
Ketogenic Diet Consumption Inhibited Mitochondrial One-Carbon Metabolism

 and
and
Abstract
1. Introduction
2. Results
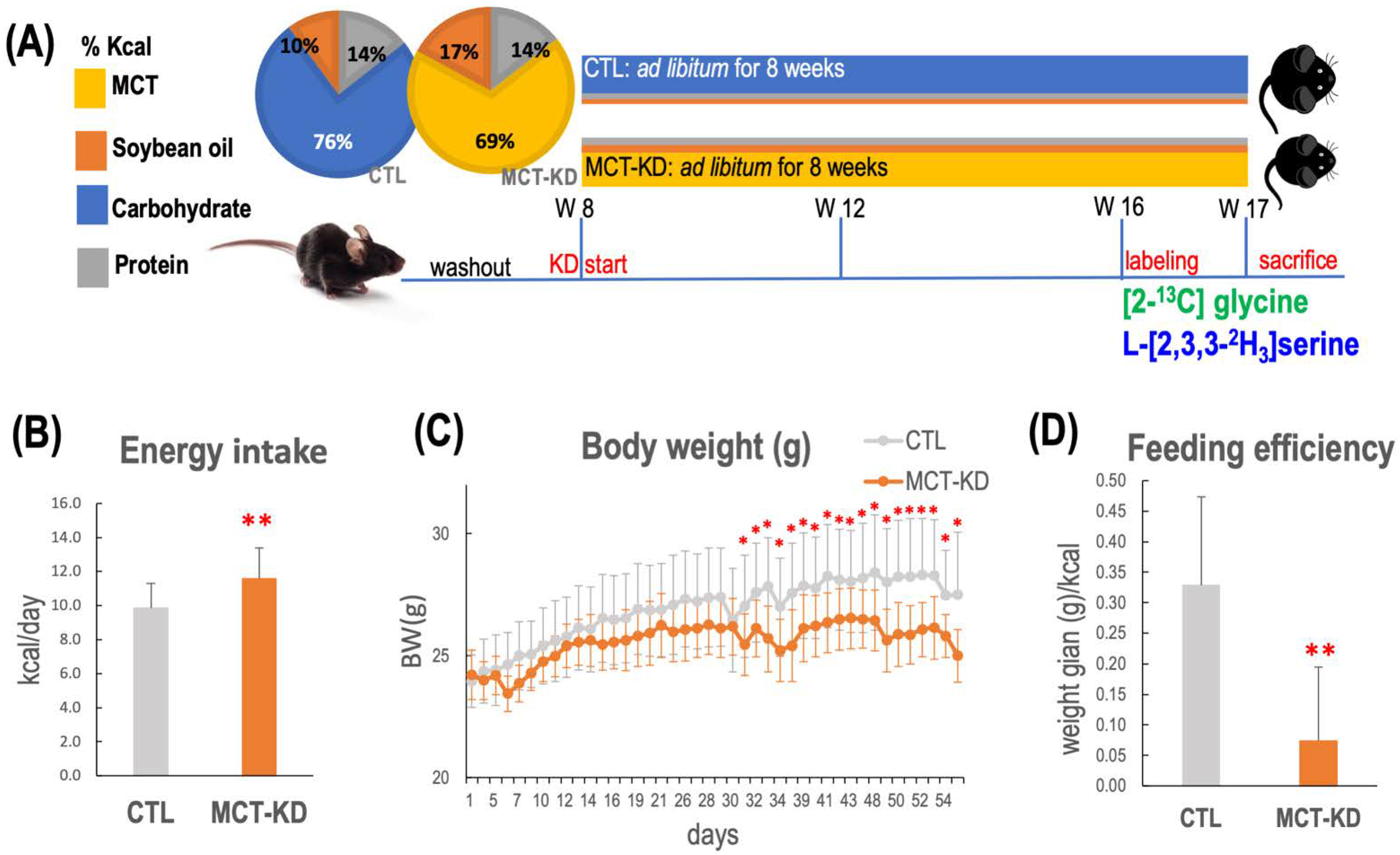
2.1. Consumption of MCT-KD Decreased Body Weight and Fat Mass but Increased Muscle Mass In Vivo
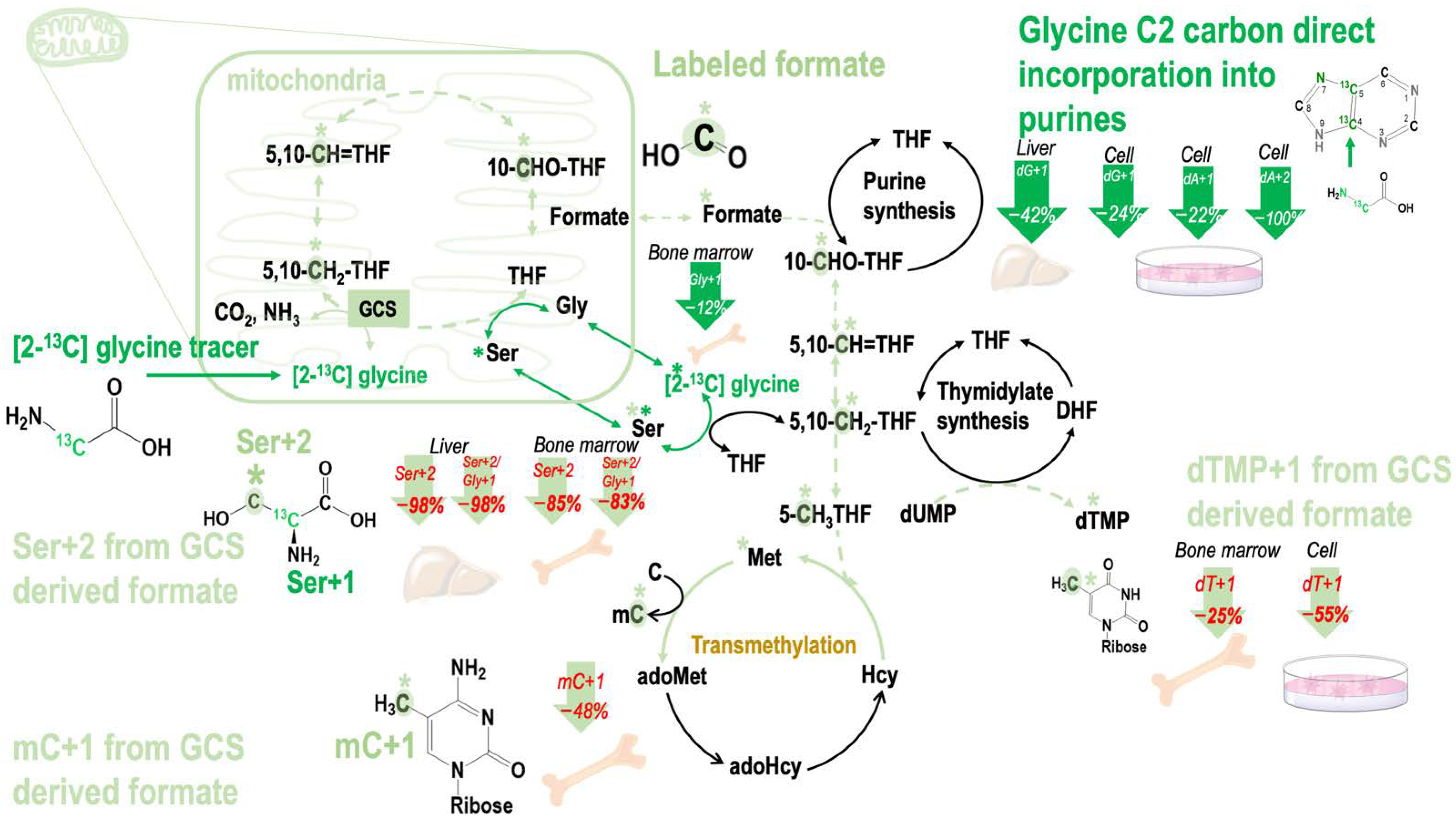
2.2. Consumption of MCT-KD Decreased Plasma Formate Concentrations and Suppressed Formate Carbon Flow from Glycine and Serine
2.3. β-Hydroxybutyrate Inhibited Mitochondrial Formate Generation from Glycine Cleavage System In Vitro
2.4. Consumption of MCT-KD Inhibited Mitochondrial Formate Generation from Glycine Cleavage System in Mouse Bone Marrow
2.5. Effects of βHB on the Partitioning between Mitochondrial and Cytosolic 1C Metabolic Fluxes in Thymidine Biosynthesis In Vitro
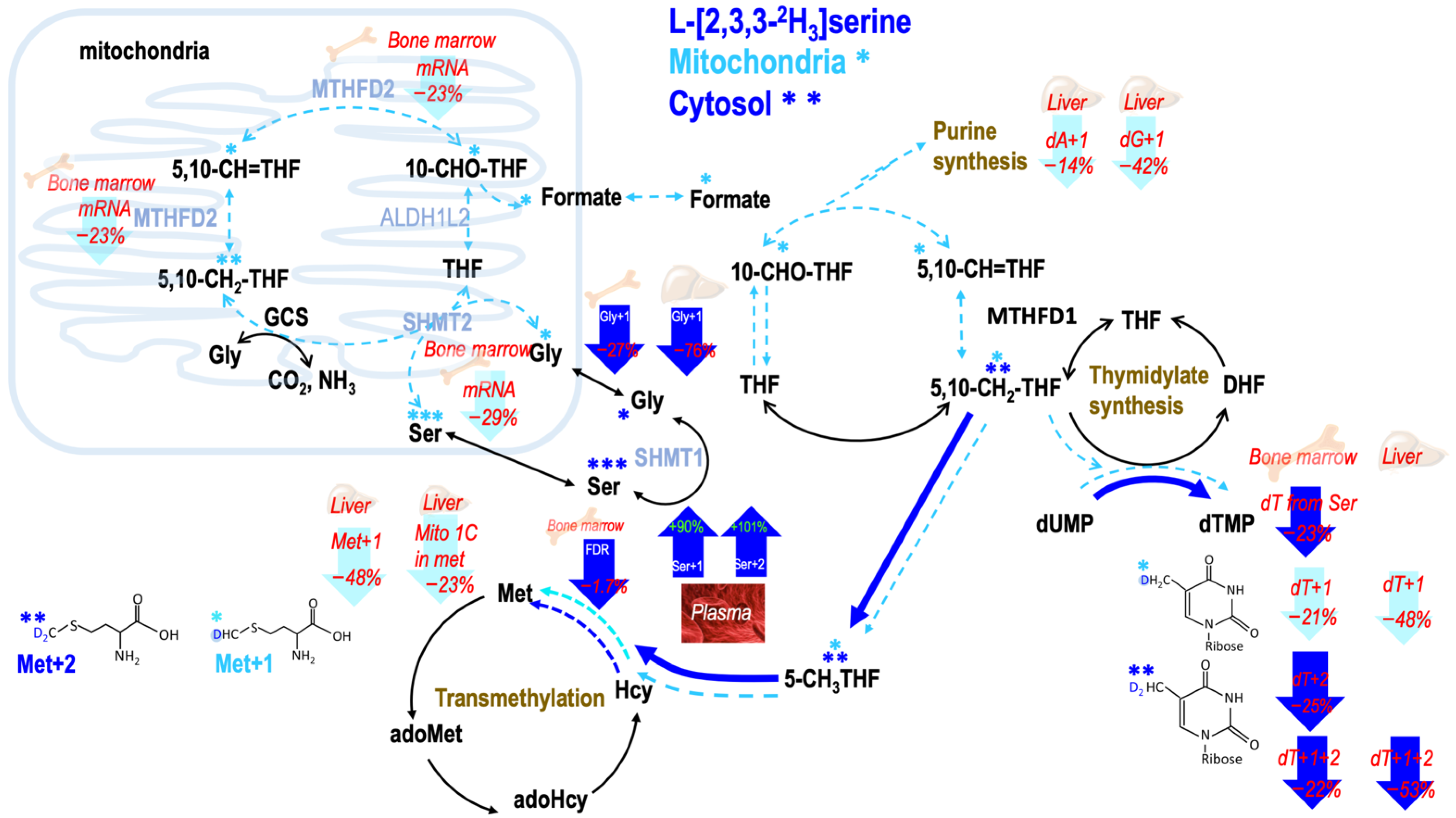
2.6. Consumption of MCT-KD Decreased Metabolic Fluxes from Mitochondrial Derived Formate In Vivo (Healthy Mouse Bone Marrow)
2.7. Consumption of MCT-KD Altered Fractional Mitochondrial Formate Supply for Methionine Synthesis In Vivo
2.8. Consumption of MCT-KD Altered One-Carbon Metabolic Fluxes between 5-Methyl THF Dependent Methionine Synthesis and 5,10-Methylene THF Dependent dTMP Synthesis
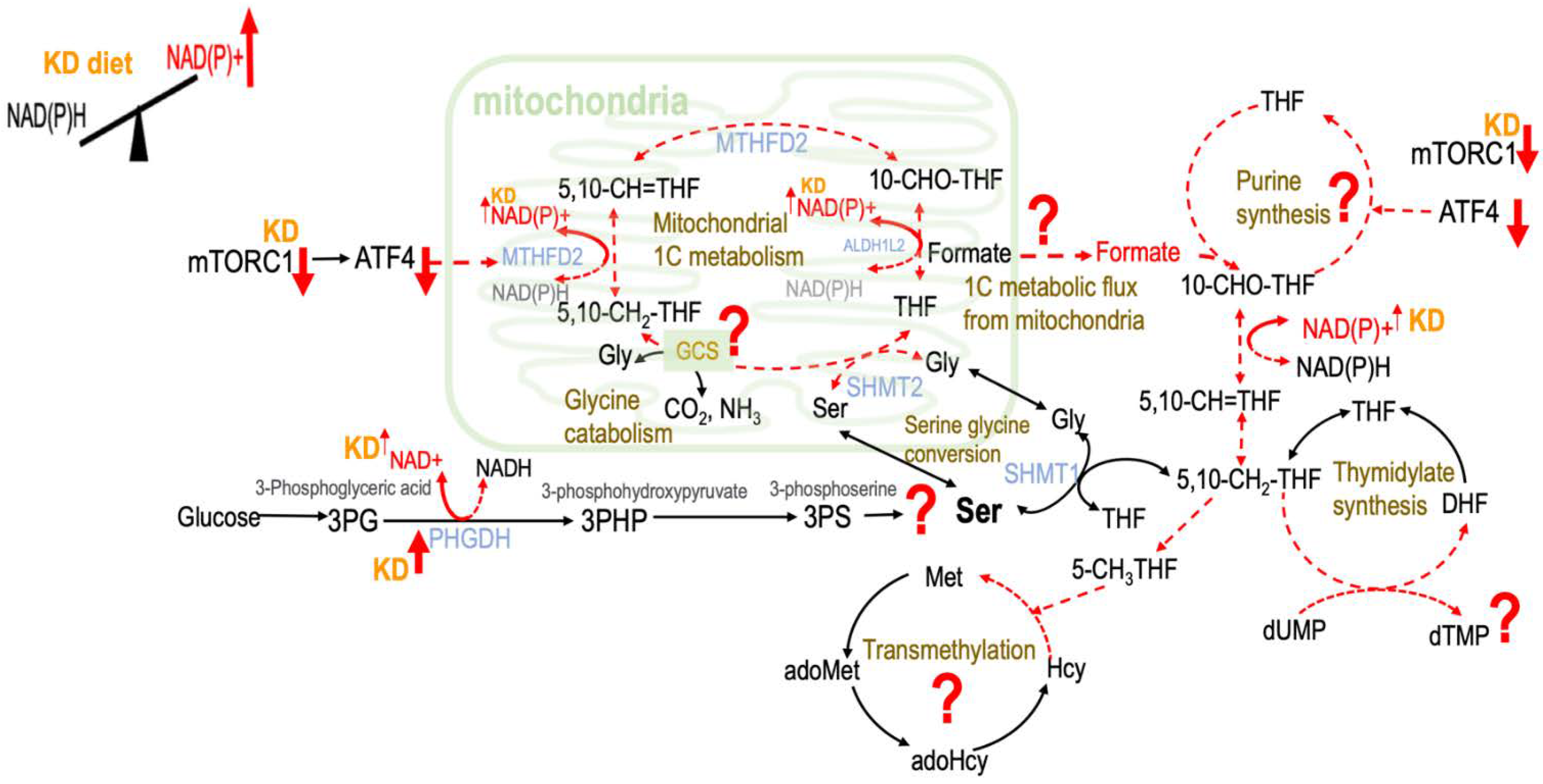
3. Discussion
3.1. MCT-Enriched Ketogenic Diet with Normal Protein Content Was Effective in Body Weight and Fat Mass Reduction
3.2. Ketogenesis Affects One-Carbon Metabolism in a Tissue-Specific Manner
3.3. Mild Ketogenic State Suppressed Mitochondrial Formate Supply in Liver and Bone Marrow via NAD+/NADP Balance
3.4. Ketogenic Diet May Exert Its Impacts on 1C Metabolism through Mitochondria Turnover
3.5. Ketogenic Diet May Exert Its Impacts on 1C Metabolism through mTOR Signaling
4. Materials and Methods
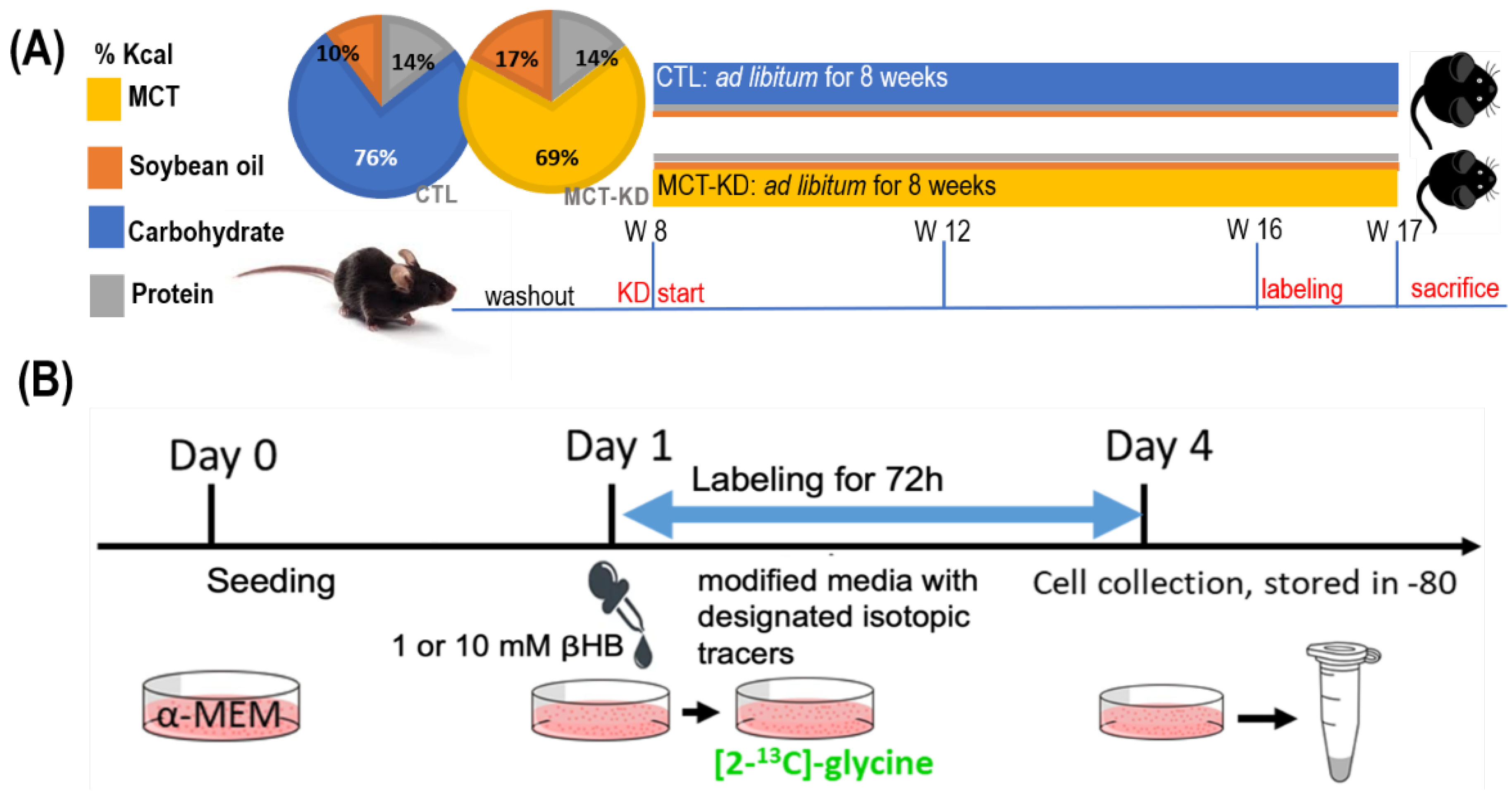
4.1. Ketogenic Cell Model
4.2. In Vitro Isotopic Tracer Studies
4.3. Ketogenic Animal Model in Mice
4.4. In Vivo Metabolic Tracer Studies
4.5. Determination of Formate Concentrations and Enrichments from Glycine and Serine
4.6. Determination of Amino Acid Concentrations and Enrichments from Glycine and Serine
4.7. Determination of Nucleotide Enrichments from Glycine and Serine
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kennedy, A.R.; Pissios, P.; Otu, H.; Roberson, R.; Xue, B.; Asakura, K.; Furukawa, N.; Marino, F.E.; Liu, F.F.; Kahn, B.B.; et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1724–E1739. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Suzuki, K. Potential application of ketogenic diet to metabolic status and exercise performance: A review. EC Nutr. 2018, 13, 496–499. [Google Scholar]
