
Necroptosis as a Novel Facet of Mitotic Catastrophe

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
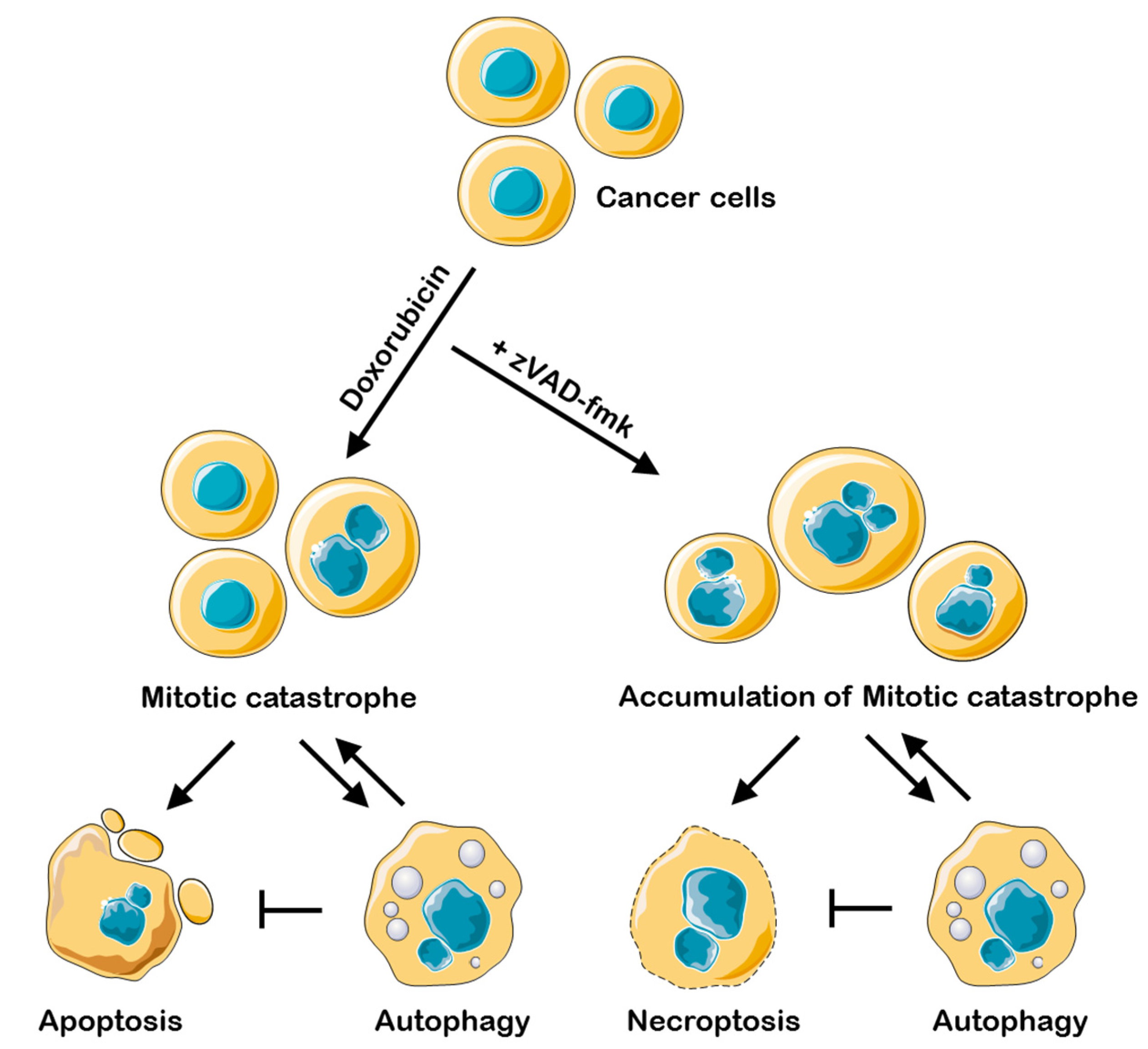
2.1. Appearance of Mitotic Catastrophe upon Treatment with a Sublethal Dose of Doxorubicin
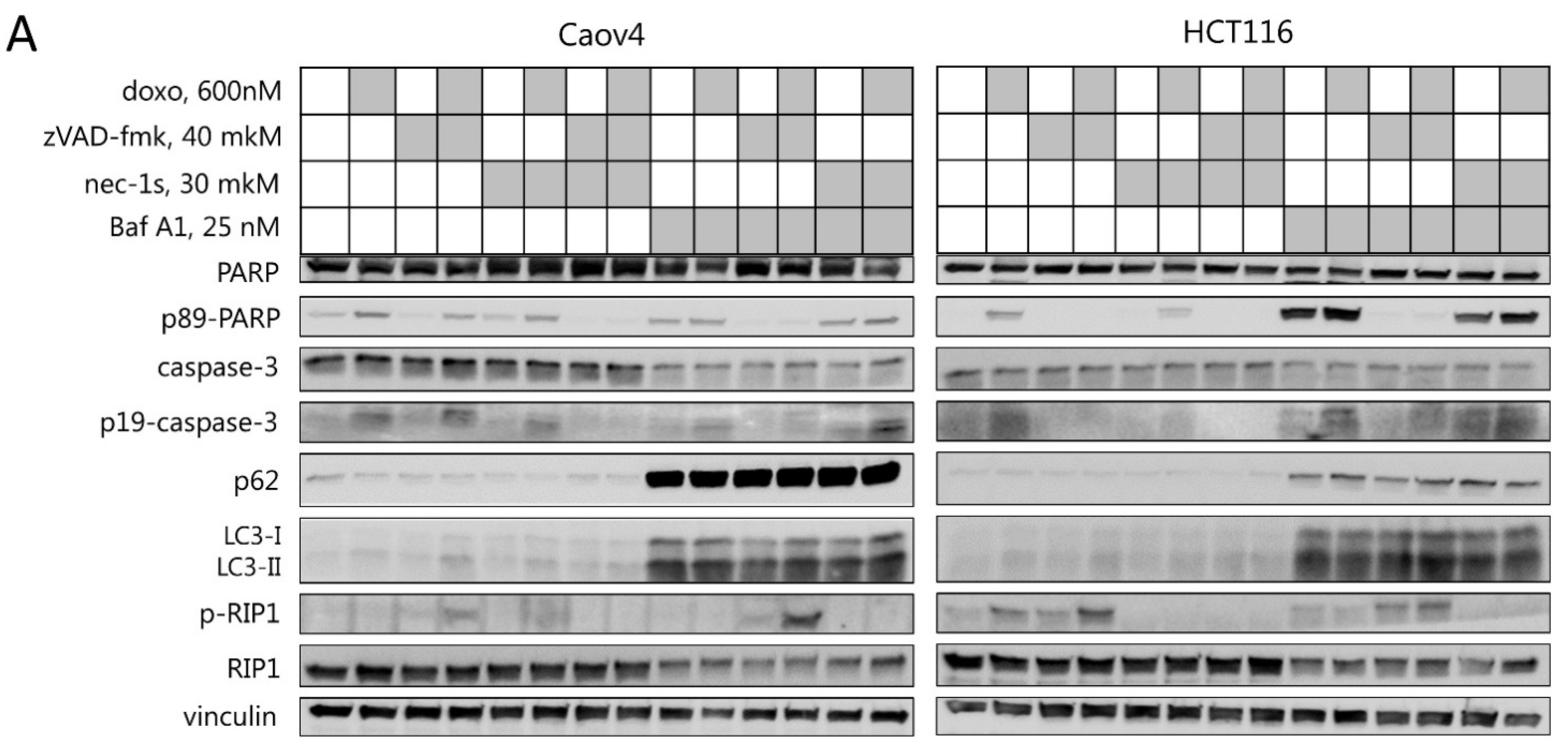
2.2. Necroptosis as a Consequence of Mitotic Catastrophe
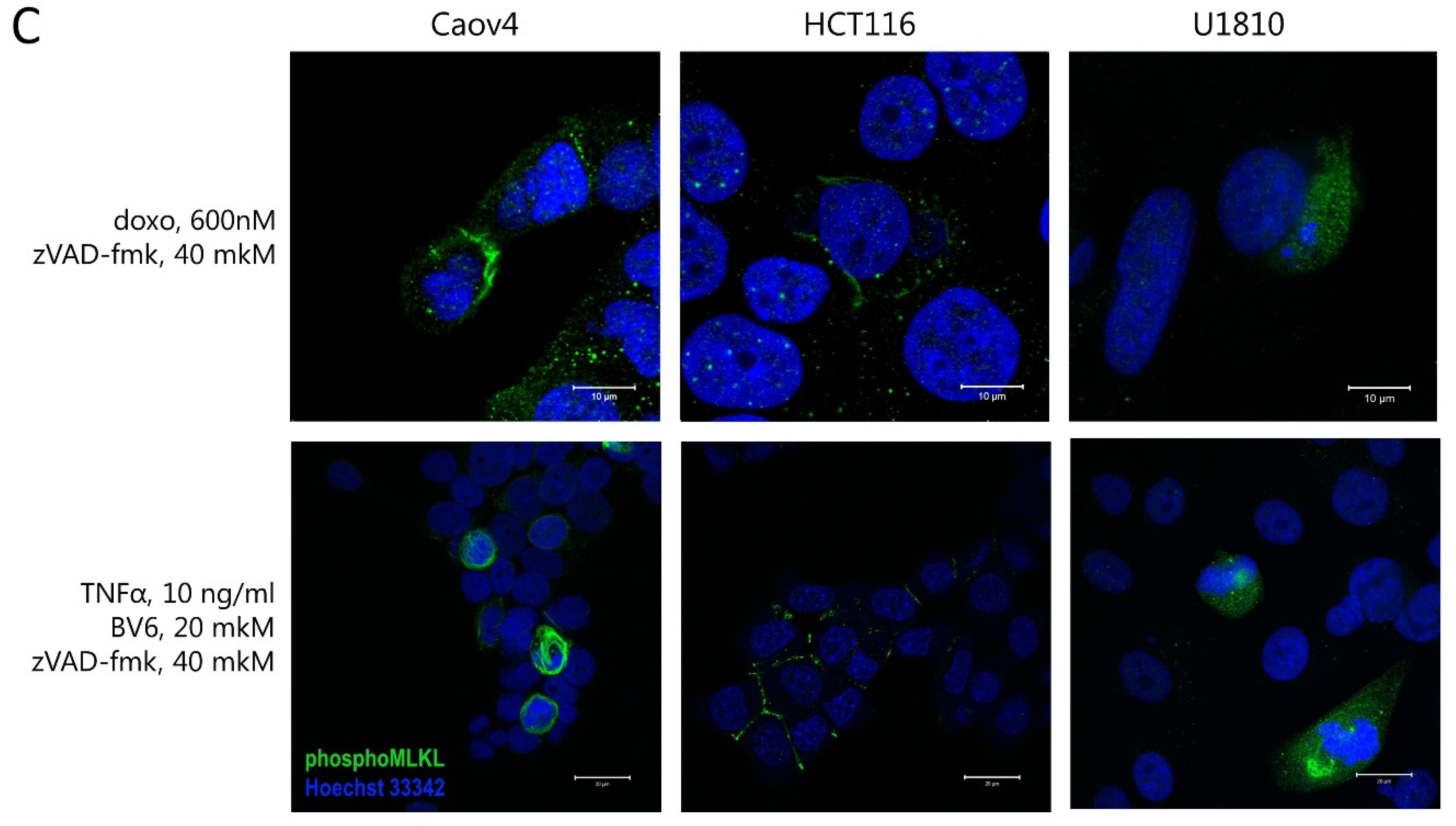
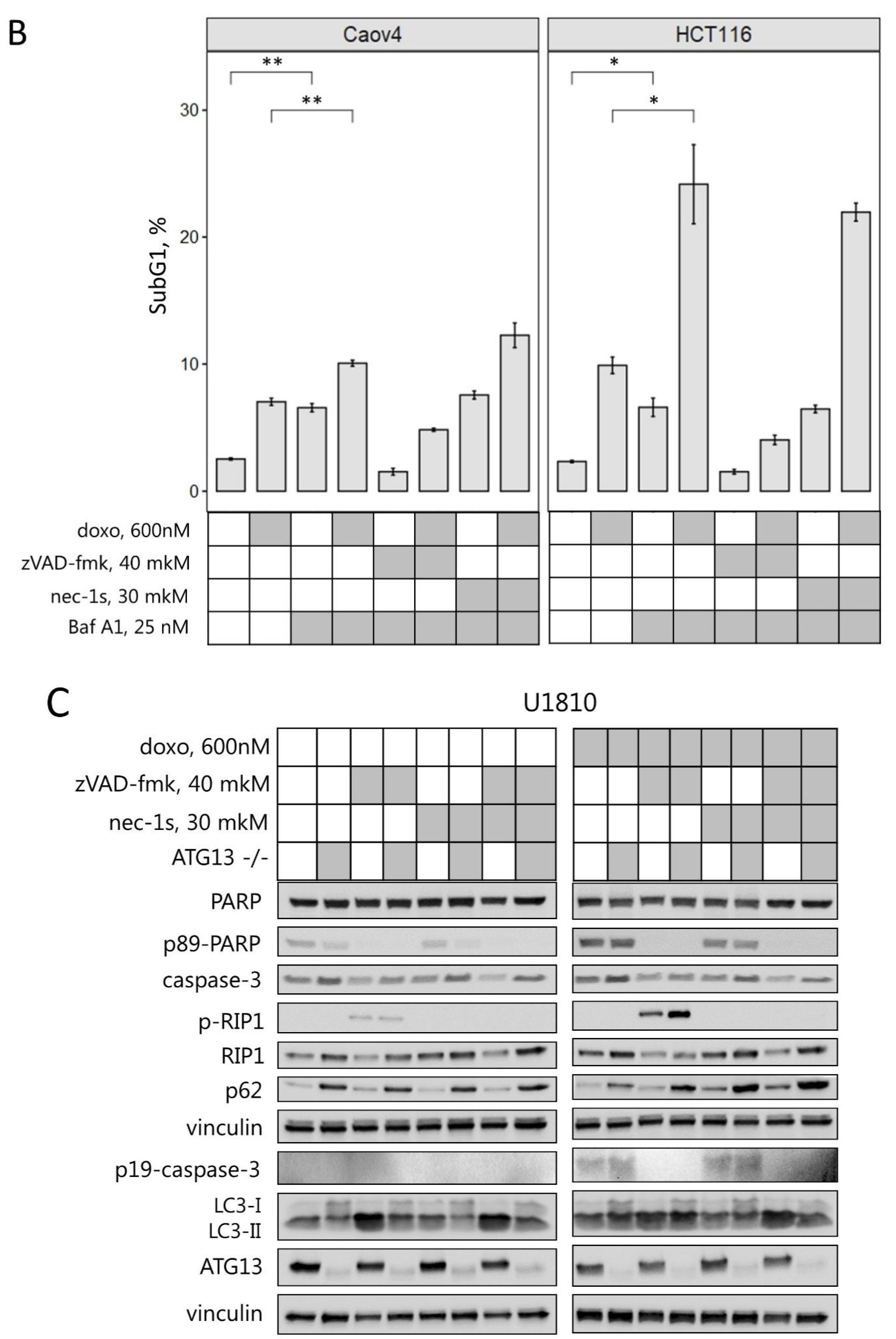
2.3. Interplay between Necroptosis, Autophagy, and Apoptosis under Conditions of Mitotic Catastrophe
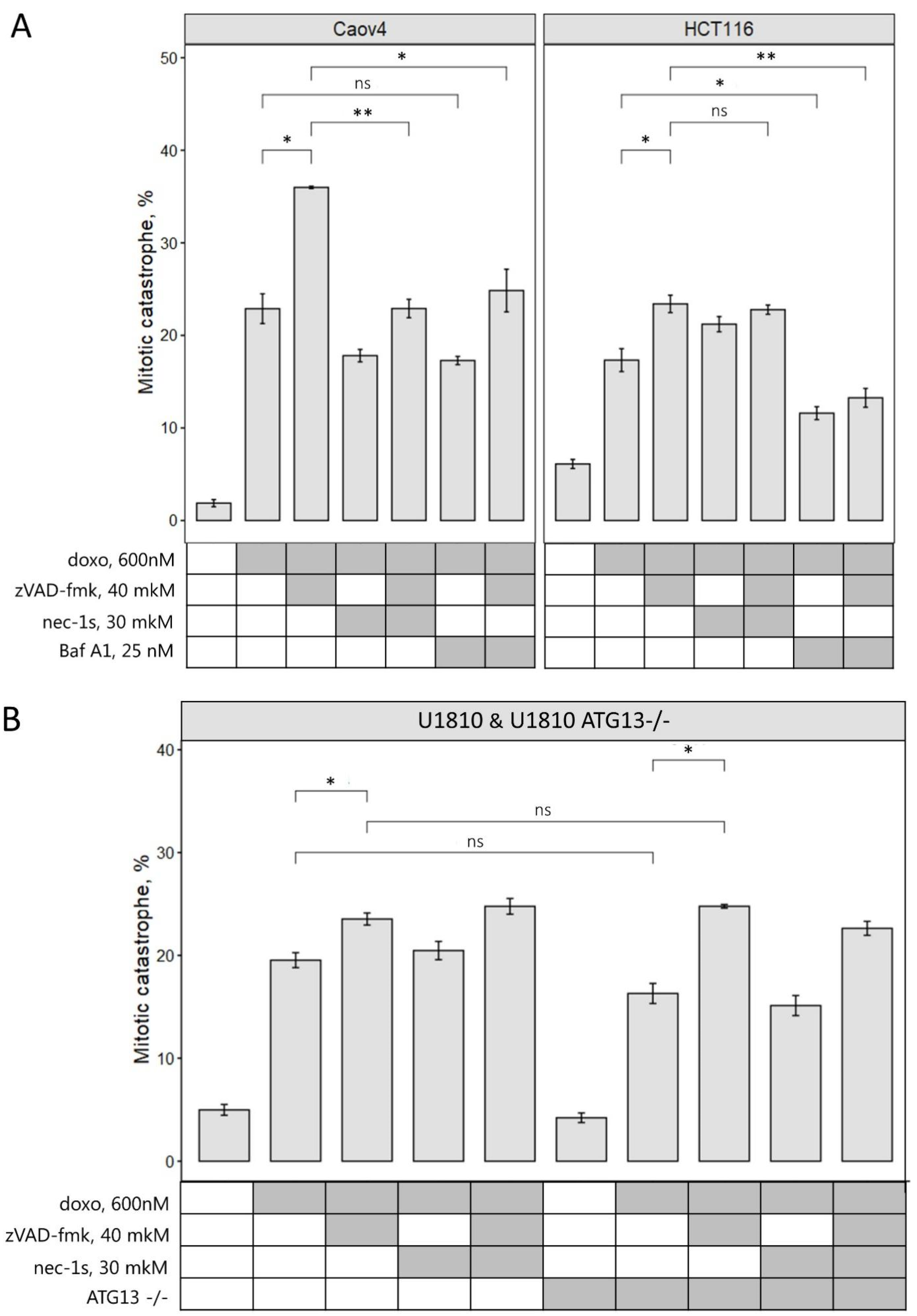
2.4. Inhibition of Different Types of PCD Modulates Mitotic Catastrophe
3. Discussion
4. Material and Methods
4.1. Cell Culture and Treatments
4.2. Gel Electrophoresis and Western Blot
4.3. Immunoprecipitation
4.4. Sub-G1 Test
4.5. Immunofluorescence Microscopy
4.6. Data Processing, Statistical Analysis, and Graphic Illustrations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prokhorova, E.A.; Egorshina, A.Y.; Zhivotovsky, B.; Kopeina, G.S. The DNA-damage response and nuclear events as regulators of nonapoptotic forms of cell death. Oncogene 2020, 39, 8. [Google Scholar] [CrossRef] [PubMed]
