
Mitochondrial Dysfunction and Acute Fatty Liver of Pregnancy



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Acute Fatty Liver of Pregnancy (AFLP)
2.1. Clinical Presentation
2.2. Complications
2.3. Diagnosis
2.4. Management
3. Mitochondrial Dysfunction and Pathogenesis of AFLP
3.1. Mitochondrial Dynamics and Biogenesis
3.2. Mitochondrial Fatty-Acid Oxidation (FAO)
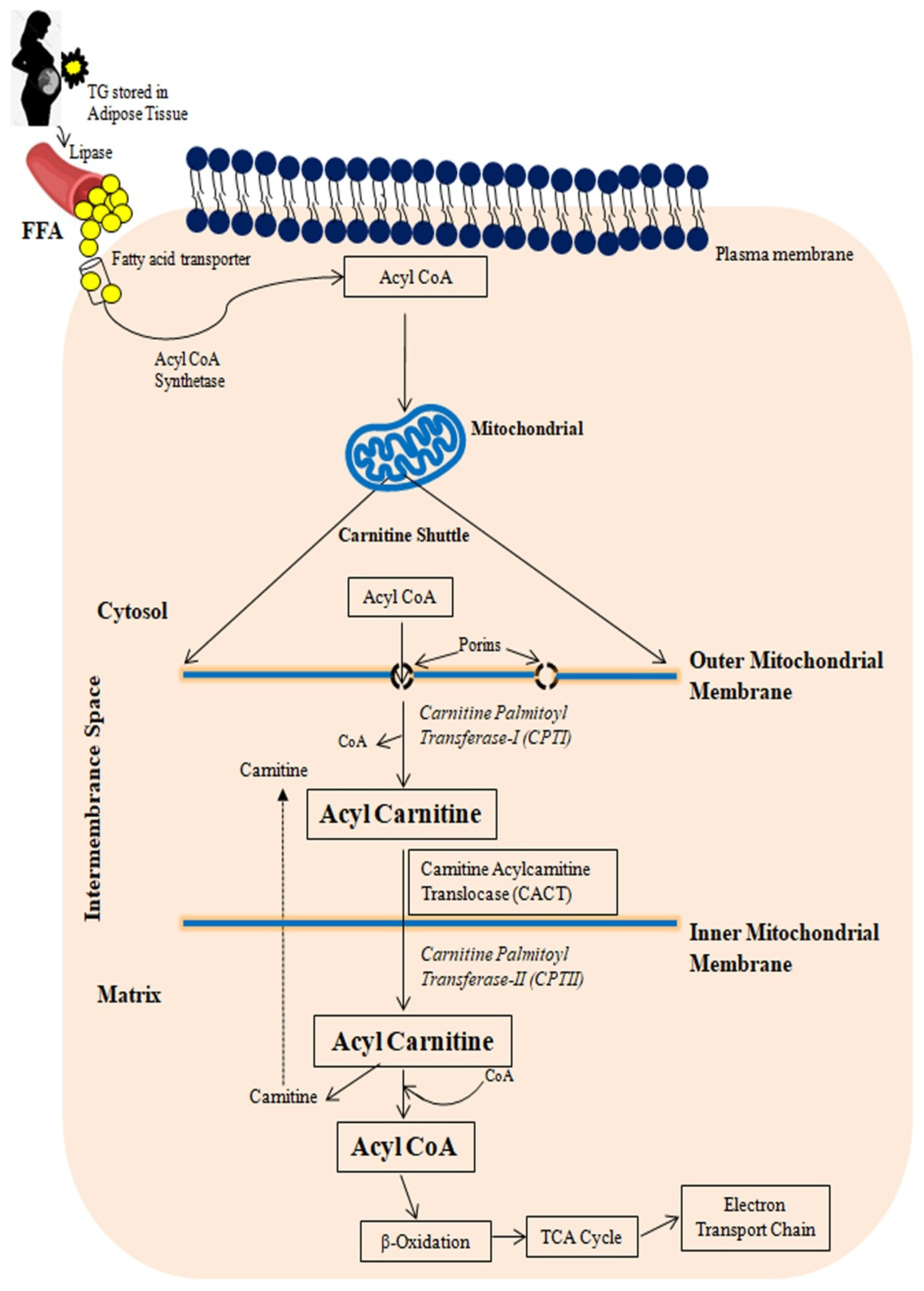
3.2.1. Fatty Acid Transport to Mitochondria
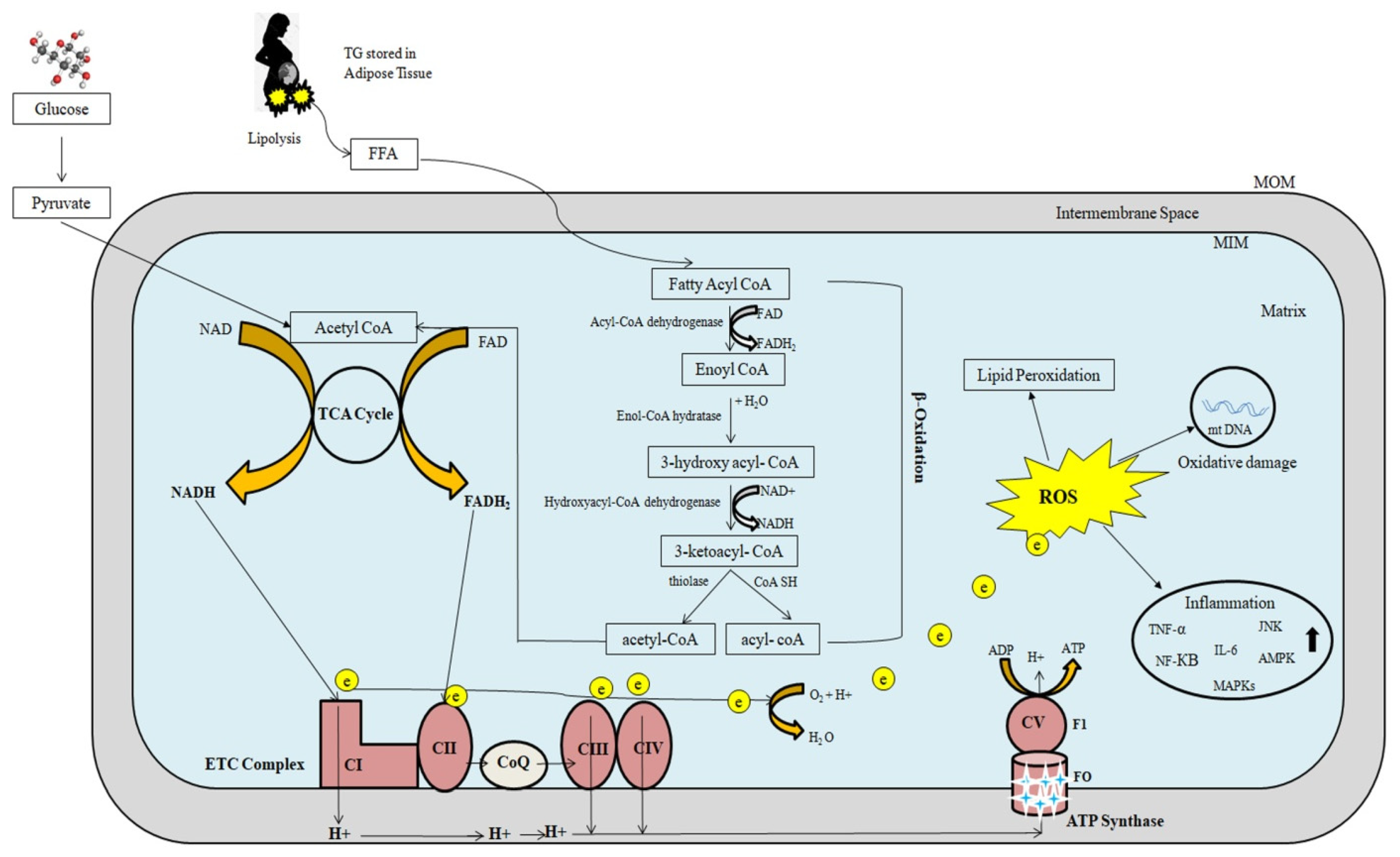
3.2.2. Mitochondrial β-Oxidation Cycle
3.2.3. Oxidative Phosphorylation
3.3. Regulation of Mitochondrial Fatty-Acid Oxidation and Reactive Oxygen Species Formation
3.4. Mitochondrial Fatty-Acid Oxidation Defects
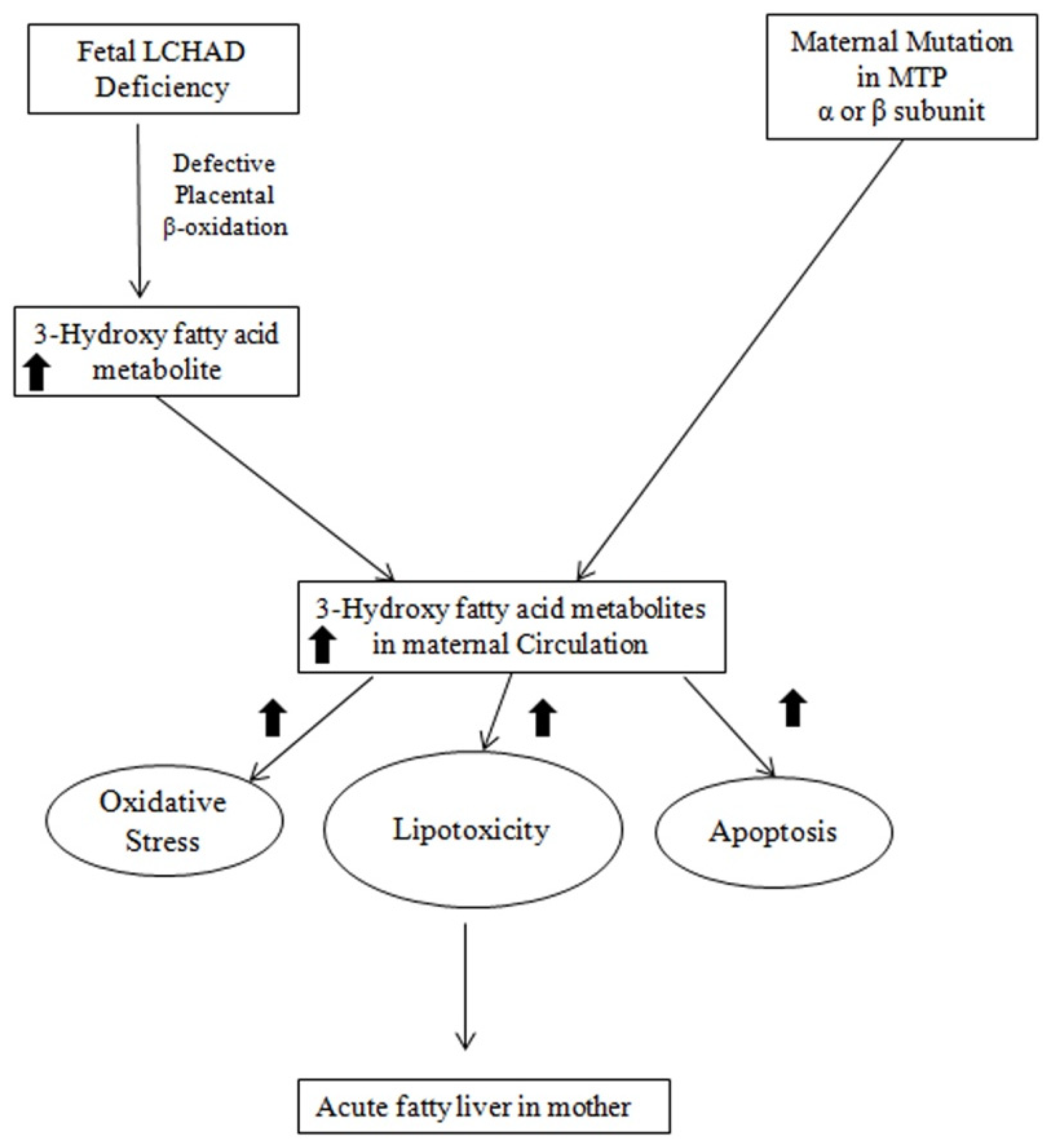
4. Fetal Mitochondrial Trifunctional Protein Defects and AFLP
5. Mechanism of the Association between Fetal LCHAD Deficiency and AFLP
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mailloux, R.J.; Lemire, J.; Appanna, V.D. Hepatic response to aluminum toxicity: Dyslipidemia and liver diseases. Exp. Cell Res. 2011, 317, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Grattagliano, I.; Caraceni, P.; Calamita, G.; Ferri, D.; Gargano, I.; Palasciano, G.; Portincasa, P. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur. J. Clin. Investig. 2008, 38, 523–530. [Google Scholar] [CrossRef]
- Cao, L.; Quan, X.B.; Zeng, W.J.; Yang, X.O.; Wang, M.J. Mechanism of Hepatocyte Apoptosis. J. Cell Death 2016, 9, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Nelson, D.B.; Yost, N.P.; Cunningham, F.G. Acute fatty liver of pregnancy: Clinical outcomes and expected duration of recovery. Am. J. Obstet. Gynecol. 2013, 209, 456.e1–456.e7. [Google Scholar] [CrossRef]
- Gao, Q.; Qu, X.; Chen, X.; Zhang, J.; Liu, F.; Tian, S.; Wang, C. Outcomes and risk factors of patients with acute fatty liver of pregnancy: A multicentre retrospective study. Singap. Med. J. 2018, 59, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Ibdah, J.A. Acute fatty liver of pregnancy: An update on pathogenesis and clinical implications. World J. Gastroenterol. 2006, 12, 7397–7404. [Google Scholar] [CrossRef]
