
Vascular Permeability in Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanism of Permeability
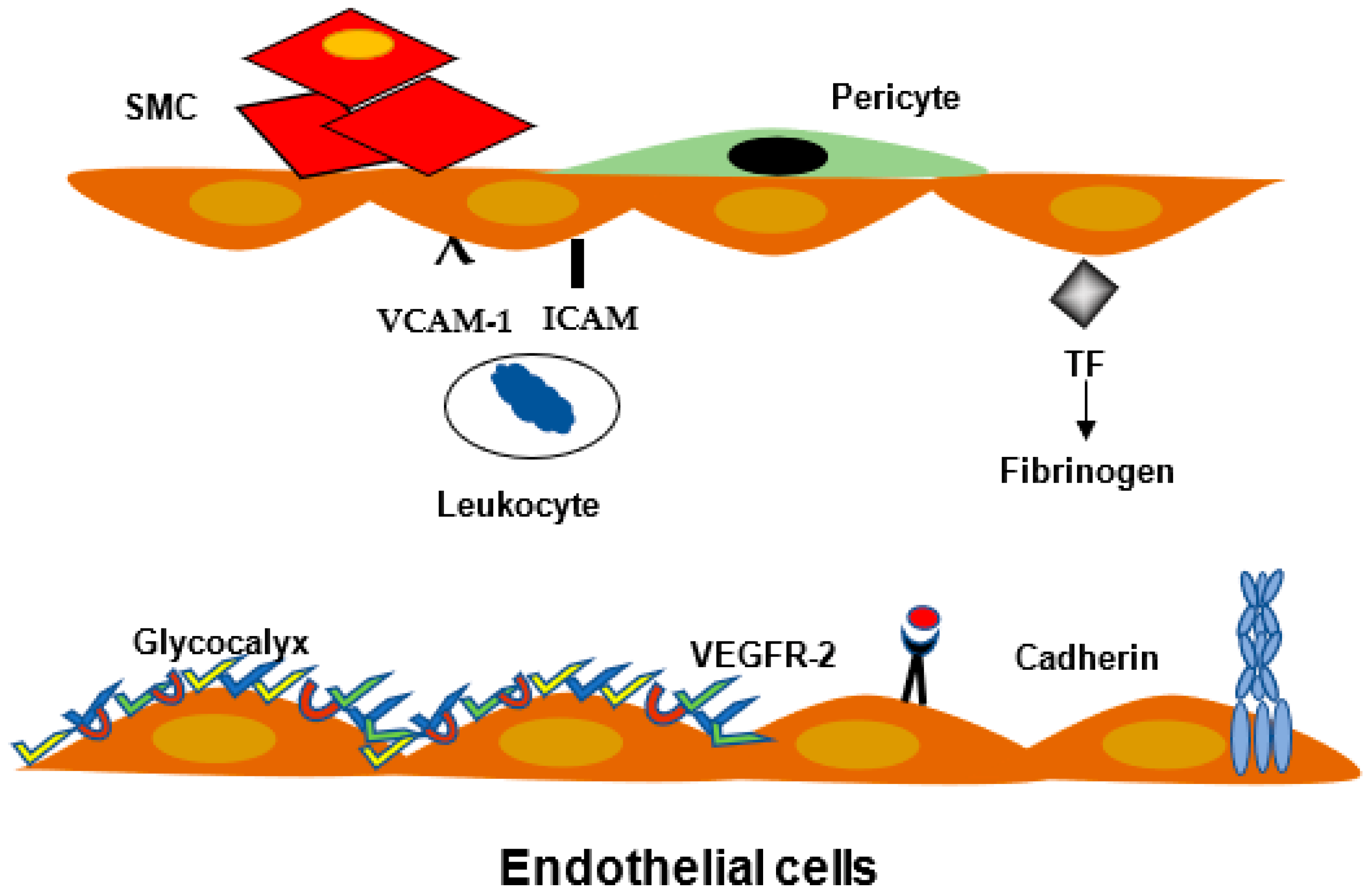
2.1. Definition
- -
- The glycocalyx covered the endothelial cell surface, and it is in contact with blood components.