- Elamin, M.; Ruskin, D.N.; Masino, S.A.; Sacchetti, P. Ketone-based metabolic therapy: Is increased NAD+ a primary mechanism? Front. Mol. Neurosci. 2017, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, S.S.; Rensing, N.R.; Thio, L.L.; Yamada, K.A.; Wong, M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 2011, 52, e7–e11. [Google Scholar] [CrossRef] [PubMed]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Vazquez, A. Dietary and pharmacological induction of serine synthesis genes. bioRxiv 2020. preprint. [Google Scholar]
- Meiser, J.; Vazquez, A. Give it or take it: The flux of one-carbon in cancer cells. FEBS J. 2016, 283, 3695–3704. [Google Scholar] [CrossRef]
- Meiser, J.; Schuster, A.; Pietzke, M.; Vande Voorde, J.; Athineos, D.; Oizel, K.; Burgos-Barragan, G.; Wit, N.; Dhayade, S.; Morton, J.P.; et al. Increased formate overflow is a hallmark of oxidative cancer. Nat. Commun. 2018, 9, 1368. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.F.; Appling, D.R. Characterization of the folate-dependent mitochondrial oxidation of carbon 3 of serine. Biochemistry 1993, 32, 4671–4676. [Google Scholar] [CrossRef]
- Kikuchi, G. The glycine cleavage system: Composition, reaction mechanism, and physiological significance. Mol. Cell. Biochem. 1973, 1, 169–187. [Google Scholar] [CrossRef]
- Tan, Y.L.; Sou, N.L.; Tang, F.Y.; Ko, H.A.; Yeh, W.T.; Peng, J.H.; Chiang, E.I. Tracing Metabolic Fate of Mitochondrial Glycine Cleavage System Derived Formate In Vitro and In Vivo. Int. J. Mol. Sci. 2020, 21, 8808. [Google Scholar] [CrossRef] [PubMed]
- Motokawa, Y.; Kikuchi, G. Glycine Metabolism by Rat Liver Mitochondria: II. Methylene Tetrahydrofolate as the Direct One Carbon Donor in the Reaction of Glycine Synthesis. J. Biochem. 1969, 65, 71–75. [Google Scholar] [PubMed]
- Peri, K.G.; Belanger, C.; Mackenzie, R.E. Nucleotide sequence of the human NAD-dependent methylene tetrahydrofolate dehydrogenase-cyclohydrolase. Nucleic Acids Res. 1989, 17, 8853. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Williams, R.W.; Auwerx, J. Metabolic networks of longevity. Cell 2010, 142, 9–14. [Google Scholar] [CrossRef]
- Wang, Y.C.; Wu, M.T.; Lin, Y.J.; Tang, F.Y.; Ko, H.A.; Chiang, E.P. Regulation of Folate-Mediated One-Carbon Metabolism by Glycine N-Methyltransferase (GNMT) and Methylenetetrahydrofolate Reductase (MTHFR). J. Nutr. Sci. Vitaminol. 2015, 61, S148–S150. [Google Scholar] [CrossRef]
- Chu, P.-Y.; Wu, H.-J.; Wang, S.-M.; Chen, P.-M.; Tang, F.-Y.; Chiang, E.-P.I. MAT2A Localization and Its Independently Prognostic Relevance in Breast Cancer Patients. Int. J. Mol. Sci. 2021, 22, 5382. [Google Scholar]
- Chen, P.-M.; Tsai, C.-H.; Huang, C.-C.; Hwang, H.-H.; Li, J.-R.; Liu, C.-C.; Ko, H.-A.; Chiang, E.-P.I. Downregulation of Methionine Cycle Genes MAT1A and GNMT Enriches Protein-Associated Translation Process and Worsens Hepatocellular Carcinoma Prognosis. Int. J. Mol. Sci. 2022, 23, 481. [Google Scholar]
- Lee, T.Y.; Chiang, E.P.; Shih, Y.T.; Lane, H.Y.; Lin, J.T.; Wu, C.Y. Lower serum folate is associated with development and invasiveness of gastric cancer. World J. Gastroenterol. 2014, 20, 11313–11320. [Google Scholar]
