
The Merkel Cell Polyomavirus T-Antigens and IL-33/ST2-IL1RAcP Axis: Possible Role in Merkel Cell Carcinoma

 , , ,
, , ,  ,
,  and
and 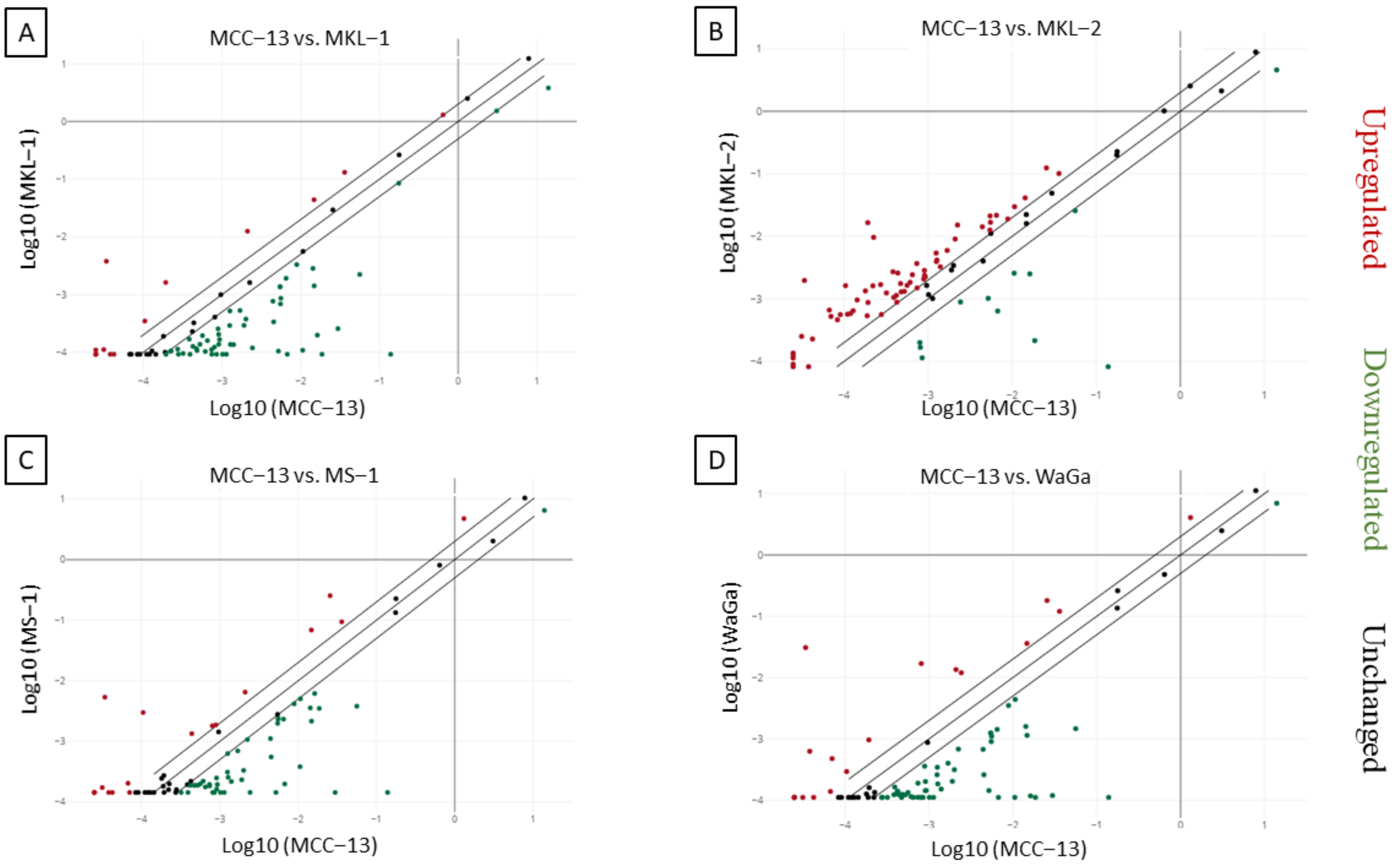{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Differential Expression of Inflammatory Cytokines and Receptors in MCC Cell Lines
2.2. Increased IL-33 Expression in MCPyV Positive-MCC Cell Lines Compared to Virus Negative-MCC Cell Lines
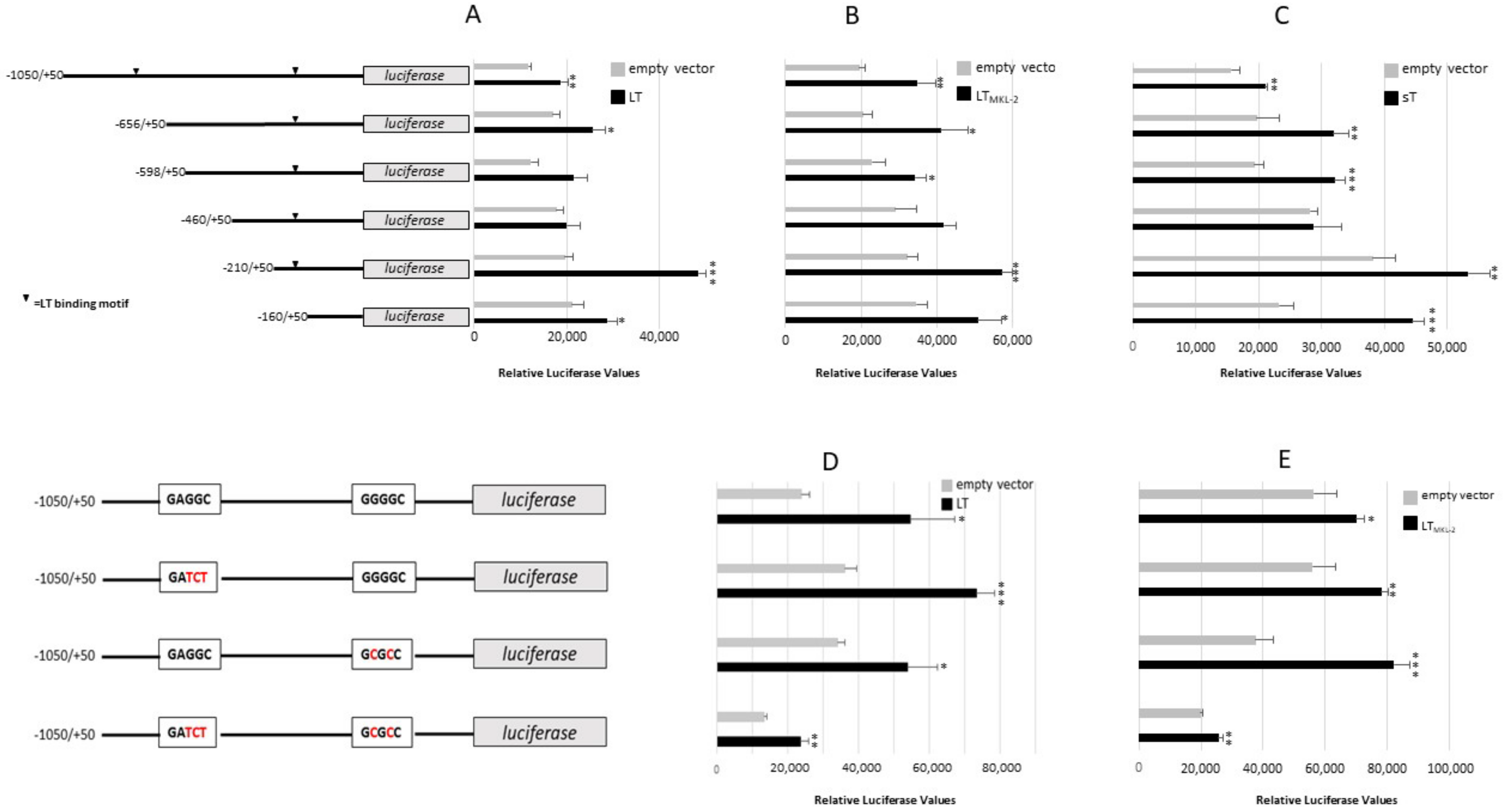
2.3. MCPyV T-Antigens Stimulates IL-33 Promoter Activity and Increases IL-33 Expression
2.4. MCPyV T-Ags Stimulate ST2/IL1RL1 and IL1RAcP Promoter Activity
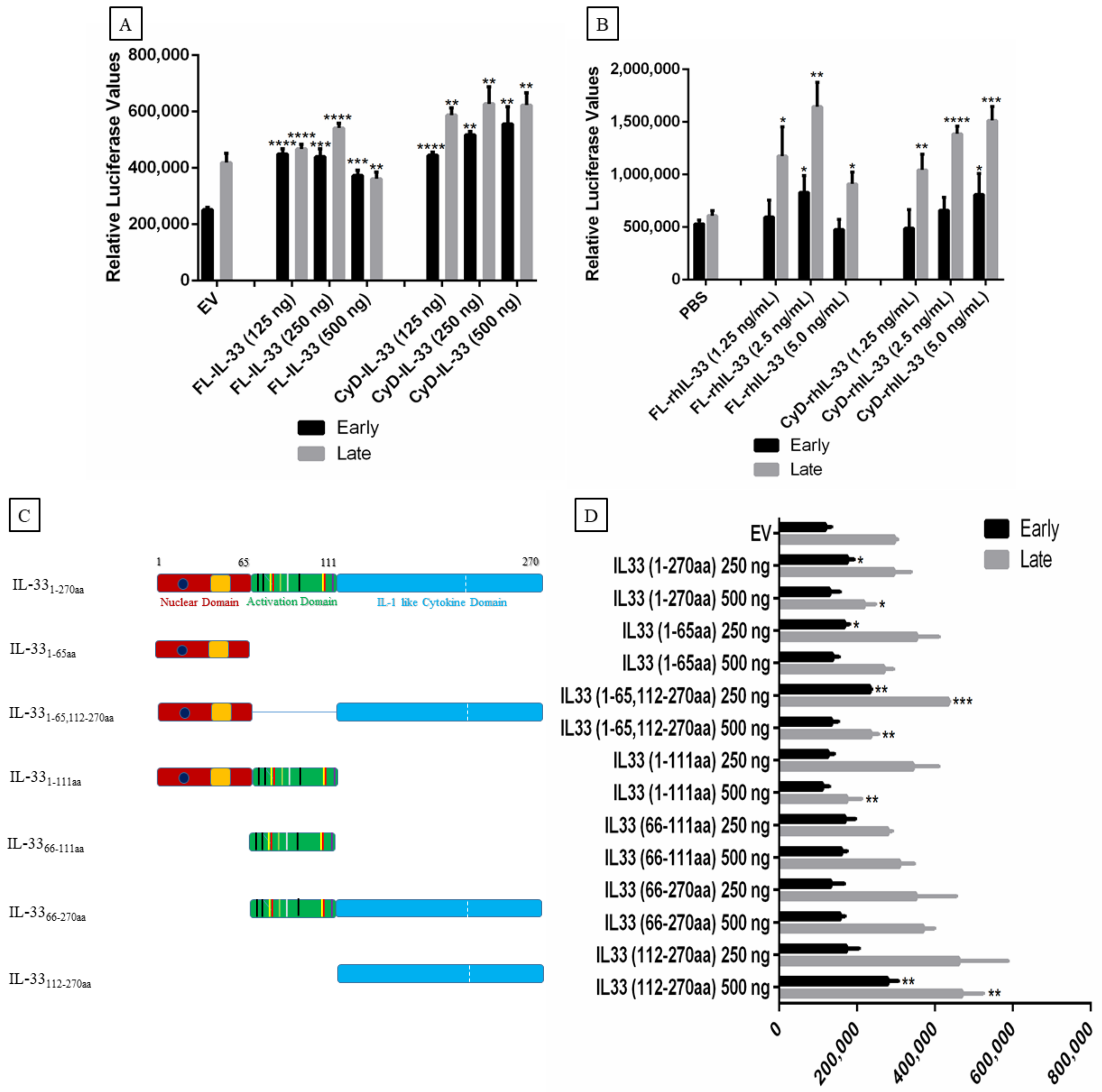
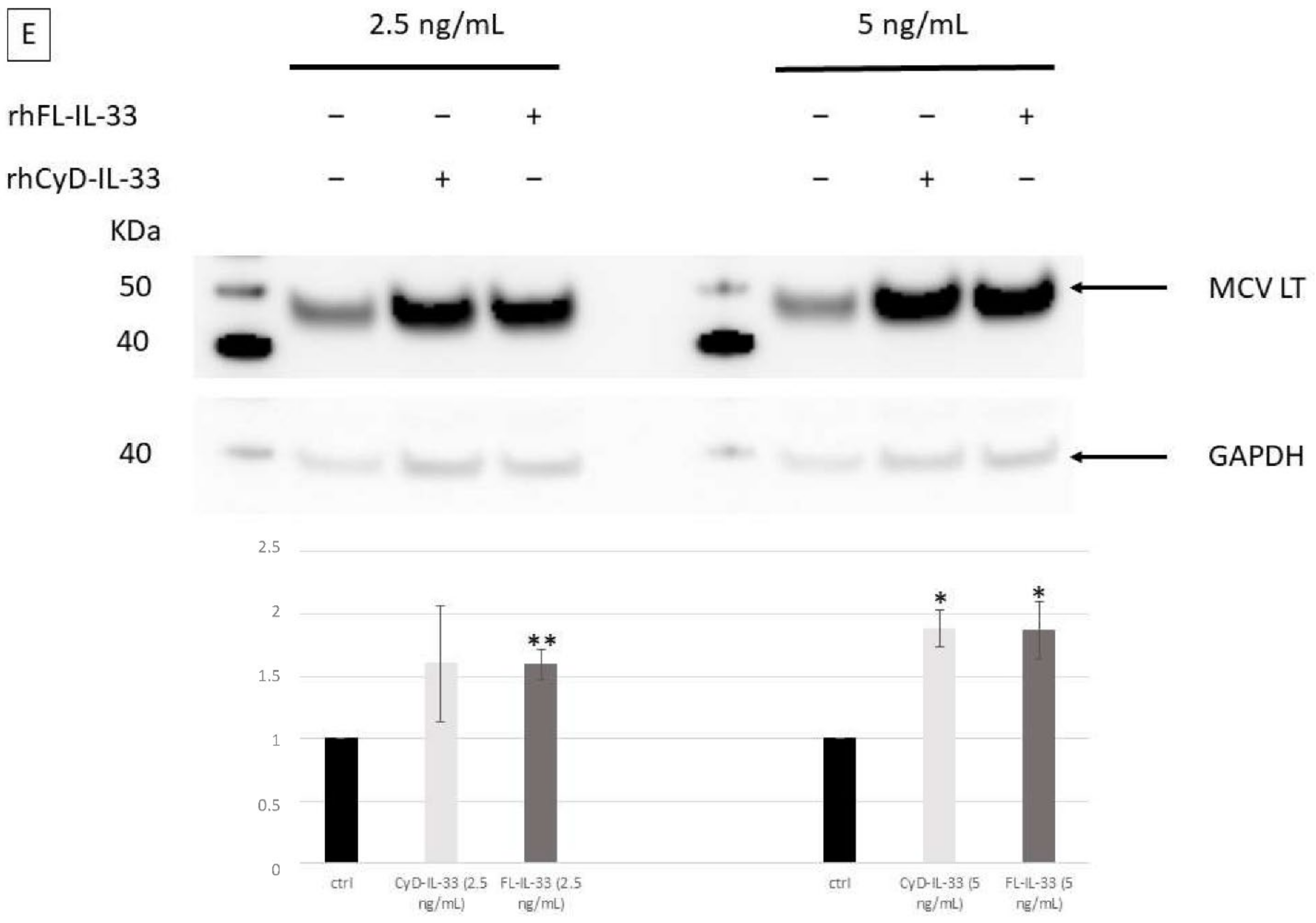
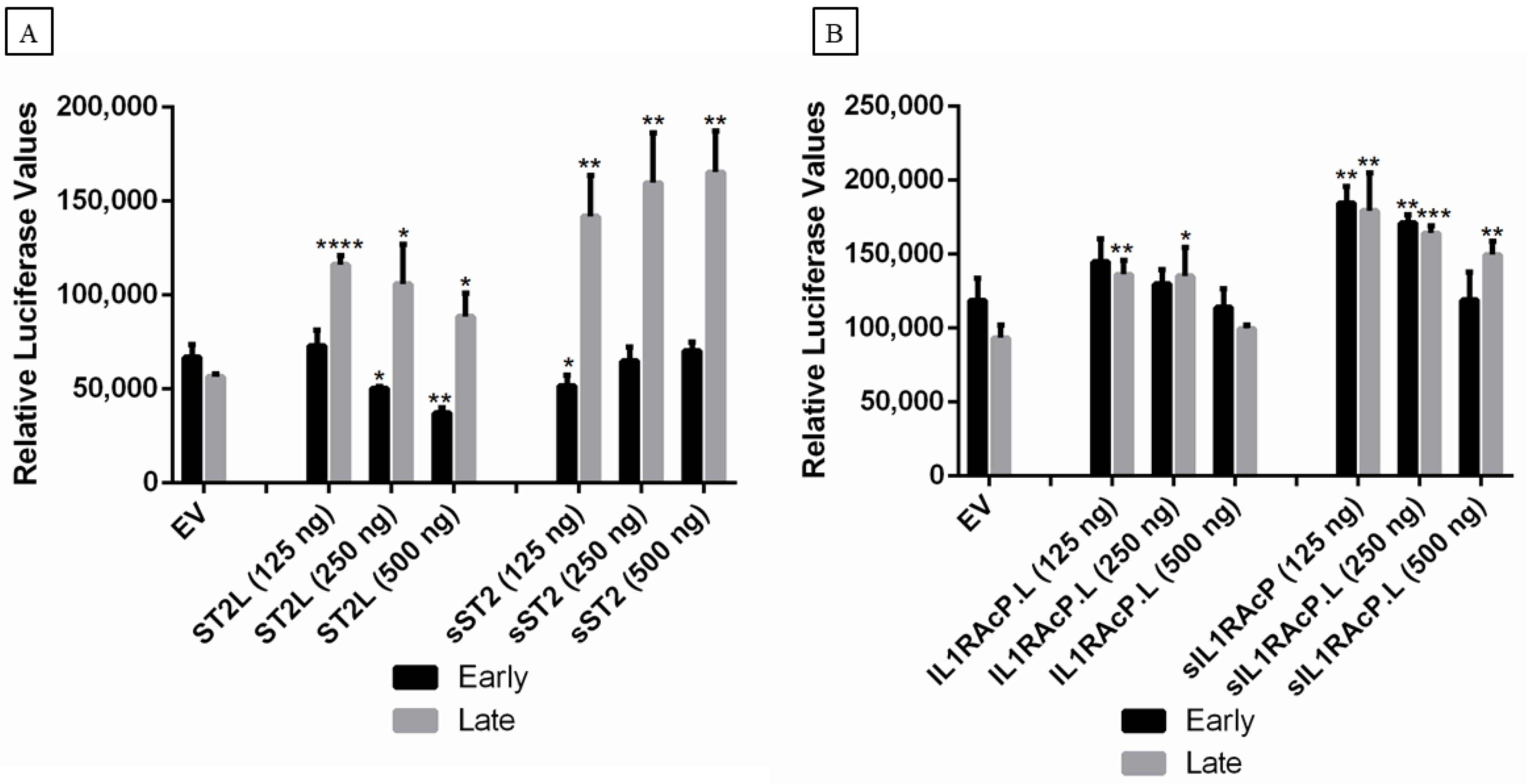
2.5. Effect of IL-33, ST2, and IL1RAcP on the Activity of the MCPyV Early and Late Promoter
2.6. Effect of IL-33, ST2/IL1RL1, and IL1RAcP on IL-33/ST2-IL1RAcP Complex Promoters
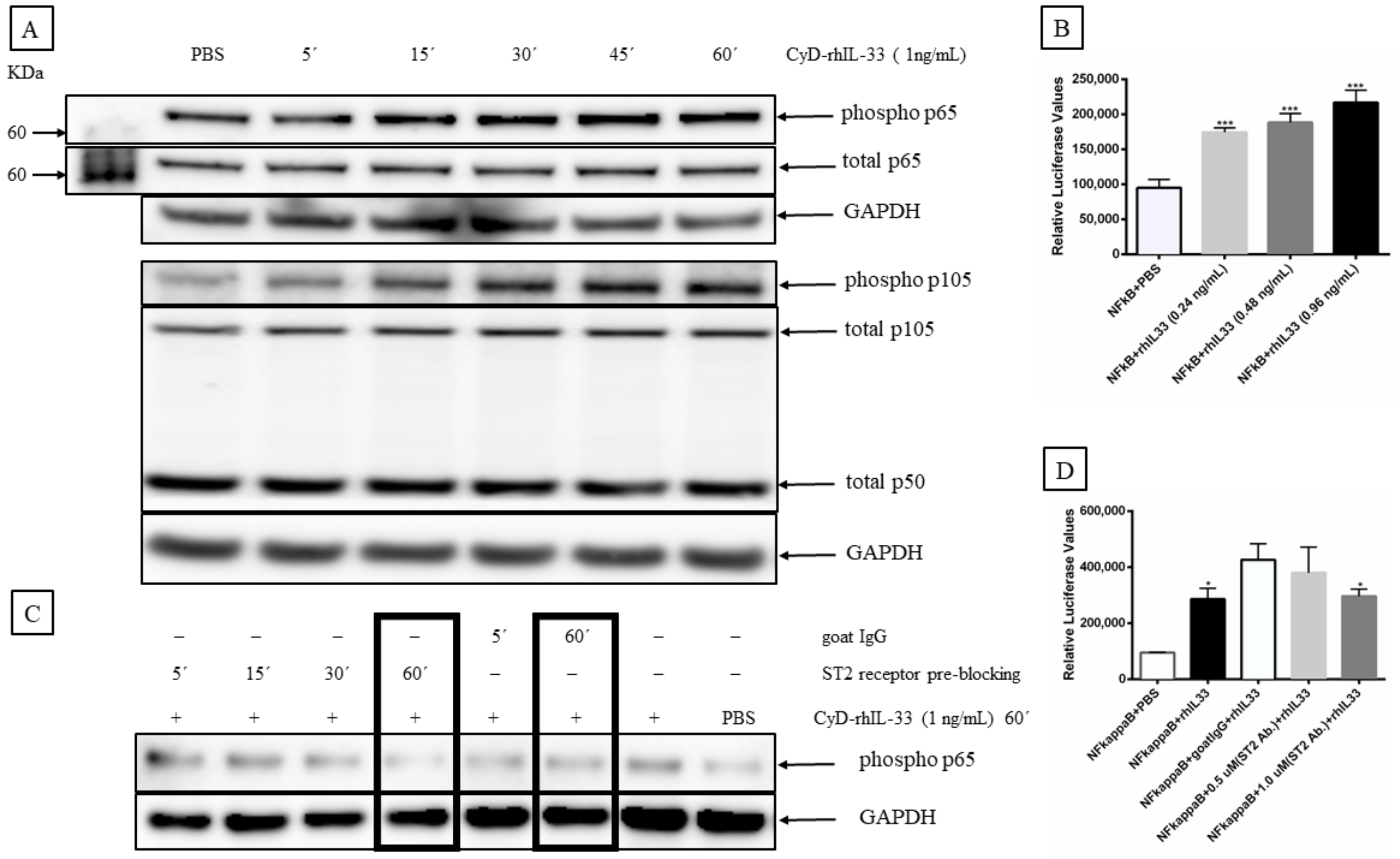
2.7. IL-33 Activates Mitogen-Activated Protein Kinase (MAP Kinase) and NF-κB Signaling Pathways in MCC Cells
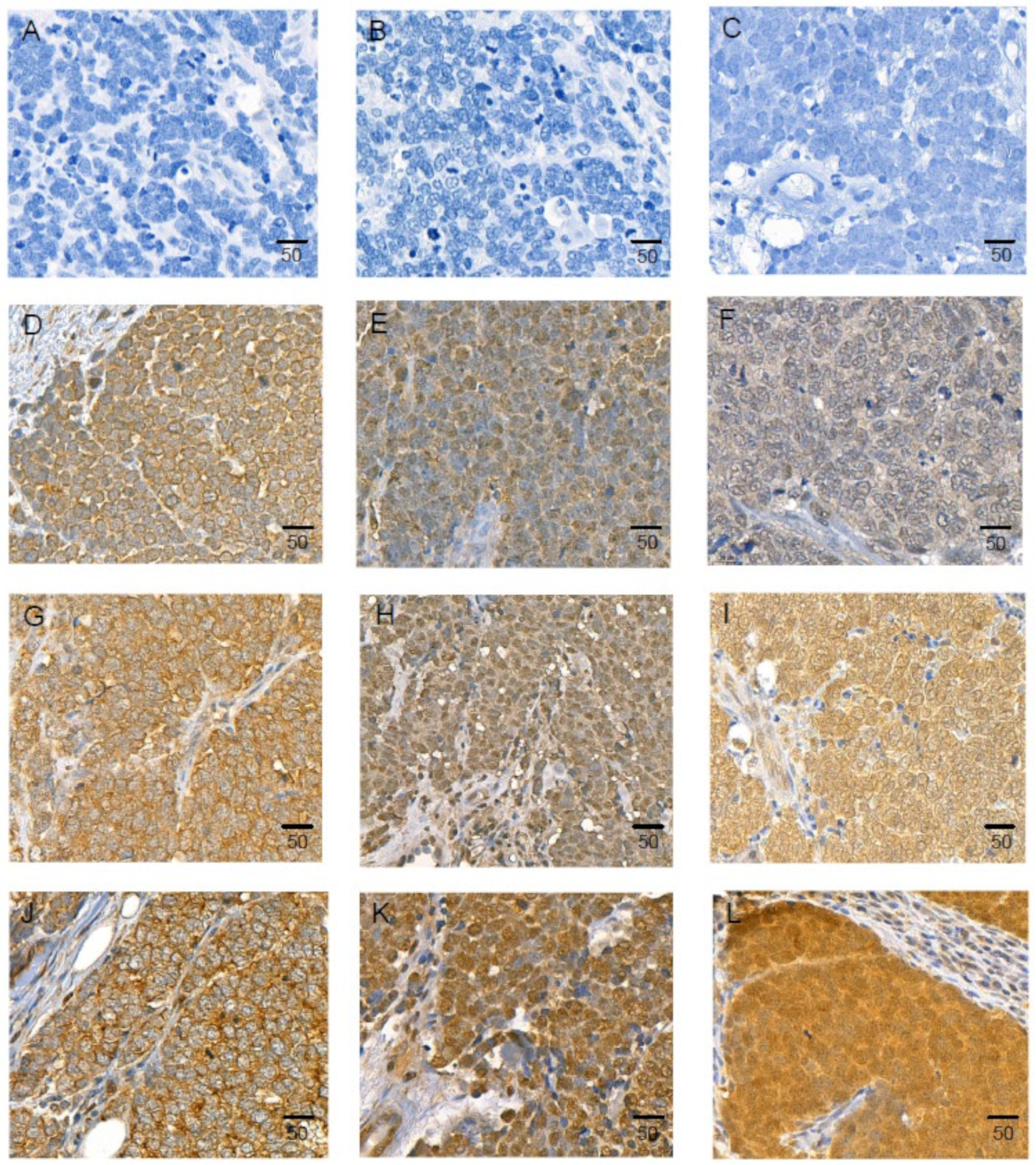
2.8. IL-33 and Its Receptors, ST2/IL1RL1, and IL1RAcP Are Expressed in MCC Tissue Samples
2.9. IL-33, sST2, and sIL1RAcP Measurement in Human Patient Plasma Samples
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Plasmids
4.3. Cell Lines and Human Tissue Samples
4.4. Transfection
4.5. RNA Extraction
4.6. cDNA Construction and Quality Control
4.7. RT2 Profile PCR Array
4.8. Luciferase Assays
4.9. Immunoblotting
4.10. Immunohistochemistry
4.11. Plasma Levels of IL33, ST2/IL1RL1, and IL1RAcP in MCC Patients and Healthy Controls
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Peñas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef]
- Lahman, M.C.; Paulson, K.G.; Nghiem, P.T.; Chapuis, A.G. Quality Is King: Fundamental Insights into Tumor Antigenicity from Virus-Associated Merkel Cell Carcinoma. J. Investig. Dermatol. 2021, 141, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Gheit, T.; Dutta, S.; Oliver, J.; Robitaille, A.; Hampras, S.; Combes, J.-D.; McKay-Chopin, S.; Le Calvez-Kelm, F.; Fenske, N.; Cherpelis, B.; et al. Isolation and characterization of a novel putative human polyomavirus. Virology 2017, 506, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Gjoerup, O.; Chang, Y. Update on Human Polyomaviruses and Cancer. Adv. Cancer Res. 2010, 106, 1–51. [Google Scholar] [CrossRef]
