
A Low Dose of Pure Cannabidiol Is Sufficient to Stimulate the Cytotoxic Function of CIK Cells without Exerting the Downstream Mediators in Pancreatic Cancer Cells


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Cytokine-Induced Killer Cells
2.2. Cell Line and Cell Culture
2.3. Cytotoxicity Assay Based on CCK-8
2.4. LDH Assay
2.5. Immunocytochemistry
2.6. Imaging and 3D Reconstruction Modeling
2.7. Fluorescence-Activated Cell Sorting (FACS) Analysis
2.8. CREB Phosphorylation Assay (p-CREB)
3. Results
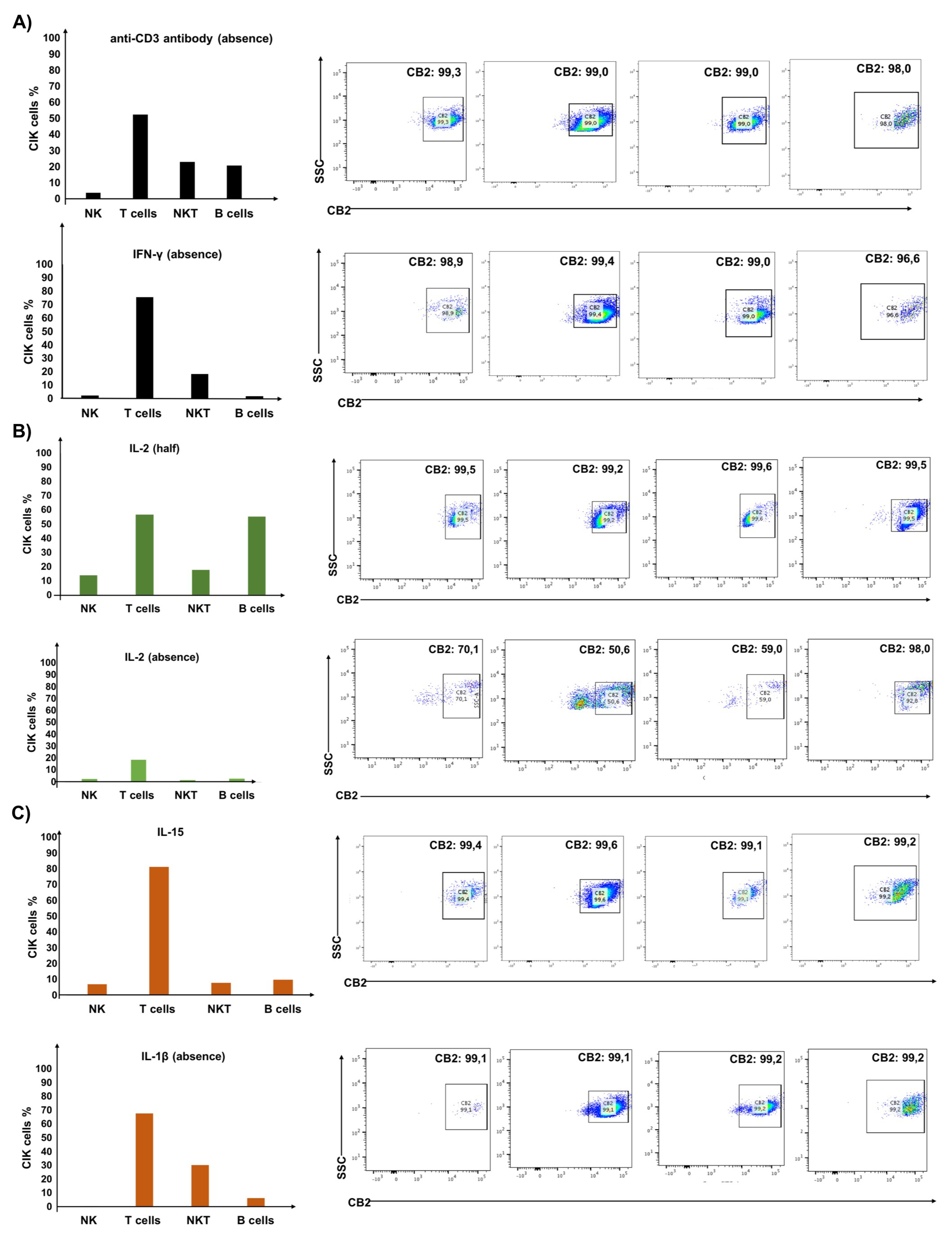
3.1. IL-2 Primarily Determines the Expression of CB2 Receptor on CIK Cells
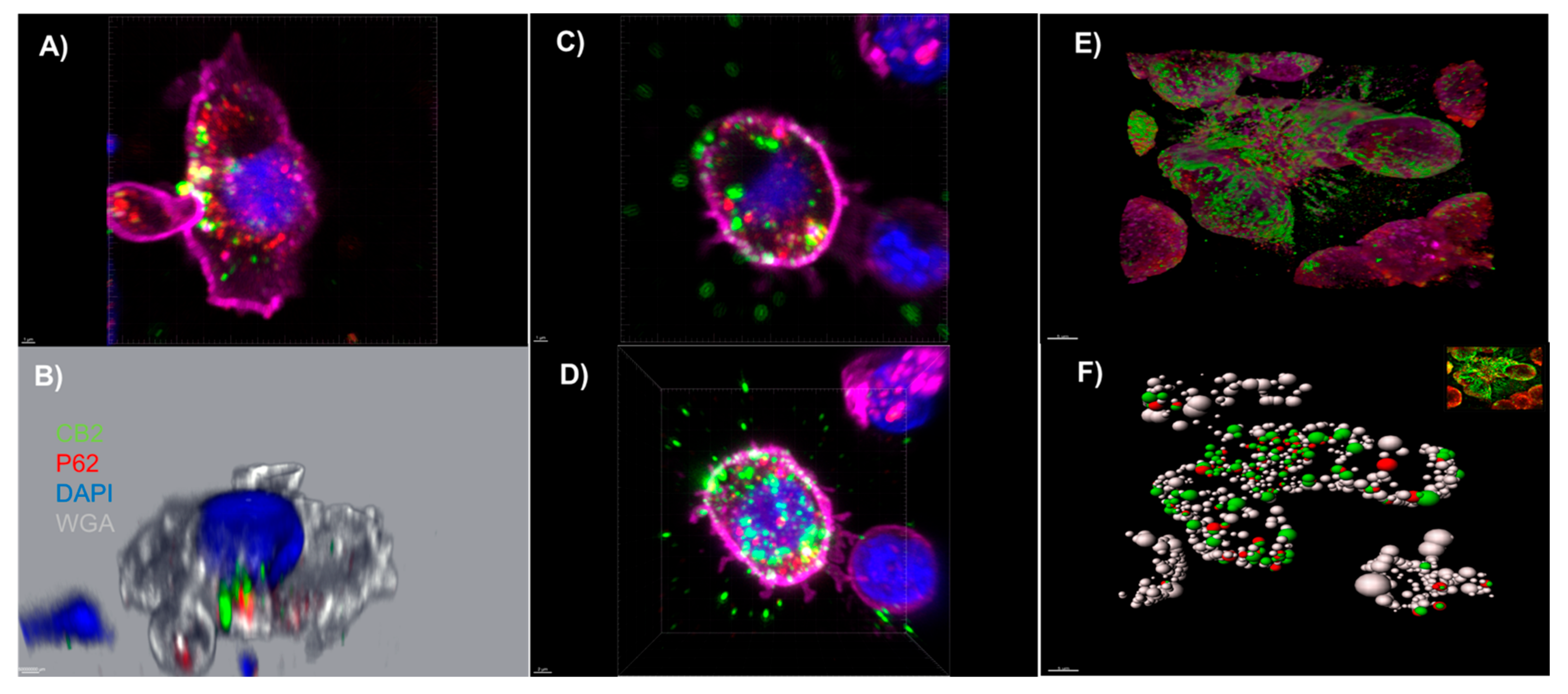
3.2. An Autophagosomal-Associated Scaffold Protein p62 Also Colocalizes with CB2 Receptors in CIK Cells and the PANC-1 Cell Line
3.3. CIK Cells Showed a Low Level of Intracellular p-p38 and Donor Specific Variability in p-CREB
3.4. Cannabidiol Significantly Decreases the Viability of PANC-1 Cells and Significantly Increases the Cytotoxicity of CIK Cells against PANC-1 at Low Concentrations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, A.; Liu, H.; Herwig-Carl, M.C.; Chand Dakal, T.; Schmidt-Wolf, I.G.H. Epigenetic Regulatory Enzymes: Mutation Prevalence and Coexistence in Cancers. Cancer Investig. 2021, 39, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Schmidt-Wolf, I.G.H. 30 years of CIK cell therapy: Recapitulating the key breakthroughs and future perspective. J. Exp. Clin. Cancer Res. 2021, 40, 388. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sharma, A.; Bloemendal, M.; Schmidt-Wolf, R.; Kornek, M.; Schmidt-Wolf, I.G.H. PD-1 blockade enhances cytokine-induced killer cell-mediated cytotoxicity in B-cell non-Hodgkin lymphoma cell lines. Oncol. Lett. 2021, 22, 613. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.; Weiher, H.; Schmidt-Wolf, I.G.H. CIK Cells and HDAC Inhibitors in Multiple Myeloma. Int. J. Mol. Sci. 2017, 18, 945. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, D., Jr.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a Promising Anti-Cancer Drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Kovalchuk, I. Cannabinoids as anticancer therapeutic agents. Cell Cycle 2020, 19, 961–989. [Google Scholar] [CrossRef]
