P. Formation and stability of inorganic complexes in soution. Ann. Chim. 1928, 9, 113–203. [Google Scholar]
- Solution Equilibria Analysis with the OPIUM Computer Program. Available online: http://web.natur.cuni.cz/~kyvala/opium.html (accessed on 9 January 2022).
- Fielding, L. Determination of association constants (Ka) from solution NMR data. Tetrahedron 2000, 56, 6151–6170. [Google Scholar] [CrossRef]
- Dignam, C.F.; Randall, L.A.; Blacken, R.D.; Cunningham, P.R.; Lester, S.K.G.; Brown, M.J.; French, S.C.; Aniagyei, S.E.; Wenzel, T.J. Carboxymethylated cyclodextrin derivatives as chiral NMR discriminating agents. Tetrahedron Asymmetry 2006, 17, 1199–1208. [Google Scholar] [CrossRef]
- De Carvalho, L.B.; Burusco, K.K.; Jaime, C.; Venâncio, T.; de Carvalho, A.F.S.; Murgas, L.D.S.; de Matos Alves Pinto, L. Complexes between methyltestosterone and β-cyclodextrin for application in aquaculture production. Carbohydr. Polym. 2018, 179, 386–393. [Google Scholar] [CrossRef]
- Ramanathan, S.; Parthasarathy, S.; Murugaiyah, V.; Magosso, E.; Tan, S.C.; Mansor, S.M. Understanding the physicochemical properties of mitragynine, a principal alkaloid of Mitragyna speciosa, for preclinical evaluation. Molecules 2015, 20, 4915–4927. [Google Scholar] [CrossRef] [Green Version]
- Várnai, B.; Malanga, M.; Sohajda, T.; Béni, S. Molecular interactions in remdesivir-cyclodextrin systems. J. Pharm. Biomed. Anal. 2022, 209, 114482. [Google Scholar] [CrossRef] [PubMed]
- Snatzke, G. Circular dichroism and absolute conformation: Application of qualitative MO theory to chiroptical phenomena. Angew. Chem. Int. Ed. Engl. 1979, 18, 363–377. [Google Scholar] [CrossRef]
- Flores-Bocanegra, L.; Raja, H.A.; Graf, T.N.; Augustinović, M.; Wallace, E.D.; Hematian, S.; Kellogg, J.J.; Todd, D.A.; Cech, N.B.; Oberlies, N.H. The chemistry of kratom [Mitragyna speciosa]: Updated characterization data and methods to elucidate indole and oxindole alkaloids. J. Nat. Prod. 2020, 83, 2165–2177. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Trager, W.F.; Beckett, A.H. Corynantheidine-type alkaloids—II: Absolute configuration of mitragynine, speciociliatine, mitraciliatine and speciogynine. Tetrahedron 1967, 23, 375–385. [Google Scholar] [CrossRef]
- Albinsson, B.; Nordén, B. Excited-state properties of the indole chromophore. Electronic transition moment directions from linear dichroism measurements: Effect of methyl and methoxy substituents. J. Phys. Chem. 1992, 96, 6204–6212. [Google Scholar] [CrossRef]
- Amann, R.; Arnold, K.; Spitzner, D.; Majer, Z.; Snatzke, G. Reaction of chiral nucleophiles with pyridinium compounds. Total Synthesis of the indole alkaloids (−)-isovallesiachotamine and (+)-vallesiachotamine. Liebigs Ann. 1996, 1996, 349–355. [Google Scholar] [CrossRef]
- Raoelison, G.E.; Terreaux, C.; Queiroz, E.F.; Zsila, F.; Simonyi, M.; Antus, S.; Randriantsoa, A.; Hostettmann, K. Absolute configuration of two new 6-alkylated a-pyrones (=2H-pyran-2-ones) from Ravensara crassifolia. Helv. Chim. Acta 2001, 84, 3470–3476. [Google Scholar] [CrossRef]
- Kirk, D.N.; Klyne, W.; Wallis, S.R. Optical rotatory dispersion. Part LXIV. Influence of solvents upon the Cotton effects of some ketones. J. Chem. Soc. C Org. 1970, 350–360. [Google Scholar] [CrossRef]
- Vsetecka, V.; Pecka, J.; Prochazka, M. (E-Z)-Isomerization of 3-substituted methyl 2-propenoates. Collect. Czech. Chem. Commun. 1978, 43, 2395–2402. [Google Scholar]