- Mjahed, K.; Charra, B.; Hamoudi, D.; Noun, M.; Barrou, L. Acute fatty liver of pregnancy. Arch. Gynecol. Obstet. 2006, 274, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, D.B.; Ishak, K.G. Acute fatty liver of pregnancy: A clinicopathologic study of 35 cases. Hepatology 1985, 5, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Yoshida, E.M. Acute fatty liver of pregnancy. Can. J. Gastroenterol. 2006, 20, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treem, W.R. Mitochondrial fatty acid oxidation and acute fatty liver of pregnancy. Semin. Gastrointest. Dis. 2002, 13, 55–66. [Google Scholar] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shi, C.; Hu, S.; Zhu, H.; Ren, J.; Chen, Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis 2018, 21, 599–615. [Google Scholar] [CrossRef]
- Jin, Q.; Li, R.; Hu, N.; Xin, T.; Zhu, P.; Hu, S.; Ma, S.; Zhu, H.; Ren, J.; Zhou, H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018, 14, 576–587. [Google Scholar] [CrossRef]
- Sumneang, N.; Siri-Angkul, N.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 2020, 680, 108241. [Google Scholar] [CrossRef]
- Ding, M.; Ning, J.; Feng, N.; Li, Z.; Liu, Z.; Wang, Y.; Li, X.; Huo, C.; Jia, X.; Xu, R.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J. Pineal Res. 2018, 64, e12447. [Google Scholar] [CrossRef]
- Feng, S.T.; Wang, Z.Z.; Yuan, Y.H.; Wang, X.L.; Sun, H.M.; Chen, N.H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553. [Google Scholar] [CrossRef]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Tsuyoshi, H.; Orisaka, M.; Shieh, D.B.; Yoshida, Y.; Tsang, B.K. Mitochondrial dynamics regulating chemoresistance in gynecological cancers. Ann. N. Y. Acad. Sci. 2015, 1350, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Liu, C.; Shi, R.; Yu, M.; Zeng, K.; Kang, J.; Fu, F.; Mi, M. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol. 2020, 229, e13428. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef]
- Cerveny, K.L.; Tamura, Y.; Zhang, Z.; Jensen, R.E.; Sesaki, H. Regulation of mitochondrial fusion and division. Trends Cell Biol. 2007, 17, 563–569. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Zhang, L.; Han, L.; Ma, R.; Hou, X.; Yu, Y.; Sun, S.; Xu, Y.; Schedl, T.; Moley, K.H.; Wang, Q. Sirt3 prevents maternal obesity-associated oxidative stress and meiotic defects in mouse oocytes. Cell Cycle 2015, 14, 2959–2968. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Ramachandran, A.; McGill, M.R.; Mansouri, A.; Asselah, T.; Farhood, A.; Woolbright, B.L.; Ding, W.X.; Jaeschke, H. Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem. Toxicol. 2017, 108, 339–350. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B.; Berson, A.; Robin, M.A.; Lettéron, P.; Moreau, R.; Mansouri, A. Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 34–87. [Google Scholar] [CrossRef]