- Garofano, F.; Schmidt-Wolf, I.G.H. High Expression of Cannabinoid Receptor 2 on Cytokine-Induced Killer Cells and Multiple Myeloma Cells. Int. J. Mol. Sci. 2020, 21, 3800. [Google Scholar] [CrossRef]
- Zhang, Y.; Schmidt-Wolf, I.G.H. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J. Cell. Physiol. 2020, 235, 9291–9303. [Google Scholar] [CrossRef]
- Garofano, F.; Gonzalez-Carmona, M.A.; Skowasch, D.; Schmidt-Wolf, R.; Abramian, A.; Hauser, S.; Strassburg, C.P.; Schmidt-Wolf, I.G.H. Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 4307. [Google Scholar] [CrossRef] [Green Version]
- Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. [Google Scholar] [CrossRef]
- Nüsgen, N.; Goering, W.; Dauksa, A.; Biswas, A.; Jamil, M.A.; Dimitriou, I.; Sharma, A.; Singer, H.; Fimmers, R.; Fröhlich, H.; et al. Inter-locus as well as intra-locus heterogeneity in LINE-1 promoter methylation in common human cancers suggests selective demethylation pressure at specific CpGs. Clin. Epigenetics 2015, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlick, K.; Kiem, D.; Greil, R. Recent Advances in Pancreatic Cancer: Novel Prognostic Biomarkers and Targeted Therapy-A Review of the Literature. Biomolecules 2021, 11, 1469. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hu, K.; Bailey, P.; Springfeld, C.; Roth, S.; Kurilov, R.; Brors, B.; Gress, T.; Buchholz, M.; An, J.; et al. Clinical Impact of Molecular Subtyping of Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 743908. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Fogli, S.; Nieri, P.; Chicca, A.; Adinolfi, B.; Mariotti, V.; Iacopetti, P.; Breschi, M.C.; Pellegrini, S. Cannabinoid derivatives induce cell death in pancreatic MIA PaCa-2 cells via a receptor-independent mechanism. FEBS Lett. 2006, 580, 1733–1739. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzmán, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef] [Green Version]