- Palacio Lapuente, F.; Hernandez Galindo, M.; Amezqueta Goni, C.; Lapuente Heppe, I.; Sola Saravia, C. Managing the atrial fibrillation process: An integral approach. Rev. Calid. Asist. 2013, 28, 19–27. [Google Scholar] [CrossRef]
- Pietzke, M.; Meiser, J.; Vazquez, A. Formate metabolism in health and disease. Mol. Metab. 2020, 33, 23–37. [Google Scholar] [CrossRef]
- Tarasenko, T.N.; Rosas, O.R.; Singh, L.N.; Kristaponis, K.; Vernon, H.; McGuire, P.J. A new mouse model of mild ornithine transcarbamylase deficiency (spf-j) displays cerebral amino acid perturbations at baseline and upon systemic immune activation. PLoS ONE 2015, 10, e0116594. [Google Scholar] [CrossRef]
- Paoli, A.; Grimaldi, K.; Toniolo, L.; Canato, M.; Bianco, A.; Fratter, A. Nutrition and acne: Therapeutic potential of ketogenic diets. Skin Pharmacol. Physiol. 2012, 25, 111–117. [Google Scholar] [CrossRef]
- Sou, N.L.; Huang, Y.H.; Chen, D.Y.; Chen, Y.M.; Tang, F.Y.; Ko, H.A.; Fan, Y.H.; Lin, Y.Y.; Wang, Y.C.; Chih, H.M.; et al. Folinate Supplementation Ameliorates Methotrexate Induced Mitochondrial Formate Depletion In Vitro and In Vivo. Int. J. Mol. Sci. 2021, 22, 1350. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Baba, N.; Bracco, E.F.; Hashim, S.A. Enhanced thermogenesis and diminished deposition of fat in response to overfeeding with diet containing medium chain triglyceride. Am. J. Clin. Nutr. 1982, 35, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hamilton, J.A.; Kirkland, J.L.; Corkey, B.E.; Guo, W. Medium-chain oil reduces fat mass and down-regulates expression of adipogenic genes in rats. Obes. Res. 2003, 11, 734–744. [Google Scholar] [CrossRef]
- St-Onge, M.P.; Ross, R.; Parsons, W.D.; Jones, P.J. Medium-chain triglycerides increase energy expenditure and decrease adiposity in overweight men. Obes. Res. 2003, 11, 395–402. [Google Scholar] [CrossRef]
- Turner, N.; Hariharan, K.; TidAng, J.; Frangioudakis, G.; Beale, S.M.; Wright, L.E.; Zeng, X.Y.; Leslie, S.J.; Li, J.-Y.; Kraegen, E.W. Enhancement of muscle mitochondrial oxidative capacity and alterations in insulin action are lipid species dependent: Potent tissue-specific effects of medium-chain fatty acids. Diabetes 2009, 58, 2547–2554. [Google Scholar] [CrossRef]
- Pissios, P.; Hong, S.; Kennedy, A.R.; Prasad, D.; Liu, F.-F.; Maratos-Flier, E. Methionine and choline regulate the metabolic phenotype of a ketogenic diet. Mol. Metab. 2013, 2, 306–313. [Google Scholar] [CrossRef]
- Borghjid, S.; Feinman, R.D. Response of C57Bl/6 mice to a carbohydrate-free diet. Nutr. Metab. 2012, 9, 69. [Google Scholar] [CrossRef]