- Vacca, M.; Allison, M.; Griffin, J.L.; Vidal-Puig, A. Fatty Acid and Glucose Sensors in Hepatic Lipid Metabolism: Implications in NAFLD. Semin. Liver Dis. 2015, 35, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Song, K.X.; Qu, S. Nonalcoholic Fatty liver disease: Pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Scott, I.; Zhu, L.; Wu, K.; Han, K.; Chen, Y.; Gucek, M.; Sack, M.N. GCN5L1 modulates cross-talk between mitochondria and cell signaling to regulate FoxO1 stability and gluconeogenesis. Nat. Commun. 2017, 8, 523. [Google Scholar] [CrossRef]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [Green Version]
- Skoda, J.; Borankova, K.; Jansson, P.J.; Huang, M.L.; Veselska, R.; Richardson, D.R. Pharmacological targeting of mitochondria in cancer stem cells: An ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 2019, 139, 298–313. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between Autophagy and the Ubiquitin-Proteasome System and Its Role in the Pathogenesis of Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Lemasters, J.J. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am. J. Physiol. Cell Physiol. 2011, 300, C308–C317. [Google Scholar] [CrossRef] [Green Version]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001, 2, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Asp. Med. 2004, 25, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Walsh, C.T.; Tu, B.P.; Tang, Y. Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism. Chem. Rev. 2018, 118, 1460–1494. [Google Scholar] [CrossRef]
- Sun, F.; Zhou, Q.; Pang, X.; Xu, Y.; Rao, Z. Revealing various coupling of electron transfer and proton pumping in mitochondrial respiratory chain. Curr. Opin. Struct. Biol. 2013, 23, 526–538. [Google Scholar] [CrossRef]
- Prentki, M.; Joly, E.; El-Assaad, W.; Roduit, R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: Role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes 2002, 51 (Suppl. S3), S405–S413. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Larosche, I.; Lettéron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldstein, A.E. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin. Liver Dis. 2010, 30, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Li, T. Ripk3 mediates cardiomyocyte necrosis through targeting mitochondria and the JNK-Bnip3 pathway under hypoxia-reoxygenation injury. J. Recept. Signal Transduct. Res. 2019, 39, 331–340. [Google Scholar] [CrossRef]