- Sharaf, A.; Mensching, L.; Keller, C.; Rading, S.; Scheffold, M.; Palkowitsch, L.; Djogo, N.; Rezgaoui, M.; Kestler, H.A.; Moepps, B.; et al. Systematic Affinity Purification Coupled to Mass Spectrometry Identified p62 as Part of the Cannabinoid Receptor CB2 Interactome. Front. Mol. Neurosci. 2019, 12, 224. [Google Scholar] [CrossRef] [Green Version]
- Saroz, Y.; Kho, D.T.; Glass, M.; Graham, E.S.; Grimsey, N.L. Cannabinoid Receptor 2 (CB(2)) Signals via G-alpha-s and Induces IL-6 and IL-10 Cytokine Secretion in Human Primary Leukocytes. ACS Pharmacol. Transl. Sci. 2019, 2, 414–428. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, H.; Luo, K.; Sharma, A.; Sun, X. Prognostic gene expression signature revealed the involvement of mutational pathways in cancer genome. J. Cancer 2020, 11, 4510–4520. [Google Scholar] [CrossRef]
- Sharma, A.; Reutter, H.; Ellinger, J. DNA Methylation and Bladder Cancer: Where Genotype does not Predict Phenotype. Curr. Genom. 2020, 21, 34–36. [Google Scholar] [CrossRef]
- Sharma, A.; Biswas, A.; Liu, H.; Sen, S.; Paruchuri, A.; Katsonis, P.; Lichtarge, O.; Chand Dakal, T.; Maulik, U.; Gromiha, M.M.; et al. Mutational Landscape of the BAP1 Locus Reveals an Intrinsic Control to Regulate the miRNA Network and the Binding of Protein Complexes in Uveal Melanoma. Cancers 2019, 11, 1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirjang, S.; Alizadeh, N.; Mansoori, B.; Mahmoodpoor, A.; Kafil, H.S.; Hojjat-Farsangi, M.; Yousefi, M. Promising immunotherapy: Highlighting cytokine-induced killer cells. J. Cell. Biochem. 2019, 120, 8863–8883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ellinger, J.; Ritter, M.; Schmidt-Wolf, I.G.H. Clinical Studies Applying Cytokine-Induced Killer Cells for the Treatment of Renal Cell Carcinoma. Cancers 2020, 12, 2471. [Google Scholar] [CrossRef]