- Johne, R.; Buck, C.; Allander, T.; Atwood, W.J.; Garcea, R.L.; Imperiale, M.J.; Major, E.O.; Ramqvist, T.; Norkin, L.C. Taxonomical developments in the family Polyomaviridae. Arch. Virol. 2011, 156, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Buck, C.B. The Merkel Cell Polyomavirus Minor Capsid Protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef]
- Liu, W.; MacDonald, M.; You, J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr. Opin. Virol. 2016, 20, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Wieland, U.; Kreuter, A.; Pawlita, M. C-terminal deletions of Merkel cell polyomavirus large T-antigen, a highly specific surrogate marker for virally induced malignancy. Int. J. Cancer 2012, 131, 2863–2868. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Cheng, J.; Wardzala, J.; Dorosario, A.; Scanlon, J.J.; Laga, A.C.; Martinez-Fernandez, A.; Barletta, J.A.; Bellizzi, A.; Sadasivam, S.; et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J. Clin. Investig. 2012, 122, 4645–4653. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Korniluk, A.; Koper-Lenkiewicz, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.-X.; Liu, J.; Zhang, G.-J. Interleukin-33 in Malignancies: Friends or Foes? Front. Immunol. 2018, 9, 3051. [Google Scholar] [CrossRef]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Peña, M.M.O. The Role of IL-33/ST2 Pathway in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef] [PubMed]
- Lefrançais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.-P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Kim, S.; Lin, P.C. Interleukin-33 and ST2 Signaling in Tumor Microenvironment. J. Interferon Cytokine Res. 2019, 39, 61–71. [Google Scholar] [CrossRef]
- Wu, C.-W.; Wu, Y.-G.; Cheng, C.; Hong, Z.-D.; Shi, Z.-M.; Lin, S.-Q.; Li, J.; He, X.-Y.; Zhu, A.-Y. Interleukin-33 Predicts Poor Prognosis and Promotes Renal Cell Carcinoma Cell Growth Through its Receptor ST2 and the JNK Signaling Pathway. Cell. Physiol. Biochem. 2018, 47, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Barbour, M.; Hou, K.; Gao, C.; Cao, S.; Zheng, J.; Zhao, Y.; Mu, R.; Jiang, H.-R. Interleukin-33 predicts poor prognosis and promotes ovarian cancer cell growth and metastasis through regulating ERK and JNK signaling pathways. Mol. Oncol. 2016, 10, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Liu, J.; Xie, K.-G.; Lan, C.-G.; Lu, L.; Tang, Y.-J. Association between functional polymorphisms in IL-33/ST2 pathway and risk of osteosarcoma. J. Cell. Mol. Med. 2018, 22, 3808–3815. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-P.; Hu, M.-H.; Hsiao, Y.-P.; Wang, Y.