- Bielohuby, M.; Menhofer, D.; Kirchner, H.; Stoehr, B.J.; Müller, T.D.; Stock, P.; Hempel, M.; Stemmer, K.; Pfluger, P.T.; Kienzle, E.; et al. Induction of ketosis in rats fed low-carbohydrate, high-fat diets depends on the relative abundance of dietary fat and protein. Am. J. Physiol. Metab. 2011, 300, E65–E76. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Ipek, Ö.; Beaumont, M.; Shevlyakova, M.; Christinat, N.; Masoodi, M.; Greenberg, N.; Gruetter, R.; Cuenoud, B. Nutritional ketosis increases NAD+/NADH ratio in healthy human brain: An in vivo study by 31P-MRS. Front. Nutr. 2018, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.P.; MacMillan, L.; Lamarre, S.G.; Young, S.K.; MacFarlane, A.J.; Brosnan, M.E.; Brosnan, J.T. In vivo kinetics of formate metabolism in folate-deficient and folate-replete rats. J. Biol. Chem. 2015, 290, 2244–2250. [Google Scholar] [CrossRef]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef]
- Hampson, R.K.; Taylor, M.; Olson, M. Regulation of the glycine cleavage system in the isolated perfused rat liver. J. Biol. Chem. 1984, 259, 1180–1185. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. beta-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Newell, C.; Shutt, T.E.; Ahn, Y.; Hittel, D.; Khan, A.; Rho, J.M.; Shearer, J. Tissue specific impacts of a ketogenic diet on mitochondrial dynamics in the BTBRT+ tf/j mouse. Front. Physiol. 2016, 7, 654. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.R.; Ong, S.-E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 2016, 5, e10575. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Wang, Y.C.; Tang, F.Y.; Chen, S.Y.; Chen, Y.M.; Chiang, E.P. Glycine-N methyltransferase expression in HepG2 cells is involved in methyl group homeostasis by regulating transmethylation kinetics and DNA methylation. J. Nutr. 2011, 141, 777–782. [Google Scholar] [CrossRef]
- Santra, S.; Gilkerson, R.W.; Davidson, M.; Schon, E.A. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann. Neurol. 2004, 56, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Chiang, E.P.; Wang, Y.C.; Chen, W.W.; Tang, F.Y. Effects of insulin and glucose on cellular metabolic fluxes in homocysteine transsulfuration, remethylation, S-adenosylmethionine synthesis, and global deoxyribonucleic acid methylation. J. Clin. Endocrinol. Metab. 2009, 94, 1017–1025. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lin, W.L.; Lin, Y.J.; Tang, F.Y.; Chen, Y.M.; Chiang, E.P. A novel role of the tumor suppressor GNMT in cellular defense against DNA damage. Int. J. Cancer 2014, 134, 799–810. [Google Scholar] [CrossRef]
- Chiang, E.P.; Wang, Y.C.; Tang, F.Y. Folate restriction and methylenetetrahydrofolate reductase 677T polymorphism decreases adoMet synthesis via folate-dependent remethylation in human-transformed lymphoblasts. Leukemia 2007, 21, 651–658. [Google Scholar] [CrossRef]