- -
- The transendothelial trafficking is energy dependent pathway.
- -
- Opening of cell/cell modification.
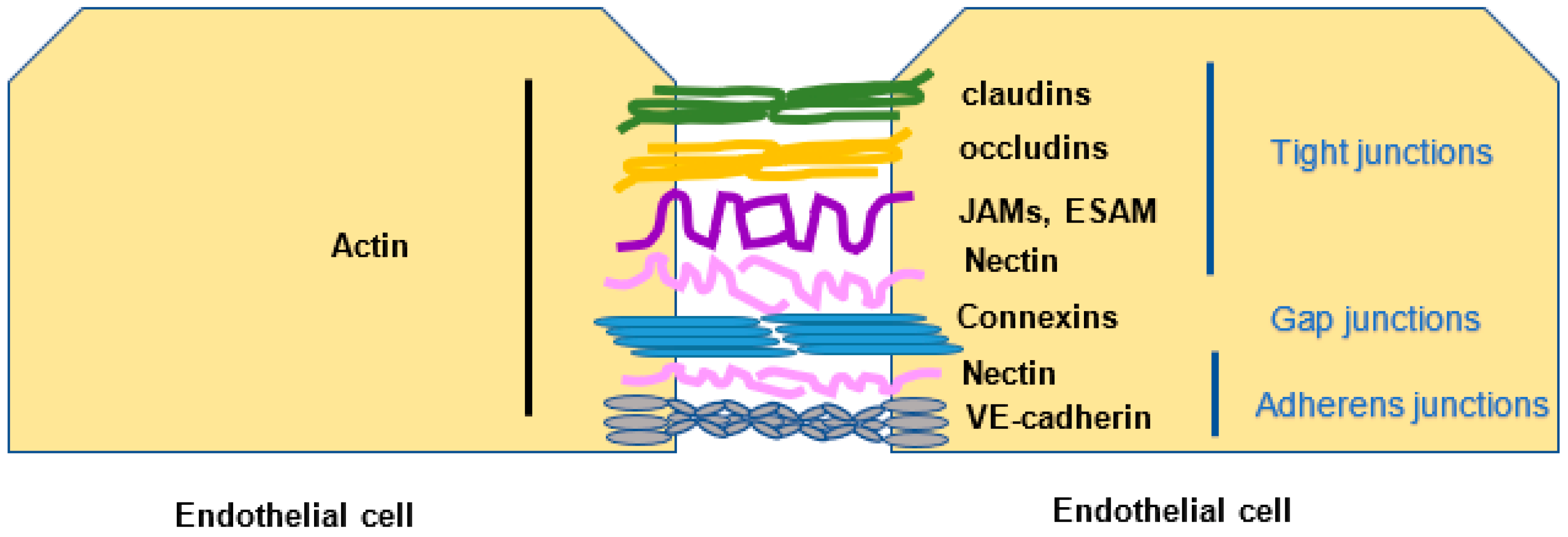
2.2. Endothelial Cell Junctions
2.3. Leukocyte Migration and Inflammation
2.4. Blood Brain Barrier
3. Modulation of Vascular Permeability
3.1. Prostaglandins
3.2. VEGF
4. Vascular Permeability in Diseases
4.1. Pulmonary Edema
4.2. Cerebral Edema
4.3. Infectious Diseases
4.4. Cancer
4.5. Diabetes Mellitus
4.5.1. Vascular Permeability in Kidney
4.5.2. Retinopathy
4.6. Permeability and Atherogenesis
5. Conclusions, Treatment, and Perspectives
Funding
Informed Consent Statement
Conflicts of Interest
References
- Broman, L.M.; Wittberg, L.P.; Westlund, C.J.; Gilbers, M.; Da Câmara, L.P.; Swol, J.; Taccone, F.S.; Malfertheiner, M.V.; Di Nardo, M.; Vercaemst, L.; et al. Pressure and flow properties of cannulae for extracorporeal membrane oxygenation I: Return (arterial) cannulae. Perfusion 2019, 34, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.E. Capillary fluid filtration. Starling forces and lymph flow. Circ. Res. 1981, 49, 557–575. [Google Scholar] [CrossRef]
- Grotte, G. Passage of dextran molecules across the blood-lymph barrier. Acta Chir. Scand. Suppl. 1956, 211, 1–84. [Google Scholar] [PubMed]
- Levick, J.R.; Michel, C.C. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010, 87, 198–210. [Google Scholar] [CrossRef]
- Claesson-Welsh, L. Vascular permeability—The essentials. Ups J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Hautefort, A.; Pfenniger, A.; Kwak, B.R. Endothelial connexins in vascular function. Vasc. Biol. 2019, 1, H117–H124. [Google Scholar] [CrossRef]
- Butler, M.J.; Down, C.J.; Foster, R.; Satchell, S.C. The Pathological Relevance of Increased Endothelial Glycocalyx Permeability. Am. J. Pathol. 2020, 190, 742–751. [Google Scholar] [CrossRef]
- Wautier, J.-L.; Wautier, M.-P. Cellular and Molecular Aspects of Blood Cell–Endothelium Interactions in Vascular Disorders. Int. J. Mol. Sci. 2020, 21, 5315. [Google Scholar] [CrossRef]
- Miao, H.; Li, S.; Hu, Y.L.; Yuan, S.; Zhao, Y.; Chen, B.P.; Puzon-McLaughlin, W.; Tarui, T.; Shyy, J.Y.; Takada, Y.; et al. Differential regulation of Rho GTPases by beta1 and beta3 integrins: The role of an extracellular domain of integrin in intracellular signaling. J. Cell Sci. 2002, 115, 2199–2206. [Google Scholar] [CrossRef]
- Wang, Y.; Miao, H.; Li, S.; Chen, K.-D.; Li, Y.-S.; Yuan, S.; Shyy, J.Y.-J.; Chien, S. Interplay between integrins and FLK-1 in shear stress-induced signaling. Am. J. Physiol. Physiol. 2002, 283, C1540–C1547. [Google Scholar] [CrossRef]
- Meuwese, M.C.; Stroes, E.S.; Hazen, S.L.; van Miert, J.N.; Kuivenhoven, J.A.; Schaub, R.G.; Wareham, N.J.; Luben, R.; Kastelein, J.J.; Khaw, K.-T.; et al. Serum Myeloperoxidase Levels Are Associated with the Future Risk of Coronary Artery Disease in Apparently Healthy Individuals: The EPIC-Norfolk Prospective Population Study. J. Am. Coll. Cardiol. 2007, 50, 159–165. [Google Scholar] [CrossRef]