-C. ST2 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, K.; Abdulsalam, I.; Fismen, S.; Grimstad, Ø.; Sveinbjørnsson, B.; Moens, U. CCL17/TARC and CCR4 expression in Merkel cell carcinoma. Oncotarget 2018, 9, 31432–31447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mehraj, V.; Ponte, R.; Routy, J.-P. The Dynamic Role of the IL-33/ST2 Axis in Chronic Viral-infections: Alarming and Adjuvanting the Immune Response. EBioMedicine 2016, 9, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Kulhan, N.G.; Kulhan, M.; Aydin, M.; Nayki, U.; Nayki, C.; Ulug, P.; Ata, N.; Mertoglu, C.; Cikman, A.; Sayar, I.; et al. Could interleukin-33 and its suppressor of tumorigenicity 2 (ST2) receptor have a role in cervical human papillomavirus (HPV) infections? Gynecol. Endocrinol. 2019, 35, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, W.; Zeng, P.; Xu, J.; Diao, H. The Contradictory Role of Interleukin-33 in Immune Cells and Tumor Immunity. Cancer Manag. Res. 2020, 12, 7527–7537. [Google Scholar] [CrossRef] [PubMed]
- Andreone, S.; Gambardella, A.R.; Mancini, J.; Loffredo, S.; Marcella, S.; La Sorsa, V.; Varricchi, G.; Schiavoni, G.; Mattei, F. Anti-Tumorigenic Activities of IL-33: A Mechanistic Insight. Front. Immunol. 2020, 11, 571593. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.E.; Besnard, A.-G.; Maillet, I.; Fagundes, C.T.; Souza, D.G.; Ryffel, B.; Teixeira, M.M.; Liew, F.Y.; Guabiraba, R. Interleukin-33 contributes to disease severity in Dengue virus infection in mice. Immunology 2018, 155, 477–490. [Google Scholar] [CrossRef]
- Moens, U.; Krumbholz, A.; Ehlers, B.; Zell, R.; Johne, R.; Calvignac-Spencer, S.; Lauber, C. Biology, evolution, and medical importance of polyomaviruses: An update. Infect. Genet. Evol. 2017, 54, 18–38. [Google Scholar] [CrossRef]
- Iwahana, H.; Yanagisawa, K.; Ito-Kosaka, A.; Kuroiwa, K.; Tago, K.; Komatsu, N.; Katashima, R.; Itakura, M.; Tominaga, S.-I. Different promoter usage and multiple transcription initiation sites of the interleukin-1 receptor-related human ST2 gene in UT-7 and TM12 cells. Eur. J. Biochem. 1999, 264, 397–406. [Google Scholar] [CrossRef]
- Bergers, G.; Reikerstorfer, A.; Braselmann, S.; Graninger, P.; Busslinger, M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J. 1994, 13, 1176–1188. [Google Scholar] [CrossRef]