- Wang, Y.C.; Wu, M.T.; Tang, F.Y.; Chen, D.Y.; Ko, H.A.; Shane, B.; Huang, W.N.; Chiang, E.P. MTHFR C677T polymorphism increases MTX sensitivity via the inhibition of S-adenosylmethionine and de novo purine synthesis. Clin. Sci. 2019, 133, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Sharples, A.P.; Hughes, D.C.; Deane, C.S.; Saini, A.; Selman, C.; Stewart, C.E. Longevity and skeletal muscle mass: The role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell 2015, 14, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.F., III; Cuskelly, G.J.; Shane, B.; Toth, J.P.; Baumgartner, T.G.; Stacpoole, P.W. Primed, constant infusion with [2H3] serine allows in vivo kinetic measurement of serine turnover, homocysteine remethylation, and transsulfuration processes in human one-carbon metabolism. Am. J. Clin. Nutr. 2000, 72, 1535–1541. [Google Scholar] [CrossRef]
- Anguera, M.C.; Field, M.S.; Perry, C.; Ghandour, H.; Chiang, E.-P.; Selhub, J.; Shane, B.; Stover, P.J. Regulation of folate-mediated one-carbon metabolism by 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 2006, 281, 18335–18342. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Chen, Y.-M.; Lin, Y.-J.; Liu, S.-P.; Chiang, E.-P.I. GNMT expression increases hepatic folate contents and folate-dependent methionine synthase-mediated homocysteine remethylation. Mol. Med. 2011, 17, 486–494. [Google Scholar]
- Wang, Y.C.; Chiang, E.P. Low-dose methotrexate inhibits methionine S-adenosyltransferase in vitro and in vivo. Mol. Med. 2012, 18, 423–432. [Google Scholar] [CrossRef]
- Lamarre, S.G.; MacMillan, L.; Morrow, G.P.; Randell, E.; Pongnopparat, T.; Brosnan, M.E.; Brosnan, J.T. An isotope-dilution, GC–MS assay for formate and its application to human and animal metabolism. Amino Acids 2014, 46, 1885–1891. [Google Scholar] [CrossRef]
- Chang, H.Y.; Tzen, J.T.; Lin, S.J.; Wu, Y.T.; Chiang, E.P. Long-term prednisolone treatments increase bioactive vitamin B6 synthesis in vivo. J. Pharmacol. Exp. Ther. 2011, 337, 102–109. [Google Scholar] [CrossRef]
- Wu, M.T.; Ye, W.T.; Wang, Y.C.; Chen, P.M.; Liu, J.Y.; Tai, C.K.; Tang, F.Y.; Li, J.R.; Liu, C.C.; Chiang, E.I. MTHFR Knockdown Assists Cell Defense against Folate Depletion Induced Chromosome Segregation and Uracil Misincorporation in DNA. Int. J. Mol. Sci. 2021, 22, 9392. [Google Scholar] [CrossRef]
- Lusardi, T.A.; Akula, K.K.; Coffman, S.Q.; Ruskin, D.N.; Masino, S.A.; Boison, D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 2015, 99, 500–509. [Google Scholar] [CrossRef]
- Hernandez, A.R.; Hernandez, C.M.; Campos, K.; Truckenbrod, L.; Federico, Q.; Moon, B.; McQuail, J.A.; Maurer, A.P.; Bizon, J.L.; Burke, S.N. A Ketogenic Diet Improves Cognition and Has Biochemical Effects in Prefrontal Cortex That Are Dissociable from Hippocampus. Front. Aging Neurosci. 2018, 10, 391. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| βHB 3 (mM) | BW 3 (g) | BAT/BW 3 | VAT/BW 3 | RAT/BW 3 | Muscle/BW 3 | |
|---|---|---|---|---|---|---|
| CTL 2 | 0.37 ± 0.11 | 27.64 ± 2.3 | 0.003 ± 0.001 | 0.005 ± 0.003 | 0.02 ± 0.009 | 0.010 ± 0.001 |
| MCT-KD 2 | 0.58 ± 0.16 | 25.48 ± 1.2 | 0.002 ± 0.000 | 0.001 ± 0.000 | 0.01 ± 0.002 | 0.011 ± 0.001 |