- Karakas, M.; Koenig, W.; Zierer, A.; Herder, C.; Rottbauer, W.; Baumert, J.; Meisinger, C.; Thorand, B. Myeloperoxidase is associated with incident coronary heart disease independently of traditional risk factors: Results from the MONICA/KORA Augsburg study. J. Intern. Med. 2011, 271, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Rom, S.; Ramirez, S.; Persidsky, Y. Emerging Roles of Pericytes in the Regulation of the Neurovascular Unit in Health and Disease. J. Neuroimmune Pharmacol. 2014, 9, 591–605. [Google Scholar] [CrossRef]
- Caporarello, N.; D’Angeli, F.; Cambria, M.T.; Candido, S.; Giallongo, C.; Salmeri, M.; Lombardo, C.; Longo, A.; Giurdanella, G.; Anfuso, C.D.; et al. Pericytes in Microvessels: From “Mural” Function to Brain and Retina Regeneration. Int. J. Mol. Sci. 2019, 20, 6351. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.-P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef]
- Omori, K.; Kida, T.; Hori, M.; Ozaki, H.; Murata, T. Multiple roles of the PGE2-EP receptor signal in vascular permeability. J. Cereb. Blood Flow Metab. 2014, 171, 4879–4889. [Google Scholar] [CrossRef]
- Lee, S.; Chen, T.T.; Barber, C.L.; Jordan, M.C.; Murdock, J.; Desai, S.; Ferrara, N.; Nagy, A.; Roos, K.P.; Iruela-Arispe, M.L. Autocrine VEGF Signaling Is Required for Vascular Homeostasis. Cell 2007, 130, 691–703. [Google Scholar] [CrossRef]
- Han, J.; Zhang, G.; Welch, E.J.; Liang, Y.; Fu, J.; Vogel, S.M.; Lowell, C.A.; Du, X.; Cheresh, D.A.; Malik, A.B.; et al. A critical role for Lyn kinase in strengthening endothelial integrity and barrier function. Blood 2013, 122, 4140–4149. [Google Scholar] [CrossRef] [PubMed]
- McAllister, R.M.; Albarracin, I.; Jasperse, J.L.; Price, E.M. Thyroid status and endothelium-dependent vasodilation in skeletal muscle. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R284–R291. [Google Scholar] [CrossRef] [PubMed]
- Mullick, A.E.; Zaid, U.B.; Athanassious, C.N.; Lentz, S.R.; Rutledge, J.C.; Symons, J.D. Hyperhomocysteinemia increases arterial permeability and stiffness in mice. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R1349–R1354. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.C.; Lominadze, D.; Roberts, A.M. Homocysteine in Microvascular Endothelial Cell Barrier Permeability. Cell Biophys. 2005, 43, 37–44. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef]
- Maggiorini, M.; Melot, C.; Pierre, S.; Pfeiffer, F.; Greve, I.; Sartori, C.; Lepori, M.; Hauser, M.; Scherrer, U.; Naeije, R. High-Altitude Pulmonary Edema Is Initially Caused by an Increase in Capillary Pressure. Circulation 2001, 103, 2078–2083. [Google Scholar] [CrossRef]
- Mammoto, A.; Mammoto, T.; Kanapathipillai, M.; Yung, C.W.; Jiang, E.; Jiang, A.; Lofgren, K.; Gee, E.P.; Ingber, D.E. Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat. Commun. 2013, 4, 1759. [Google Scholar] [CrossRef]