- Abend, J.R.; Imperiale, M.J. Transforming growth factor-beta-mediated regulation of BK virus gene expression. Virology 2008, 378, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Enam, S.; Sweet, T.M.; Amini, S.; Khalili, K.; Del Valle, L. Evidence for involvement of transforming growth factor beta1 signaling pathway in activation of JC virus in human immunodeficiency virus 1-associated progressive multifocal leukoencephalopathy. Arch. Pathol. Lab. Med. 2004, 128, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Choi, E.-C.; Jo, Y.W.; Henson, J.W.; Kim, H.-S. Transcriptional activation of JC virus early promoter by phorbol ester and interleukin-1β: Critical role of nuclear factor-1. Virology 2004, 327, 60–69. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Atwood, W.J.; Wang, L.; Durham, L.C.; Amemiya, K.; Traub, R.G.; Major, E.O. Evaluation of the role of cytokine activation in the multiplication of JC virus (JCV) in human fetal glial cells. J. Neurovirol. 1995, 1, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.-L.; Zhao, Y.-R.; Weng, G.-B.; Chen, Y.-C.; Wei, X.-N.; Shao, J.-P.; Ji, H. IL-33-induced JNK pathway activation confers gastric cancer chemotherapy resistance. Oncol. Rep. 2015, 33, 2746–2752. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.L.; Williams, J.O.; Chan, Y.-H.; Laubertová, L.; Gallagher, H.; Moss, J.; Ramji, D.P. The interleukin-33-mediated inhibition of expression of two key genes implicated in atherosclerosis in human macrophages requires MAP kinase, phosphoinositide 3-kinase and nuclear factor-κB signaling pathways. Sci. Rep. 2019, 9, 11317. [Google Scholar] [CrossRef] [PubMed]
- Mine, Y.; Makihira, S.; Yamaguchi, Y.; Tanaka, H.; Nikawa, H. Involvement of ERK and p38 MAPK pathways on Interleukin-33-induced RANKL expression in osteoblastic cells. Cell Biol. Int. 2014, 38, 655–662. [Google Scholar] [CrossRef]
- Fang, M.; Li, Y.; Huang, K.; Qi, S.; Zhang, J.; Zgodzinski, W.; Majewski, M.; Wallner, G.; Gozdz, S.; Macek, P.; et al. IL33 Promotes Colon Cancer Cell Stemness via JNK Activation and Macrophage Recruitment. Cancer Res. 2017, 77, 2735–2745. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-S.; Park, J.A.; Kim, J.; Rho, S.-S.; Park, H.; Kim, Y.-M.; Kwon, Y.-G. Nuclear IL-33 is a transcriptional regulator of NF-κB p65 and induces endothelial cell activation. Biochem. Biophys. Res. Commun. 2012, 421, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF- B Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Griffiths, D.A.; Abdul-Sada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.S.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel Cell Polyomavirus Small T Antigen Targets the NEMO Adaptor Protein To Disrupt Inflammatory Signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef]