| p-value | 0.009 | 0.014 | 0.039 | 0.0001 | 0.001 | 0.017 |
| % change | +54% | −8% | −25% | −79% | −61% | +11% |
| Serine 1 (μM) | Glycine 1 (μM) | Formate 1 (μM) | Formate + 1 from L-[2,3,3-2H3]-Serine 2 | Formate + 1 from [2-13C]-Glycine 2 | |
|---|---|---|---|---|---|
| CTL 4 | 180 ± 36 | 584 ± 149 | 27.0 ± 7.0 | 0.023 ± 0.002 | 0.002 ± 0.001 |
| MCT-KD 4 | 214 ± 158 | 764 ± 244 | 21.7 ± 7.1 | 0.014 ± 0.001 | Undetectable |
| % change 3 | +31% # | −19% # | −38% | −100% |
| (A) BHB at 10 mM Inhibited dTMP Synthesis and Purine Synthesis from [2-13C]-Glycine In Vitro 1 | |||||||
| From direct incorporation into DNA | From GCS derived formate | ||||||
| dA + 1 2 | dG + 1 2 | dA + 2 2 | dTMP + 1 2 | ||||
| CTL 3 | 0.323 ± 0.002 | 0.350 ± 0.003 | 0.011 | 0.027 ± 0.002 | |||
| BHB low 3 | 0.321 ± 0.010 | 0.340 ± 0.001 | 0.009 | 0.025 ± 0.001 | |||
| BHB high 3 | 0.253 ± 0.001 | 0.267 ± 0.018 | 0.000 | 0.012 ± 0.001 | |||
| % low BHB 4 | −3% # | ||||||
| % high BHB 4 | −22% | −24%% | −100 | −55% | |||
| (B) MCT-KD Inhibited the Utilization of GCS Derived Formate in Thymidine Synthesis in the Bone Marrow 1 | |||||||
| From direct incorporation into protein/DNA | From GCS derived formate | ||||||
| Ser + 1 2 | Gly + 1 2 | dA + 1 2 | dG + 1 2 | dTMP + 1 2 | Ser + 2 2 | Ser + 2/Gly + 1 2 | |
| CTL 5 | 0.046 ± 0.002 | 0.057 ± 0.001 | 0.085 ± 0.008 | 0.089 ± 0.013 | 0.011 ± 0.001 | 0.0008 ± 0.000 | 0.015 ± 0.003 |
| MCT-KD 5 | 0.049 ± 0.003 | 0.051 ± 0.003 # | 0.072 ± 0.001 | 0.081 ± 0.001 | 0.008 ± 0.000 | 0.0001 ± 0.000 | 0.002 ± 0.001 |
| % change 4 | 4% | −12% # | −16% | −9% # | −25% # | −85% | −83% # |
| (C) MCT-KD Inhibited the Utilization of GCS Derived Formate on Synthesizing Serine in the Liver 1 | |||||||
| From direct incorporation into protein/DNA | From GCS derived formate | ||||||
| Ser + 1 2 | Gly + 1 2 | Ser + 1 2 | Gly + 1 2 | Ser + 1 2 | |||
| CTL 5 | 0.088 ± 0.005 | 0.070 ± 0.005 | 0.034 ± 0.005 | 0.058 ± 0.006 | 0.002 ± 0.000 | 0.001 ± 0.000 | 0.019 ± 0.002 |
| MCT-KD 5 | 0.078 ± 0.008 | 0.061 ± 0.016 | 0.025 ± 0.001 | 0.034 ± 0.002 | 0.002 ± 0.000 | undetectable | undetectable |
| % change 4 | −11% | −13% | −25% | −42% # | 12% | −100% | −100% |
| (A) Cell Model | ||||
| dTMP + 1 3 | dTMP + 2 3 | Total 3 | % Mito 3 | |
| CTL 4 | 0.295 ± 0.008 | 0.050 ± 0.000 | 0.345 ± 0.008 | 85% |
| βHB low 4 | 0.299 ± 0.012 | 0.049 ± 0.001 | 0.349 ± 0.012 | 86% |
| βHB high 4 | 0.296 ± 0.009 | 0.047 ± 0.001 | 0.343 ± 0.010 | 86% |
| % low βHB 4 | ||||
| % high βHB 4 | −7% # | |||
| (B) Mouse Bone Marrow DNA | ||||
| dTMP + 1 3 | dTMP + 2 3 | Total 3 | % Mito 3 | |
| CTL 3 | 0.086 ± 0.002 | 0.010 ± 0.00 | 0.096 ± 0.002 | 90% |
| MCT-KD 3 | 0.068 ± 0.005 | 0.008 ± 0.001 | 0.075 ± 0.005 | 90% |
| % change 2 | −21% # | −25% # | −22% # | |
| (C) Mouse Liver DNA | ||||
| dTMP + 1 3 | dTMP + 2 3 | Total 3 | % Mito 3 | |