- Upadhyay, K.K.; Jadeja, R.N.; Vyas, H.S.; Pandya, B.; Joshi, A.; Vohra, A.; Thounaojam, M.C.; Martin, P.M.; Bartoli, M.; Devkar, R.V. Carbon monoxide releasing molecule-A1 improves nonalcoholic steatohepatitis via Nrf2 activation mediated improvement in oxidative stress and mitochondrial function. Redox Biol. 2020, 28, 101314. [Google Scholar] [CrossRef]
- Xu, J.; Peng, Y.; Zeng, Y.; Hua, Y.Q.; Xu, X.L. 2, 3, 4’, 5-tetrahydroxystilbene-2-0-β-d Glycoside Attenuates Age- and Diet-Associated Non-Alcoholic Steatohepatitis and Atherosclerosis in LDL Receptor Knockout Mice and Its Possible Mechanisms. Int. J. Mol. Sci. 2019, 20, 1617. [Google Scholar] [CrossRef] [Green Version]
- Knebel, B.; Fahlbusch, P.; Dille, M.; Wahlers, N.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Herebian, D.; Koellmer, C.; et al. Fatty Liver Due to Increased. Front. Cell Dev. Biol. 2019, 7, 248. [Google Scholar] [CrossRef] [Green Version]
- Erland, L.A.E.; Shukla, M.R.; Singh, A.S.; Murch, S.J.; Saxena, P.K. Melatonin and serotonin: Mediators in the symphony of plant morphogenesis. J. Pineal Res. 2018, 64, e12452. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Liu, J.; Lu, W.; Shi, B.; Klein, S.; Su, X. Peroxisomal regulation of redox homeostasis and adipocyte metabolism. Redox Biol. 2019, 24, 101167. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Fang, L.; Liu, F.; Jiang, S.; Wu, L.; Zhang, J. Leptin and chronic kidney diseases. J. Recept. Signal Transduct. Res. 2018, 38, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.; Gorla, M.; Bisoyi, V.; Tammineni, P.; Sepuri, N.B.V. Rotenone-induced reactive oxygen species signal the recruitment of STAT3 to mitochondria. FEBS Lett. 2020, 594, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Van Opbergen, C.J.M.; den Braven, L.; Delmar, M.; van Veen, T.A.B. Mitochondrial Dysfunction as Substrate for Arrhythmogenic Cardiomyopathy: A Search for New Disease Mechanisms. Front. Physiol. 2019, 10, 1496. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024. [Google Scholar] [CrossRef] [Green Version]
- Fromenty, B.; Pessayre, D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, T.; Wanders, R.J.; Saudubray, J.M.; Aoyama, T.; Komiyama, A.; Hashimoto, T. Mitochondrial trifunctional protein deficiency. Catalytic heterogeneity of the mutant enzyme in two patients. J. Clin. Investig. 1994, 93, 1740–1747. [Google Scholar] [CrossRef]
- Aoyama, T.; Wakui, K.; Orii, K.E.; Hashimoto, T.; Fukushima, Y. Fluorescence in situ hybridization mapping of the alpha and beta subunits (HADHA and HADHB) of human mitochondrial fatty acid beta-oxidation multienzyme complex to 2p23 and their evolution. Cytogenet. Genome Res. 1997, 79, 221–224. [Google Scholar] [CrossRef]