- Friedrich, E.E.; Hong, Z.; Xiong, S.; Di, A.; Rehman, J.; Komarova, Y.A.; Malik, A.B. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc. Natl. Acad. Sci. USA 2019, 116, 12980–12985. [Google Scholar] [CrossRef]
- Nassar, T.; Yarovoi, S.; Abu Fanne, R.; Akkawi, S.; Jammal, M.; Allen, T.C.; Idell, S.; Cines, D.B.; Higazi, A.A.-R. Regulation of Airway Contractility by Plasminogen Activators through N-Methyl-D-Aspartate Receptor–1. Am. J. Respir. Cell Mol. Biol. 2010, 43, 703–711. [Google Scholar] [CrossRef]
- Huang, L.; Cao, W.; Deng, Y.; Zhu, G.; Han, Y.; Zeng, H. Hypertonic saline alleviates experimentally induced cerebral oedema through suppression of vascular endothelial growth factor and its receptor VEGFR2 expression in astrocytes. BMC Neurosci. 2016, 17, 64. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A.; et al. Effect of SARS-CoV-2 proteins on vascular permeability. eLife 2021, 10, e69314. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M. Vascular Endothelial Glycocalyx Damage in COVID-19. Int. J. Mol. Sci. 2020, 21, 9712. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Rupprecht, C.; Haque, A.; Pattanaik, D.; Yusin, J.; Krishnaswamy, G. Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond. Int. J. Mol. Sci. 2021, 22, 7785. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Cui, J.; Barnes, L.; Cheresh, D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J. Cell Biol. 2004, 167, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Kato, M.; Hiratsuka, S. Regulation of vascular permeability in cancer metastasis. Cancer Sci. 2021, 112, 2966–2974. [Google Scholar] [CrossRef] [PubMed]
- Tannock, L.R.; De Beer, M.C.; Ji, A.; Shridas, P.; Noffsinger, V.P.; Hartigh, L.D.; Chait, A.; De Beer, F.C.; Webb, N.R. Serum amyloid A3 is a high density lipoprotein-associated acute-phase protein. J. Lipid Res. 2018, 59, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ma, X.; Jiang, D.; Wang, L.; Zhan, Q.; Zhao, J. CXC chemokine ligand 5 (CXCL5) disrupted the permeability of human brain microvascular endothelial cells via regulating p38 signal. Microbiol. Immunol. 2021, 65, 40–47. [Google Scholar] [CrossRef]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef]
- Wautier, M.-P.; Boulanger, E.; Guillausseau, P.-J.; Massin, P.; Wautier, J.-L. AGEs, macrophage colony stimulating factor and vascular adhesion molecule blood levels are increased in patients with diabetic microangiopathy. Thromb. Haemost. 2004, 91, 879–885. [Google Scholar] [CrossRef]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef]