- Richards, K.F.; Guastafierro, A.; Shuda, M.; Toptan, T.; Moore, P.; Chang, Y. Merkel cell polyomavirus T antigens promote cell proliferation and inflammatory cytokine gene expression. J. Gen. Virol. 2015, 96, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Krump, N.A.; Wang, R.; Liu, W.; Yang, J.F.; Ma, T.; You, J. Merkel Cell Polyomavirus Infection Induces an Antiviral Innate Immune Response in Human Dermal Fibroblasts. J. Virol. 2021, 95, 0221120. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, E.; Heo, J.-S.; Bae, D.-J.; Lee, J.-U.W.; Lee, T.-H.; Lee, H.J.; Chang, H.S.; Park, J.S.; Jang, A.S.; et al. Circulating IL-33 level is associated with the progression of lung cancer. Lung Cancer 2015, 90, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-P.; Ling, D.-Y.; Xie, Y.-H.; Wu, W.-X.; Li, J.-R.; Jiang, J.; Zheng, J.-L.; Fan, Y.-H.; Zhang, Y. The Association of Serum IL-33 and sST2 with Breast Cancer. Dis. Markers 2015, 2015, 516895. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Minaee, K.; Farsinejad, A.R.; Nemati, M.; Khosravimashizi, A.; Daneshvar, H.; Mohammadi, M.M.; Sheikhi, A.; Ghaderi, A. Evaluation of the circulating levels of IL-12 and IL-33 in patients with breast cancer: Influences of the tumor stages and cytokine gene polymorphisms. Iran. J. Basic Med. Sci. 2015, 18, 1189–1198. [Google Scholar]
- Sun, P.; Ben, Q.; Tu, S.; Dong, W.; Qi, X.; Wu, Y. Serum Interleukin-33 Levels in Patients with Gastric Cancer. Dig. Dis. Sci. 2011, 56, 3596–3601. [Google Scholar] [CrossRef] [PubMed]
- Kieler, M.; Unseld, M.; Wojta, J.; Kaider, A.; Bianconi, D.; Demyanets, S.; Prager, G.W. Plasma levels of interleukin-33 and soluble suppression of tumorigenicity 2 in patients with advanced pancreatic ductal adenocarcinoma undergoing systemic chemotherapy. Med. Oncol. 2018, 36, 95. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, C.; Amako, Y.; Harold, A.; Toptan, T.; Chang, Y.; Shuda, M. Characterization of a Merkel Cell Polyomavirus-Positive Merkel Cell Carcinoma Cell Line CVG-1. Front. Microbiol. 2018, 9, 713. [Google Scholar] [CrossRef]
- Moens, U.; Macdonald, A. Effect of the Large and Small T-Antigens of Human Polyomaviruses on Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 3914. [Google Scholar] [CrossRef]
- Messeguer, X.; Escudero, R.; Farre, D.; Núñez, O.; Martınez, J.; Albà, M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef]