- Orii, K.E.; Orii, K.O.; Souri, M.; Orii, T.; Kondo, N.; Hashimoto, T.; Aoyama, T. Genes for the human mitochondrial trifunctional protein alpha- and beta-subunits are divergently transcribed from a common promoter region. J. Biol. Chem. 1999, 274, 8077–8084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibdah, J.A.; Bennett, M.J.; Rinaldo, P.; Zhao, Y.; Gibson, B.; Sims, H.F.; Strauss, A.W. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 1999, 340, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, P.; Raymond, K.; al-Odaib, A.; Bennett, M.J. Clinical and biochemical features of fatty acid oxidation disorders. Curr. Opin. Pediatr. 1998, 10, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Pons, R.; Roig, M.; Riudor, E.; Ribes, A.; Briones, P.; Ortigosa, L.; Baldellou, A.; Gil-Gibernau, J.; Olesti, M.; Navarro, C.; et al. The clinical spectrum of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr. Neurol. 1996, 14, 236–243. [Google Scholar] [CrossRef]
- Ibdah, J.A.; Tein, I.; Dionisi-Vici, C.; Bennett, M.J.; IJlst, L.; Gibson, B.; Wanders, R.J.; Strauss, A.W. Mild trifunctional protein deficiency is associated with progressive neuropathy and myopathy and suggests a novel genotype-phenotype correlation. J. Clin. Investig. 1998, 102, 1193–1199. [Google Scholar] [CrossRef]
- Sims, H.F.; Brackett, J.C.; Powell, C.K.; Treem, W.R.; Hale, D.E.; Bennett, M.J.; Gibson, B.; Shapiro, S.; Strauss, A.W. The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc. Natl. Acad. Sci. USA 1995, 92, 841–845. [Google Scholar] [CrossRef] [Green Version]
- IJlst, L.; Wanders, R.J.; Ushikubo, S.; Kamijo, T.; Hashimoto, T. Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim. Biophys. Acta 1994, 1215, 347–350. [Google Scholar] [CrossRef]
- Schoeman, M.N.; Batey, R.G.; Wilcken, B. Recurrent acute fatty liver of pregnancy associated with a fatty-acid oxidation defect in the offspring. Gastroenterology 1991, 100, 544–548. [Google Scholar] [CrossRef]
- Wilcken, B.; Leung, K.C.; Hammond, J.; Kamath, R.; Leonard, J.V. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet 1993, 341, 407–408. [Google Scholar] [CrossRef]
- Treem, W.R.; Rinaldo, P.; Hale, D.E.; Stanley, C.A.; Millington, D.S.; Hyams, J.S.; Jackson, S.; Turnbull, D.M. Acute fatty liver of pregnancy and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Hepatology 1994, 19, 339–345. [Google Scholar]