| CTL 4 | 0.005 ± 0.000 | 0.0005 ± 0.000 | 0.005 ± 0.000 | 91% |
| MCT-KD 4 | 0.002 ± 0.000 | 0.0002 ± 0.000 | 0.003 ± 0.001 | 93% |
| % change 2 | −48% | −55% # | −49% | |
| (A) in Plasma Free Amino Acid | ||||
| Met + 1 3 | Met + 2 3 | Total | % Mito 2 | |
| CTL 4 | 0.011 ± 0.000 | 0.003 ± 0.000 | 0.016 ± 0.000 | 80% ± 2% |
| MCT-KD 4 | 0.013 ± 0.003 | 0.002 ± 0.002 | 0.014 ± 0.005 | 88% ± 12% |
| (B) in Liver Cellular Protein | ||||
| Met + 1 3 | Met + 2 3 | Total | % Mito 2 | |
| CTL 4 | 0.012 ± 0.001 | 0.004 ± 0.000 | 0.017 ± 0.002 | 74% ± 1% |
| MCT-KD 4 | 0.006 ± 0.001 | 0.005 ± 0.002 | 0.011 ± 0.003 | 57% ± 4% |
| % change 3 | −48% # | −35% # | −23% | |
| (C) in Bone Marrow Cellular Protein | ||||
| Met + 1 2 | Met + 2 2 | Total | % Mito | |
| CTL 4 | 0.019 ± 0.001 | 0.007 ± 0.001 | 0.026 ± 0.002 | 75% ± 3% |
| MCT-KD 4 | 0.018 ± 0.001 | 0.006 ± 0.000 | 0.024 ± 0.002 | 75% ± 0% |
| (A) Plasma | ||||||
| Leu + 1 + 2 + 3 1 | Ser + 1 + 2 + 3 3 | Ser + 1 3 | Ser + 2 3 | Ser + 3 3 | Gly + 1 3 | |
| CTL 4 | 0.089 ± 0.000 | 0.042 ± 0.005 | 0.012 ± 0.002 | 0.020 ± 0.003 | 0.010 ± 0.001 | 0.003 ± 0.001 |
| MCT-KD 4 | 0.089 ± 0.002 | 0.08 ± 0.019 | 0.022 ± 0.004 | 0.040 ± 0.009 | 0.017 ± 0.006 | 0.004 ± 0.001 |
| % change 3 | +91% # | +90% # | +101% # | |||
| (B) Liver Protein | ||||||
| Leu + 1 + 2 + 3 1 | Ser + 1 + 2 + 3 3 | Ser + 1 3 | Ser + 2 3 | Ser + 3 3 | Gly + 1 3 | |
| CTL 4 | 0.099 ± 0.002 | 0.190 ± 0.035 | 0.046 ± 0.009 | 0.075 ± 0.003 | 0.069 ± 0.030 | 0.022 ± 0.006 |
| MCT-KD 4 | 0.069 ± 0.007 | 0.143 ± 0.012 | 0.030 ± 0.007 | 0.067 ± 0.011 | 0.046 ± 0.006 | 0.005 ± 0.001 |
| % change 3 | −30% # | −76% # | ||||
| (C) Bone Marrow Protein | ||||||
| Leu + 1 + 2 + 3 1 | Ser + 1 + 2 + 3 3 | Ser + 1 3 | Ser + 2 3 | Ser + 3 3 | Gly + 1 3 | |
| CTL 4 | 0.110 ± 0.002 | 0.0196 ± 0.012 | 0.054 ± 0.003 | 0.073 ± 0.002 | 0.069 ± 0.006 | 0.039 ± 0.004 |
| MCT-KD 4 | 0.101 ± 0.001 | 0.200 ± 0.018 | 0.050 ± 0.003 | 0.079 ± 0.008 | 0.070 ± 0.007 | 0.029 ± 0.003 |
| % change 3 | −8% # | −27% # | ||||
| (A) The Methionine Synthesis from L-[2,3,3-2H3]serine in Plasma 1 | ||||
| Ser + 1 + 2 + 3 1 | Met + 1 + 2 3 | FDR 3 | ||
| CTL 5 | 0.042 ± 0.005 | 0.016 ± 0.000 | 39.6 ± 4.9% | |
| MCT-KD 5 | 0.08 ± 0.019 | 0.014 ± 0.005 | 16.3 ± 2.9% | |
| % change 3 | +91% | −23% | ||
| (B) Methionine Synthesis from L-[2,3,3-2H3]serine in Liver Cytoplasm | ||||
| Ser + 1 + 2 + 3 4 | Met + 1 + 2 4 | FDR 5 | ||
| CTL 5 | 0.149 ± 0.045 | 0.016 ± 0.006 | 10.9 ± 0.4% | |
| MCT-KD 5 | 0.104 ± 0.013 | 0.017 ± 0.004 | 17.6 ± 5.7% | |
| (C) Utilization of L-[2,3,3-2H3]serine in Liver Protein Methionine and DNA | ||||
| via 5-methylTHF | via 5,10 methylene THF | |||
| Met + 1 + 2 4 | FDR 5 | dT + 1 + 2 4 | dT from Ser 4 | |
| CTL 5 | 0.017 ± 0.002 | 9.2 ± 2.7% | 0.0052 ± 0.000 | 2.9 ± 0.66% |
| MCT-KD 5 | 0.011 ± 0.003 | 7.8 ± 1.6% | 0.0026 ± 0.001 | 1.8 ± 0.66% |