- Gorbacheva, A.M.; Kuprash, D.V.; Mitkin, N.A. Regulation of IL33 Gene Expression by SP1 and Foxa1 in Breast and Lung Cancer Cells. Mol. Biol. 2021, 55, 107–117. [Google Scholar] [CrossRef]
- Sontag, E.; Fedorov, S.; Kamibayashi, C.; Robbins, D.; Cobb, M.; Mumby, M. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 1993, 75, 887–897. [Google Scholar] [CrossRef]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.-A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.-P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef]
- Shao, D.; Perros, F.; Caramori, G.; Meng, C.; Dormuller, P.; Chou, P.-C.; Church, C.; Papi, A.; Casolari, P.; Welsh, D.; et al. Nuclear IL-33 regulates soluble ST2 receptor and IL-6 expression in primary human arterial endothelial cells and is decreased in idiopathic pulmonary arterial hypertension. Biochem. Biophys. Res. Commun. 2014, 451, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Tao, L.; Chen, C.; Song, H.; Li, Z.; Gao, Y.; Nie, J.; Piccioni, M.; Shi, G.; Li, B. The Deubiquitinase USP17 Regulates the Stability and Nuclear Function of IL-33. Int. J. Mol. Sci. 2015, 16, 27956–27966. [Google Scholar] [CrossRef] [PubMed]
- Lefrançais, E.; Roga, S.; Gautier, V.; Gonzalez-De-Peredo, A.; Monsarrat, B.; Girard, J.-P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef]
- Smithgall, M.D.; Comeau, M.R.; Yoon, B.-R.P.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK Cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Waldman, J.D.; Swensson, R.E. Therapeutic cardiac catheterization in children. West. J. Med. 1990, 153, 288–295. [Google Scholar]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lim, S.-C.; Kim, G.; Yun, H.J.; Ahn, S.-G.; Choi, H.S. Interleukin-33/ST2 axis promotes epithelial cell transformation and breast tumorigenesis via upregulation of COT activity. Oncogene 2015, 34, 4928–4938. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-X.; Hu, Z.; Shen, X.; Dong, L.-Y.; Zhou, W.-Z.; Hu, W.-H. IL-33 Promotes Gastric Cancer Cell Invasion and Migration Via ST2-ERK1/2 Pathway. Dig. Dis. Sci. 2015, 60, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-F.; Tao, T.; Wang, K.; Zhang, G.-X.; Yan, Y.; Lin, H.-R.; Li, Y.; Guan, M.-W.; Yu, J.-J.; Wang, X.-D. IL-33/ST2 axis promotes glioblastoma cell invasion by accumulating tenascin-C. Sci. Rep. 2019, 9, 20276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shi, J.; Qi, S.; Zhang, J.; Peng, D.; Chen, Z.; Wang, G.; Wang, Z.; Wang, L. IL-33 facilitates proliferation of colorectal cancer dependent on COX2/PGE2. J. Exp. Clin. Cancer Res. 2018, 37, 196. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-H.; Sharrocks, A.D.; Whitmarsh, A.J. Transcriptional regulation by the MAP kinase signaling cascades. Gene 2003, 320, 3–21. [Google Scholar] [CrossRef]