- Yang, Z.; Zhao, Y.; Bennett, M.J.; Strauss, A.W.; Ibdah, J.A. Fetal genotypes and pregnancy outcomes in 35 families with mitochondrial trifunctional protein mutations. Am. J. Obstet. Gynecol. 2002, 187, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yamada, J.; Zhao, Y.; Strauss, A.W.; Ibdah, J.A. Prospective screening for pediatric mitochondrial trifunctional protein defects in pregnancies complicated by liver disease. JAMA 2002, 288, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Ahn, J.; Reau, N.S. ACG Clinical Guideline: Liver Disease and Pregnancy. Am. J. Gastroenterol. 2016, 111, 176–194; quiz 196. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Brady, C.W.; Fleckenstein, J.; Forde, K.A.; Khungar, V.; Molleston, J.P.; Afshar, Y.; Terrault, N.A. Reproductive Health and Liver Disease: Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2021, 73, 318–365. [Google Scholar] [CrossRef]
- De Biase, I.; Viau, K.S.; Liu, A.; Yuzyuk, T.; Botto, L.D.; Pasquali, M.; Longo, N. Diagnosis, Treatment, and Clinical Outcome of Patients with Mitochondrial Trifunctional Protein/Long-Chain 3-Hydroxy Acyl-CoA Dehydrogenase Deficiency. JIMD Rep. 2017, 31, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibdah, J.A.; Zhao, Y.; Viola, J.; Gibson, B.; Bennett, M.J.; Strauss, A.W. Molecular prenatal diagnosis in families with fetal mitochondrial trifunctional protein mutations. J. Pediatr. 2001, 138, 396–399. [Google Scholar] [CrossRef]
- Innes, A.M.; Seargeant, L.E.; Balachandra, K.; Roe, C.R.; Wanders, R.J.; Ruiter, J.P.; Casiro, O.; Grewar, D.A.; Greenberg, C.R. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr. Res. 2000, 47, 43–45. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Minami, S.; Mitani, A.; Tanizaki, Y.; Booka, M.; Okutani, T.; Yamaguchi, S.; Ino, K. Acute fatty liver of pregnancy associated with fetal mitochondrial trifunctional protein deficiency. J. Obstet. Gynaecol. Res. 2015, 41, 799–802. [Google Scholar] [CrossRef]
- Matern, D.; Schehata, B.M.; Shekhawa, P.; Strauss, A.W.; Bennett, M.J.; Rinaldo, P. Placental floor infarction complicating the pregnancy of a fetus with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Mol. Genet. Metab. 2001, 72, 265–268. [Google Scholar] [CrossRef]
- Cecatto, C.; Godoy, K.D.S.; da Silva, J.C.; Amaral, A.U.; Wajner, M. Disturbance of mitochondrial functions provoked by the major long-chain 3-hydroxylated fatty acids accumulating in MTP and LCHAD deficiencies in skeletal muscle. Toxicol. In Vitro 2016, 36, 1–9. [Google Scholar] [CrossRef]