- Ghelli Luserna Di Rorà, A.; Martinelli, G.; Simonetti, G. The balance between mitotic death and mitotic slippage in acute leukemia: A new therapeutic window? J. Hematol. Oncol. 2019, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Imreh, G.; Norberg, H.V.; Imreh, S.; Zhivotovsky, B. Chromosomal breaks during mitotic catastrophe trigger γH2AX-ATM-p53-mediated apoptosis. J. Cell Sci. 2011, 124, 2951–2963. [Google Scholar] [CrossRef] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef] [Green Version]
- Denisenko, T.V.; Sorokina, I.V.; Gogvadze, V.; Zhivotovsky, B. Mitotic catastrophe and cancer drug resistance: A link that must to be broken. Drug Resist. Updates 2016, 24, 1–12. [Google Scholar] [CrossRef]
- Florio, R.; Veschi, S.; Di Giacomo, V.; Pagotto, S.; Carradori, S.; Verginelli, F.; Cirilli, R.; Casulli, A.; Grassadonia, A.; Tinari, N.; et al. The benzimidazole-based anthelmintic parbendazole: A repurposed drug candidate that synergizes with gemcitabine in pancreatic cancer. Cancers 2019, 11, 2042. [Google Scholar] [CrossRef] [Green Version]
- Králová, V.; Hanušová, V.; Rudolf, E.; Čáňová, K.; Skálová, L. Flubendazole induces mitotic catastrophe and senescence in colon cancer cells in vitro. J. Pharm. Pharmacol. 2016, 68, 208–218. [Google Scholar] [CrossRef]