- Winterfeldt, E.; Preuss, H. Additionen an die Dreifachbindung, V. Der sterische Verlauf von Additionen an die Dreifachbindung. Chem. Ber. 1966, 99, 450–458. [Google Scholar] [CrossRef]
- Pescitelli, G. ECD exciton chirality method today: A modern tool for determining absolute configurations. Chirality 2021, 34, 333–363. [Google Scholar] [CrossRef] [PubMed]
- Auer, H.E. Far-ultraviolet absorption and circular dichroism spectra of L-tryptophan and some derivatives. J. Am. Chem. Soc. 1973, 95, 3003–3011. [Google Scholar] [CrossRef] [PubMed]
- Kajtár, M.; Horváth-Toró, C.; Kuthi, É.; Szejtli, J. A simple rule for predicting circular dichroism induced in aromatic guests by cyclodextrin hosts in inclusion complexes. Acta Chim. Acad. Sci. Hung. 1982, 110, 327–355. [Google Scholar]
- Kano, K.; Tatsumi, M.; Hashimoto, S. Cyclodextrin-induced conformational enantiomerism of dinaphthylmethanes. J. Org. Chem. 1991, 56, 6579–6585. [Google Scholar] [CrossRef]
- Buvári, A.; Barcza, L.; Kajtár, M. Complex formation of phenolphthalein and some related compounds with β-cyclodextrin. J. Chem. Soc. Perkin Trans. 2 1988, 9, 1687–1690. [Google Scholar] [CrossRef]
- Stadler-Szöke, Á.; Vikmon, M.; Szejtli, J.; Kajtár, M. Fendiline-β-cyclodextrin inclusion complex. J. Incl. Phenom. 1985, 3, 71–84. [Google Scholar] [CrossRef]
- Darcsi, A.; Szakács, Z.; Zsila, F.; Tóth, G.; Rácz, Á.; Béni, S. NMR, CD and UV spectroscopic studies reveal uncommon binding modes of dapoxetine to native cyclodextrins. RSC Adv. 2016, 6, 102315–102328. [Google Scholar] [CrossRef]
- Belica-Pacha, S.; Daśko, M.; Buko, V.; Zavodnik, I.; Miłowska, K.; Bryszewska, M. Thermodynamic studies of interactions between sertraline hydrochloride and randomly methylated β-cyclodextrin molecules supported by circular dichroism spectroscopy and molecular docking results. Int. J. Mol. Sci. 2021, 22, 12357. [Google Scholar] [CrossRef]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Krait, S.; Salgado, A.; Chankvetadze, B.; Gago, F.; Scriba, G.K.E. Investigation of the complexation between cyclodextrins and medetomidine enantiomers by capillary electrophoresis, NMR spectroscopy and molecular modeling. J. Chromatogr. A 2018, 1567, 198–210. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Várnai, B.; Zsila, F.; Szakács, Z.; Garádi, Z.; Malanga, M.; Béni, S. Sulfobutylation of Beta-Cyclodextrin Enhances the Complex Formation with Mitragynine: An NMR and Chiroptical Study. Int. J. Mol. Sci. 2022, 23, 3844. https://doi.org/10.3390/ijms23073844
Várnai B, Zsila F, Szakács Z, Garádi Z, Malanga M, Béni S. Sulfobutylation of Beta-Cyclodextrin Enhances the Complex Formation with Mitragynine: An NMR and Chiroptical Study. International Journal of Molecular Sciences. 2022; 23(7):3844. https://doi.org/10.3390/ijms23073844
Chicago/Turabian StyleVárnai, Bianka, Ferenc Zsila, Zoltán Szakács, Zsófia Garádi, Milo Malanga, and Szabolcs Béni. 2022. "Sulfobutylation of Beta-Cyclodextrin Enhances the Complex Formation with Mitragynine: An NMR and Chiroptical Study" International Journal of Molecular Sciences 23, no. 7: 3844. https://doi.org/10.3390/ijms23073844
APA StyleVárnai, B., Zsila, F., Szakács, Z., Garádi, Z., Malanga, M., & Béni, S. (2022). Sulfobutylation of Beta-Cyclodextrin Enhances the Complex Formation with Mitragynine: An NMR and Chiroptical Study. International Journal of Molecular Sciences, 23(7), 3844. https://doi.org/10.3390/ijms23073844