- Steinmann, D.; Knab, J.; Priebe, H.J. Perioperative management of a child with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Paediatr. Anaesth. 2010, 20, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Oey, N.A.; den Boer, M.E.; Ruiter, J.P.; Wanders, R.J.; Duran, M.; Waterham, H.R.; Boer, K.; van der Post, J.A.; Wijburg, F.A. High activity of fatty acid oxidation enzymes in human placenta: Implications for fetal-maternal disease. J. Inherit. Metab. Dis. 2003, 26, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Oey, N.A.; Ruiter, J.P.; Attié-Bitach, T.; Ijlst, L.; Wanders, R.J.; Wijburg, F.A. Fatty acid oxidation in the human fetus: Implications for fetal and adult disease. J. Inherit. Metab. Dis. 2006, 29, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.; McDermott, L. Long chain PUFA transport in human term placenta. J. Nutr. 2009, 139, 636–639. [Google Scholar] [CrossRef] [Green Version]
- Oey, N.A.; den Boer, M.E.; Wijburg, F.A.; Vekemans, M.; Augé, J.; Steiner, C.; Wanders, R.J.; Waterham, H.R.; Ruiter, J.P.; Attié-Bitach, T. Long-chain fatty acid oxidation during early human development. Pediatr. Res. 2005, 57, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, S.; Runmei, M. Retrospective study of seven cases with acute Fatty liver of pregnancy. ISRN Obstet. Gynecol. 2013, 2013, 730569. [Google Scholar] [CrossRef]
- Tonin, A.M.; Grings, M.; Busanello, E.N.; Moura, A.P.; Ferreira, G.C.; Viegas, C.M.; Fernandes, C.G.; Schuck, P.F.; Wajner, M. Long-chain 3-hydroxy fatty acids accumulating in LCHAD and MTP deficiencies induce oxidative stress in rat brain. Neurochem. Int. 2010, 56, 930–936. [Google Scholar] [CrossRef]
- Neuman-Łaniec, M.; Wierzba, J.; Irga, N.; Zaborowska-Sołtys, M.; Balcerska, A. LCHAD (long-chain 3-hydroxyacyl-CoA dehydrogenase) deficiency as a cause of sudden death of a three months old infant. Med. Wieku Rozw. 2002, 6, 221–226. [Google Scholar]
- Olpin, S.E.; Clark, S.; Andresen, B.S.; Bischoff, C.; Olsen, R.K.; Gregersen, N.; Chakrapani, A.; Downing, M.; Manning, N.J.; Sharrard, M.; et al. Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J. Inherit. Metab. Dis. 2005, 28, 533–544. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Thangaraj, K.R.; Eapen, C.E.; Ramachandran, A.; Mukhopadhya, A.; Mathai, M.; Seshadri, L.; Peedikayil, A.; Ramakrishna, B.; Balasubramanian, K.A. Liver injury in acute fatty liver of pregnancy: Possible link to placental mitochondrial dysfunction and oxidative stress. Hepatology 2010, 51, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, S.K.; Thomas, S.; Ramamoorthy, P.; Basivireddy, J.; Pulimood, A.B.; Ramachandran, A.; Balasubramanian, K.A. Oxidative stress in the development of liver cirrhosis: A comparison of two different experimental models. J. Gastroenterol. Hepatol. 