| % change 3 | −32% | −16% | −53% # | −34% |
| (D) Methionine Synthesis from L-[2,3,3-2H3]serine in Bone Marrow Cytoplasm | ||||
| Ser + 1 + 2 + 3 4 | Met + 1 + 2 4 | FDR 4 | ||
| CTL 5 | 0.196 ± 0.012 | 0.029 ± 0.009 | 41.5 ± 24.2% | |
| MCT-KD 5 | 0.200 ± 0.018 | 0.017 ± 0.000 | 28.5 ± 2.1% | |
| % change 3 | −12% # | |||
| (E) Utilization of L-[2,3,3-2H3]serine in Marrow Protein and DNA | ||||
| via 5-methylTHF | via 5,10 methylene THF | |||
| Met + 1 + 2 4 | FDR 5 | dT + 1 + 2 4 | dT from Ser 4 | |
| CTL 5 | 0.023 ± 0.001 | 11.9 ± 0.2% | 0.096 ± 0.002 | 122.8 ± 38.7% |
| MCT-KD 5 | 0.020 ± 0.001 | 10.2 ± 0.2% | 0.075 ± 0.005 | 124.6 ± 18% |
| % change 3 | −1.7% | −22% # | ||
| (F) Partitioning of 5,10 MethyleneTHF between Methionine and dTMP Thymidylate Synthesis in the Liver 4 | ||||
| mito 1C Met/dTMP 4 | cyto 1C Met/dTMP 4 | Total met/dTMP | ||
| CTL 5 | 2.593 ± 0.199 | 0.111 ± 0.002 | 0.316 ± 0.019 | |
| MCT-KD 5 | 2.723 ± 0.927 | 0.069 ± 0.069 | 0.270 ± 0.138 | |
| (G) Partitioning of 5,10 MethyleneTHF between Methionine and dTMP Thymidylate Synthesis in the Bone Marrow 4 | ||||
| mito 1C Met/dTMP 4 | cyto 1C Met/dTMP 4 | Total met/dTMP | ||
| CTL 5 | 0.238 ± 0.015 | 6.226 ± 4.018 | 4.143 ± 0.093 | |
| MCT-KD 5 | 0.232 ± 0.01 | 1.717 ± 0.255 | 3.717 ± 0.000 | |
| Macronutrients | CTL (AIN-93M) | MCT-KD 1 |
|---|---|---|
| Protein, % of energy | 14.4% | 14.4% |
| Carbohydrate, % of energy | 75.4% | 0.0% |
| Fat % of energy | 10.2% | 85.6% |
| Energy, kcal/g | 3.60 | 5.89 |
| Casein | 140.0 | 140.0 |
| L-Cysteine | 1.8 | 1.8 |
| Sucrose | 100.0 | 0.0 |
| Cornstarch | 465.7 | 0.0 |
| Dyetrose | 155.0 | 0.0 |
| Soybean oil | 40.0 | 67.2 |
| MCT oil | 0 | 291.64 |
| Cellulose | 50 | 50 |
| Salt Mix #210053 | 35 | 35 |
| Vitamin Mix #310025 | 10 | 10 |
| Choline bitartrate | 2.5 | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, F.-Y.; Liou, J.-Y.; Tang, F.-Y.; Sou, N.-L.; Peng, J.-H.; Chiang, E.-P.I. Ketogenic Diet Consumption Inhibited Mitochondrial One-Carbon Metabolism. Int. J. Mol. Sci. 2022, 23, 3650. https://doi.org/10.3390/ijms23073650
Hsu F-Y, Liou J-Y, Tang F-Y, Sou N-L, Peng J-H, Chiang E-PI. Ketogenic Diet Consumption Inhibited Mitochondrial One-Carbon Metabolism. International Journal of Molecular Sciences. 2022; 23(7):3650. https://doi.org/10.3390/ijms23073650
Chicago/Turabian StyleHsu, Fan-Yu, Jia-Ying Liou, Feng-Yao Tang, Nga-Lai Sou, Jian-Hau Peng, and En-Pei Isabel Chiang. 2022. "Ketogenic Diet Consumption Inhibited Mitochondrial One-Carbon Metabolism" International Journal of Molecular Sciences 23, no. 7: 3650. https://doi.org/10.3390/ijms23073650
APA StyleHsu, F.-Y., Liou, J.-Y., Tang, F.-Y., Sou, N.-L., Peng, J.-H., & Chiang, E.-P. I. (2022). Ketogenic Diet Consumption Inhibited Mitochondrial One-Carbon Metabolism. International Journal of Molecular Sciences, 23(7), 3650. https://doi.org/10.3390/ijms23073650