- Moens, U.; Prezioso, C.; Pietropaolo, V. Genetic Diversity of the Noncoding Control Region of the Novel Human Polyomaviruses. Viruses 2020, 12, 1406. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11, eaax2945. [Google Scholar] [CrossRef]
- Chinthrajah, S.; Cao, S.; Liu, C.; Lyu, S.-C.; Sindher, S.B.; Long, A.; Sampath, V.; Petroni, D.; Londei, M.; Nadeau, K.C. Phase 2a randomized, placebo-controlled study of anti-IL-33 in peanut allergy. JCI Insight 2019, 4, e131347. [Google Scholar] [CrossRef]
- Kelsen, S.G.; Agache, I.O.; Soong, W.; Israel, E.; Chupp, G.L.; Cheung, D.S.; Theess, W.; Yang, X.; Staton, T.L.; Choy, D.F.; et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J. Allergy Clin. Immunol. 2021, 148, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Y.; Tsai, T.-H.; Yang, J.-L.; Li, L.-C. Therapeutic Strategies for Targeting IL-33/ST2 Signalling for the Treatment of Inflammatory Diseases. Cell. Physiol. Biochem. 2018, 49, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Lian, J.; Yue, Y.; Zhang, Y. IL-33/ST2 as a potential target for tumor immunotherapy. Eur. J. Immunol. 2021, 51, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Knight, L.M.; Stakaityte, G.; Jennifer, J.W.; Abdul-Sada, H.; Griffiths, D.A.; Howell, G.J.; Wheat, R.; Blair, G.E.; Steven, N.M.; Macdonald, A.; et al. Merkel Cell Polyomavirus Small T Antigen Mediates Microtubule Destabilization to Promote Cell Motility and Migration. J. Virol. 2015, 89, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.H.; Dash, P.; Holland, P.; Kearsley, J.H.; Bell, J.R. Characterisation of four merkel cell carcinoma adherent cell lines. Int. J. Cancer 1995, 60, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ronan, S.G.; Green, A.D.; Shilkaitis, A.; Huang, T.-S.W.; Das Gupta, T. Merkel cell carcinoma: In vitro and in vivo characteristics of a new cell line. J. Am. Acad. Dermatol. 1993, 29, 715–722. [Google Scholar] [CrossRef]
- Rosen, S.T.; Gould, V.E.; Salwen, H.R.; Herst, C.V.; Le Beau, M.M.; Lee, I.; Bauer, K.; Marder, R.J.; Andersen, R.; Kies, M.S. Establishment and characterization of a neuroendocrine skin carcinoma cell line. Lab. Investig. 1987, 56, 302–312. [Google Scholar] [PubMed]