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: A link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Gravallese, E.; Bunn, H.F. Nonenzymatic glycosylation of erythrocyte membrane proteins. Relevance to diabetes. J. Clin. Investig. 1980, 65, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.-L.; Paton, R.C.; Wautier, M.-P.; Pintigny, D.; Abadie, E.; Passa, P.; Caen, J.P. Increased Adhesion of Erythrocytes to Endothelial Cells in Diabetes Mellitus and Its Relation to Vascular Complications. N. Engl. J. Med. 1981, 305, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Zoukourian, C.; Chappey, O.; Wautier, M.P.; Guillausseau, P.J.; Cao, R.; Hori, O.; Stern, D.; Schmidt, A.M. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J. Clin. Investig. 1996, 97, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Tamarat, R.; Silvestre, J.-S.; Huijberts, M.; Benessiano, J.; Ebrahimian, T.G.; Duriez, M.; Wautier, M.-P.; Wautier, J.L.; Lévy, B.I. Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 8555–8560. [Google Scholar] [CrossRef]
- Wautier, J.L.; Schmidt, A.M. Protein glycation: A firm link to endothelial cell dysfunction. Circ. Res. 2004, 95, 233–238. [Google Scholar] [CrossRef]
- Cipollone, F.; Iezzi, A.; Fazia, M.; Zucchelli, M.; Pini, B.; Cuccurullo, C.; De Cesare, D.; De Blasis, G.; Muraro, R.; Bei, R.; et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: Role of glycemic control. Circulation 2003, 108, 1070–1077. [Google Scholar] [CrossRef]
- Makita, Z.; Radoff, S.; Rayfield, E.J.; Yang, Z.; Skolnik, E.; Delaney, V.; Friedman, E.A.; Cerami, A.; Vlassara, H. Advanced Glycosylation End Products in Patients with Diabetic Nephropathy. N. Engl. J. Med. 1991, 325, 836–842. [Google Scholar] [CrossRef]
- Weiss, M.F.; Erhard, P.; Kader-Attia, F.A.; Wu, Y.C.; Deoreo, P.B.; Araki, A.; Glomb, M.A.; Monnier, V.M. Mechanisms for the formation of glycoxidation products in end-stage renal disease. Kidney Int. 2000, 57, 2571–2585. [Google Scholar] [CrossRef]
- Sol, M.; Kamps, J.A.A.M.; Born, J.V.D.; Heuvel, M.C.V.D.; Van Der Vlag, J.; Krenning, G.; Hillebrands, J.-L. Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 2020, 11, 573557. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kato, I.; Doi, T.; Yonekura, H.; Ohashi, S.; Takeuchi, M.; Watanabe, T.; Yamagishi, S.; Sakurai, S.; Takasawa, S.; et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J. Clin. Investig. 2001, 108, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S.; et al. RAGE Drives the Development of Glomerulosclerosis and Implicates Podocyte Activation in the Pathogenesis of Diabetic Nephropathy. Am. J. Pathol. 2003, 162, 1123–1137. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, E.; Wautier, M.-P.; Wautier, J.-L.; Boval, B.; Panis, Y.; Wernert, N.; Danze, P.-M.; Dequiedt, P. AGEs bind to mesothelial cells via RAGE and stimulate VCAM-1 expression. Kidney Int. 2002, 61, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Vidal, P.; Cabezas-Cerrato, J. The stable products of the non-enzymatic glycation of pig crystallins: New findings related to the pathogenesis of diabetic cataracts. Diabetes Res. 1988, 8, 183–187. [Google Scholar]
- Stitt, A.; Gardiner, T.A.; Anderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; et al. The AGE Inhibitor Pyridoxamine Inhibits Development of Retinopathy in Experimental Diabetes. Diabetes 2002, 51, 2826–2832. [Google Scholar] [CrossRef]