- Khing, T.M.; Choi, W.S.; Kim, D.M.; Po, W.W.; Thein, W.; Shin, C.Y.; Sohn, U.D. The effect of paclitaxel on apoptosis, autophagy and mitotic catastrophe in AGS cells. Sci. Rep. 2021, 11, 23490. [Google Scholar] [CrossRef]
- Eom, Y.W.; Kim, M.A.; Park, S.S.; Goo, M.J.; Kwon, H.J.; Sohn, S.; Kim, W.H.; Yoon, G.; Choi, K.S. Two distinct modes of cell death induced by doxorubicin: Apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005, 24, 4765–4777. [Google Scholar] [CrossRef] [Green Version]
- Sorokina, I.V.; Denisenko, T.V.; Imreh, G.; Tyurin-Kuzmin, P.A.; Kaminskyy, V.O.; Gogvadze, V.; Zhivotovsky, B. Involvement of autophagy in the outcome of mitotic catastrophe. Sci. Rep. 2017, 7, 14571. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Fulda, S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol. Ther. 2013, 14, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, K.; Chan, F.K.-M. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Yu, J.; Zhang, L. Necroptosis: An alternative cell death program defending against cancer. Biochim. Biophys. Acta 2016, 1865, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.A.; Marquis, R.W.W.; et al. MLKL compromises plasma membrane integrity upon binding to phosphatidyl inositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Leverkus, M. Programmed necrosis and necroptosis signalling. FEBS J. 2015, 282, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Hannes, S.; Abhari, B.A.; Fulda, S. Smac mimetic triggers necroptosis in pancreatic carcinoma cells when caspase activation is blocked. Cancer Lett. 2016, 380, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Rohde, K.; Kleinesudeik, L.; Roesler, S.; Löwe, O.; Heidler, J.; Schröder, K.; Wittig, I.; Dröse, S.; Fulda, S. A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ. 2017, 24, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Liccardi, G.; Ramos Garcia, L.; Tenev, T.; Annibaldi, A.; Legrand, A.J.; Robertson, D.; Feltham, R.; Anderton, H.; Darding, M.; Peltzer, N.; et al. RIPK1 and Caspase-8 Ensure Chromosome Stability Independently of Their Role in Cell Death and Inflammation. Mol. Cell 2019, 73, 413–428. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Liu, B. PLK1-mediated S369 phosphorylation of RIPK3 during G2 and M phases enables its ripoptosome incorporation and activity. iScience 2021, 24, 102320. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Sung, Y.J.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Caspase-8 gene is frequently inactivated by the frameshift somatic mutation 1225_1226delTG in hepatocellular carcinomas. Oncogene 2005, 24, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Uzunparmak, B.; Gao, M.; Lindemann, A.; Erikson, K.; Wang, L.; Lin, E.; Frank, S.J.; Gleber-Netto, F.O.; Zhao, M.; Skinner, H.D.; et al. Caspase-8 loss radiosensitizes head and neck squamous cell carcinoma to SMAC mimetic–induced necroptosis. JCI Insight 2020, 5, e139837. [Google Scholar] [CrossRef]
- Hakem, A.; El Ghamrasni, S.; Maire, G.; Lemmers, B.; Karaskova, J.; Jurisicova, A.; Sanchez, O.; Squire, J.; Hakem, R. Caspase-8 is essential for maintaining chromosomal stability and suppressing B-cell lymphomagenesis. Blood 2012, 119, 3495–3502. [Google Scholar] [CrossRef]
- Zhou, W.; Yuan, J. SnapShot: Necroptosis. Cell 2014, 158, 464. [Google Scholar] [CrossRef] [Green Version]
- Farkas, T.; Daugaard, M.; Jäättelä, M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J. Biol. Chem. 2011, 286, 38904–38912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.C.; Yu, L.; Wang, H.J.; Tashiro, S.I.; Onodera, S.; Ikejima, T. TNFα-induced necroptosis and autophagy via supression of the p38-NF-κB survival pathway in L929 cells. J. Pharmacol. Sci. 2011, 117, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Park, H.; Heisler, J.; Maculins, T.; Roose-Girma, M.; Xu, M.; McKenzie, B.; van Lookeren Campagne, M.; Newton, K.; Murthy, A. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife 2019, 8, e44452. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; De Wijngaert, P.; Vanmeerbeerk, I.; Martin, S.; Vangheluwe, P.; Schlenner, S.; Krysko, D.V.; Parys, J.B.; Bultynck, G.; Vandenabeele, P.; et al. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells 2020, 9, 1823. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Da Silva, R.B.; Reis E Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-kB signaling in dying cells determines cross-priming of CD8+ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Allavena, G.; Boyd, C.; Oo, K.S.; Maellaro, E.; Zhivotovsky, B.; Kaminskyy, V.O. Suppressed translation and ULK1 degradation as potential mechanisms of autophagy limitation under prolonged starvation. Autophagy 2016, 12, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 1 March 2022).