2006, 21, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Eapen, C.E.; Pullimood, A.B.; Balasubramanian, K.A. Oxidative stress in experimental liver microvesicular steatosis: Role of mitochondria and peroxisomes. J. Gastroenterol. Hepatol. 2006, 21, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Thangaraj, K.R.; Goel, A.; Eapen, C.E.; Balasubramanian, K.A.; Ramachandran, A. Acute fatty liver of pregnancy: An update on mechanisms. Obstet. Med. 2011, 4, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Ingham, S.A.; Mohr, A.M.; Wehrkamp, C.J.; Ray, A.; Roy, S.; Cazanave, S.C.; Phillippi, M.A.; Mott, J.L. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology 2014, 60, 1942–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akazawa, Y.; Cazanave, S.; Mott, J.L.; Elmi, N.; Bronk, S.F.; Kohno, S.; Charlton, M.R.; Gores, G.J. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J. Hepatol. 2010, 52, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, A.K.; Glaser, S.S. Cholangiocyte lipoapoptosis: Implications for biliary damage during nonalcoholic fatty liver disease. Hepatology 2014, 60, 1809–1811. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Stringham, B.A.; Mohr, A.M.; Wehrkamp, C.J.; Lu, S.; Phillippi, M.A.; Harrison-Findik, D.; Mott, J.L. FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J. Lipid Res. 2017, 58, 866–875. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramanathan, R.; Ibdah, J.A. Mitochondrial Dysfunction and Acute Fatty Liver of Pregnancy. Int. J. Mol. Sci. 2022, 23, 3595. https://doi.org/10.3390/ijms23073595
Ramanathan R, Ibdah JA. Mitochondrial Dysfunction and Acute Fatty Liver of Pregnancy. International Journal of Molecular Sciences. 2022; 23(7):3595. https://doi.org/10.3390/ijms23073595
Chicago/Turabian StyleRamanathan, Raghu, and Jamal A. Ibdah. 2022. "Mitochondrial Dysfunction and Acute Fatty Liver of Pregnancy" International Journal of Molecular Sciences 23, no. 7: 3595. https://doi.org/10.3390/ijms23073595
APA StyleRamanathan, R., & Ibdah, J. A. (2022). Mitochondrial Dysfunction and Acute Fatty Liver of Pregnancy. International Journal of Molecular Sciences, 23(7), 3595. https://doi.org/10.3390/ijms23073595