- Houben, R.; Grimm, J.; Willmes, C.; Weinkam, R.; Becker, J.C.; Schrama, D. Merkel Cell Carcinoma and Merkel Cell Polyomavirus: Evidence for Hit-and-Run Oncogenesis. J. Investig. Dermatol. 2012, 132, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.; Becker, J.C. Merkel Cell Polyomavirus-Infected Merkel Cell Carcinoma Cells Require Expression of Viral T Antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [PubMed]
- Guastafierro, A.; Feng, H.; Thant, M.; Kirkwood, J.M.; Chang, Y.; Moore, P.; Shuda, M. Characterization of an early passage Merkel cell polyomavirus-positive Merkel cell carcinoma cell line, MS-1, and its growth in NOD scid gamma mice. J. Virol. Methods 2013, 187, 6–14. [Google Scholar] [CrossRef]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Merkel Cell Polyomavirus Infection, Large T Antigen, Retinoblastoma Protein and Outcome in Merkel Cell Carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Ishida, T.; Hatake, K.; Taniwaki, M.; Ando, K.; Tobinai, K.; Fujimoto, K.; Yamamoto, K.; Miyamoto, T.; Uike, N.; et al. Multicenter Phase II Study of Mogamulizumab (KW-0761), a Defucosylated Anti-CC Chemokine Receptor 4 Antibody, in Patients With Relapsed Peripheral T-Cell Lymphoma and Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2014, 32, 1157–1163. [Google Scholar] [CrossRef] [PubMed]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasheed, K.; Moens, U.; Policastro, B.; Johnsen, J.I.; Koljonen, V.; Sihto, H.; Lui, W.-O.; Sveinbjørnsson, B. The Merkel Cell Polyomavirus T-Antigens and IL-33/ST2-IL1RAcP Axis: Possible Role in Merkel Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3702. https://doi.org/10.3390/ijms23073702
Rasheed K, Moens U, Policastro B, Johnsen JI, Koljonen V, Sihto H, Lui W-O, Sveinbjørnsson B. The Merkel Cell Polyomavirus T-Antigens and IL-33/ST2-IL1RAcP Axis: Possible Role in Merkel Cell Carcinoma. International Journal of Molecular Sciences. 2022; 23(7):3702. https://doi.org/10.3390/ijms23073702
Chicago/Turabian StyleRasheed, Kashif, Ugo Moens, Benedetta Policastro, John Inge Johnsen, Virve Koljonen, Harri Sihto, Weng-Onn Lui, and Baldur Sveinbjørnsson. 2022. "The Merkel Cell Polyomavirus T-Antigens and IL-33/ST2-IL1RAcP Axis: Possible Role in Merkel Cell Carcinoma" International Journal of Molecular Sciences 23, no. 7: 3702. https://doi.org/10.3390/ijms23073702
APA StyleRasheed, K., Moens, U., Policastro, B., Johnsen, J. I., Koljonen, V., Sihto, H., Lui, W.-O., & Sveinbjørnsson, B. (2022). The Merkel Cell Polyomavirus T-Antigens and IL-33/ST2-IL1RAcP Axis: Possible Role in Merkel Cell Carcinoma. International Journal of Molecular Sciences, 23(7), 3702. https://doi.org/10.3390/ijms23073702