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorshina, A.Y.; Zamaraev, A.V.; Kaminskyy, V.O.; Radygina, T.V.; Zhivotovsky, B.; Kopeina, G.S. Necroptosis as a Novel Facet of Mitotic Catastrophe. Int. J. Mol. Sci. 2022, 23, 3733. https://doi.org/10.3390/ijms23073733
Egorshina AY, Zamaraev AV, Kaminskyy VO, Radygina TV, Zhivotovsky B, Kopeina GS. Necroptosis as a Novel Facet of Mitotic Catastrophe. International Journal of Molecular Sciences. 2022; 23(7):3733. https://doi.org/10.3390/ijms23073733
Chicago/Turabian StyleEgorshina, Aleksandra Yu., Alexey V. Zamaraev, Vitaliy O. Kaminskyy, Tatiana V. Radygina, Boris Zhivotovsky, and Gelina S. Kopeina. 2022. "Necroptosis as a Novel Facet of Mitotic Catastrophe" International Journal of Molecular Sciences 23, no. 7: 3733. https://doi.org/10.3390/ijms23073733
APA StyleEgorshina, A. Y., Zamaraev, A. V., Kaminskyy, V. O., Radygina, T. V., Zhivotovsky, B., & Kopeina, G. S. (2022). Necroptosis as a Novel Facet of Mitotic Catastrophe. International Journal of Molecular Sciences, 23(7), 3733. https://doi.org/10.3390/ijms23073733