- Hammes, H.-P.; Du, X.; Edelstein, D.; Taguchi, T.; Matsumura, T.; Ju, Q.; Lin, J.; Bierhaus, A.; Nawroth, P.; Hannak, D.; et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat. Med. 2003, 9, 294–299. [Google Scholar] [CrossRef]
- Zoukourian, C.; Wautier, M.P.; Chappey, O.; Dosquet, C.; Rohban, T.; Schmidt, A.M.; Stern, D.; Wautier, J.L. Endothelial cell dysfunction secondary to the adhesion of diabetic erythrocytes. Modulation by iloprost. Int. Angiol. 1996, 15, 195–200. [Google Scholar]
- Miller, J.W.; Le Couter, J.; Strauss, E.C.; Ferrara, N. Vascular Endothelial Growth Factor A in Intraocular Vascular Disease. Ophthalmology 2013, 120, 106–114. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Moore, L.L.; Brinck-Johnsen, T.; Curphey, T.J. Increased collagen-linked pentosidine levels and advanced glycosylation end products in early diabetic nephropathy. J. Clin. Investig. 1993, 92, 212–217. [Google Scholar] [CrossRef] [PubMed]
- McCance, D.R.; Dyer, D.G.; Dunn, J.A.; E Bailie, K.; Thorpe, S.R.; Baynes, J.W.; Lyons, T.J. Maillard reaction products and their relation to complications in insulin-dependent diabetes mellitus. J. Clin. Investig. 1993, 91, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 1106–1124. [Google Scholar] [CrossRef] [PubMed]
- Stein-Merlob, A.F.; Hara, T.; McCarthy, J.R.; Mauskapf, A.; Hamilton, J.A.; Ntziachristos, V.; Libby, P.; Jaffer, F.A. Atheroma Susceptible to Thrombosis Exhibit Impaired Endothelial Permeability In Vivo as Assessed by Nanoparticle-Based Fluorescence Molecular Imaging. Circ. Cardiovasc. Imaging 2017, 10, e005813. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Nentwich, M.M.; Ulbig, M.W. Diabetic retinopathy—Ocular complications of diabetes mellitus. World J. Diabet. 2015, 6, 489–499. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Wautier, J.L.; Stern, D. Activation of receptor for advanced glycation end products: A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999, 84, 489–497. [Google Scholar] [CrossRef]
- Bandello, F.; Cunha-Vaz, J.; Chong, V.; Lang, G.E.; Massin, P.; Mitchell, P.; Porta, M.; Prünte, C.; Schlingemann, R.; Schmidt-Erfurth, U. New approaches for the treatment of diabetic macular oedema: Recommendations by an expert panel. Eye 2012, 26, 485–493. [Google Scholar] [CrossRef]
- Glassman, A.R.; Wells, J.A., 3rd; Josic, K.; Maguire, M.G.; Antoszyk, A.N.; Baker, C.; Beaulieu, W.T.; Elman, M.J.; Jampol, L.M.; Sun, J.K. Five-Year Outcomes after Initial Aflibercept, Bevacizumab, or Ranibizumab Treatment for Diabetic Macular Edema (Protocol T Extension Study). Ophthalmology 2020, 127, 1201–1210. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Lewin, A.; Patel, S.; Liu, D.; Kaste, R.; Woerle, H.J.; Broedl, U.C. Combination of empagliflozin and linagliptin as second-line therapy in subjects with type 2 diabetes inadequately controlled on metformin. Diabet. Care 2015, 38, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Ways, K.; Desai, M.; Shaw, W.; Capuano, G.; Alba, M.; et al. Efficacy and Safety of Canagliflozin, an Inhibitor of Sodium–Glucose Cotransporter 2, When Used in Conjunction with Insulin Therapy in Patients with Type 2 Diabetes. Diabet. Care 2014, 38, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Nakashima, S.; Nishino, Y.; Ojima, A.; Nakamura, N.; Arima, K.; Fukami, K.; Okuda, S.; Yamagishi, S.-I. Dipeptidyl peptidase-4 deficiency protects against experimental diabetic nephropathy partly by blocking the advanced glycation end products-receptor axis. Lab. Investig. 2015, 95, 525–533. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mozos, I.; Flangea, C.; Vlad, D.; Gug, C.; Mozos, C.; Stoian, D.; Luca, C.; Horbańczuk, J.; Horbańczuk, O.; Atanasov, A. Effects of Anthocyanins on Vascular Health. Biomolecules 2021, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Caprnda, M.; Zulli, A.; Shiwani, H.A.; Kubatka, P.; Filipova, S.; Valentova, V.; Gazdikova, K.; Mozos, I.; Berukstis, A.; Laucevicius, A.; et al. The therapeutic effect of B-type natriuretic peptides in acute decompensated heart failure. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1120–1133. [Google Scholar] [CrossRef]
- Taqueti, V.R.; Di Carli, M.F.; Jerosch-Herold, M.; Sukhova, G.K.; Murthy, V.L.; Folco, E.J.; Kwong, R.Y.; Ozaki, C.K.; Belkin, M.; Nahrendorf, M.; et al. Increased Microvascularization and Vessel Permeability Associate with Active Inflammation in Human Atheromata. Circ. Cardiovasc. Imaging 2014, 7, 920–929. [Google Scholar] [CrossRef]
- Huang, C.-C.; Kao, K.-C.; Hsu, K.-H.; Ko, H.-W.; Li, L.-F.; Hsieh, M.-J.; Tsai, Y.-H. Effects of hydroxyethyl starch resuscitation on extravascular lung water and pulmonary permeability in sepsis-related acute respiratory distress syndrome *. Crit. Care Med. 2009, 37, 1948–1955. [Google Scholar] [CrossRef]
- Mei, H.; Campbell, J.M.; Paddock, C.M.; Lertkiatmongkol, P.; Mosesson, M.W.; Albrecht, R.; Newman, P.J. Regulation of Endothelial Cell Barrier Function by Antibody-driven Affinity Modulation of Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1). J. Biol. Chem. 2014, 289, 20836–20844. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wautier, J.-L.; Wautier, M.-P. Vascular Permeability in Diseases. Int. J. Mol. Sci. 2022, 23, 3645. https://doi.org/10.3390/ijms23073645
Wautier J-L, Wautier M-P. Vascular Permeability in Diseases. International Journal of Molecular Sciences. 2022; 23(7):3645. https://doi.org/10.3390/ijms23073645
Chicago/Turabian StyleWautier, Jean-Luc, and Marie-Paule Wautier. 2022. "Vascular Permeability in Diseases" International Journal of Molecular Sciences 23, no. 7: 3645. https://doi.org/10.3390/ijms23073645
APA StyleWautier, J.-L., & Wautier, M.-P. (2022). Vascular Permeability in Diseases. International Journal of Molecular Sciences, 23(7), 3645. https://doi.org/10.3390/ijms23073645