- Zhang, Y.; Sharma, A.; Weiher, H.; Schmid, M.; Kristiansen, G.; Schmidt-Wolf, I.G.H. Clinical Studies on Cytokine-Induced Killer Cells: Lessons from Lymphoma Trials. Cancers 2021, 13, 6007. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Donadelli, M.; Costanzo, C.; Dalla Pozza, E.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef] [Green Version]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Dalla Pozza, E.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garofano, F.; Sharma, A.; Abken, H.; Gonzalez-Carmona, M.A.; Schmidt-Wolf, I.G.H. A Low Dose of Pure Cannabidiol Is Sufficient to Stimulate the Cytotoxic Function of CIK Cells without Exerting the Downstream Mediators in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2022, 23, 3783. https://doi.org/10.3390/ijms23073783
Garofano F, Sharma A, Abken H, Gonzalez-Carmona MA, Schmidt-Wolf IGH. A Low Dose of Pure Cannabidiol Is Sufficient to Stimulate the Cytotoxic Function of CIK Cells without Exerting the Downstream Mediators in Pancreatic Cancer Cells. International Journal of Molecular Sciences. 2022; 23(7):3783. https://doi.org/10.3390/ijms23073783
Chicago/Turabian StyleGarofano, Francesca, Amit Sharma, Hinrich Abken, Maria A. Gonzalez-Carmona, and Ingo G. H. Schmidt-Wolf. 2022. "A Low Dose of Pure Cannabidiol Is Sufficient to Stimulate the Cytotoxic Function of CIK Cells without Exerting the Downstream Mediators in Pancreatic Cancer Cells" International Journal of Molecular Sciences 23, no. 7: 3783. https://doi.org/10.3390/ijms23073783
APA StyleGarofano, F., Sharma, A., Abken, H., Gonzalez-Carmona, M. A., & Schmidt-Wolf, I. G. H. (2022). A Low Dose of Pure Cannabidiol Is Sufficient to Stimulate the Cytotoxic Function of CIK Cells without Exerting the Downstream Mediators in Pancreatic Cancer Cells. International Journal of Molecular Sciences, 23(7), 3783. https://doi.org/10.3390/ijms23073783