
Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets

 ,
,
Abstract
:1. Introduction
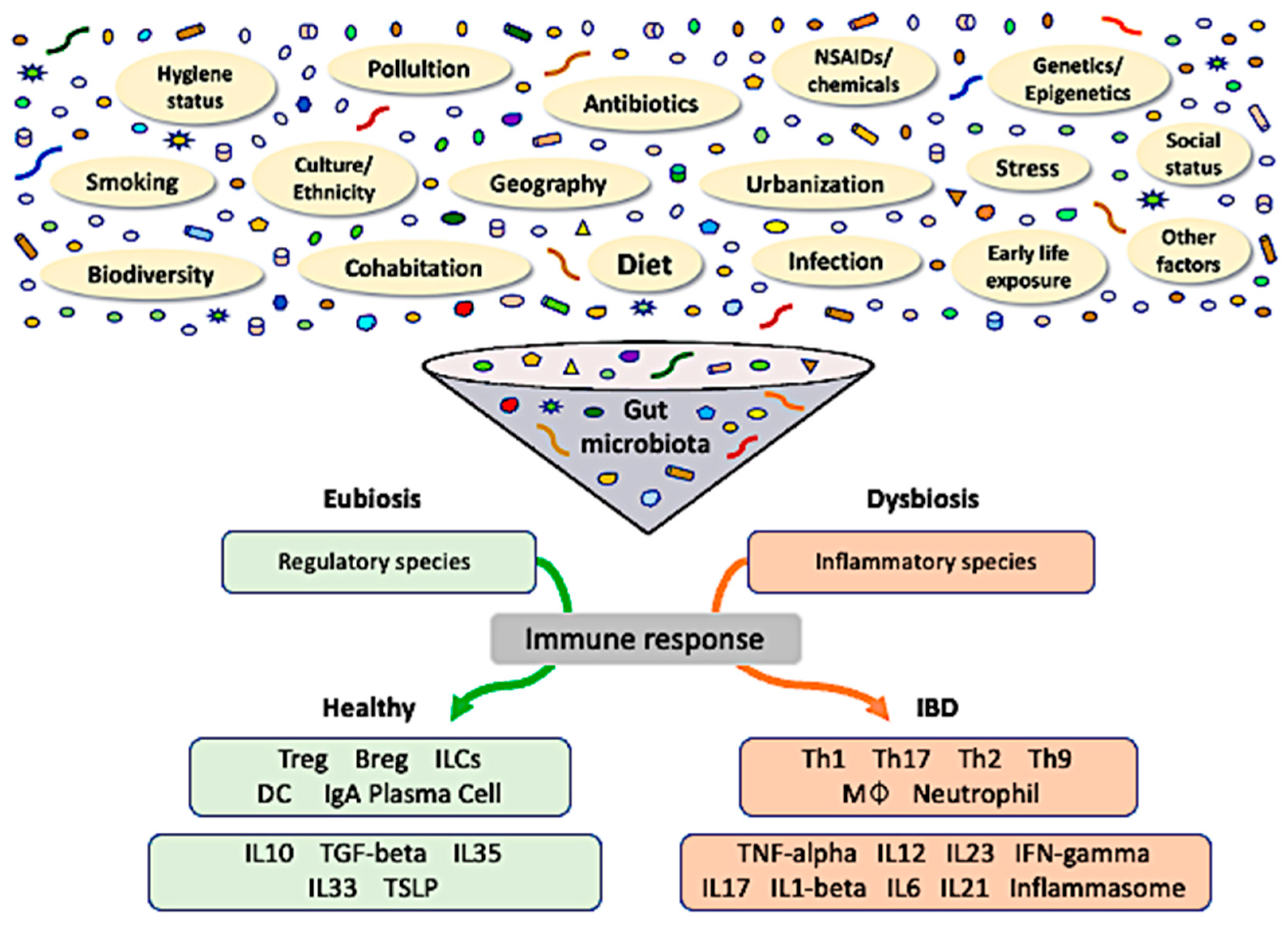
2. Intestinal Microbial Dysbiosis
3. Influence of the Exposome
3.1. Geosocial Factors
3.2. Antibiotics
3.3. Dietary Factors
4. Genetic Susceptibility
5. Epigenetic Modifications
6. Inappropriate Immune Response
7. Microbial-Based Therapies
8. Complex Genetic and Molecular Network
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caradonna, L.; Amati, L.; Magrone, T.; Pellegrino, N.M.; Jirillo, E.; Caccavo, D. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: Biological and clinical significance. J. Endotoxin Res. 2000, 6, 205–214. [Google Scholar] [PubMed]
- Mahida, Y.R.; Rolfe, V.E. Host-bacterial interactions in inflammatory bowel disease. Clin. Sci. 2004, 107, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Parkes, M. Evidence from genetics for a role of autophagy and innate immunity in IBD pathogenesis. Dig. Dis. 2012, 30, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.M.; Abreu, M.T. The innate immune system and inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 2014, 40, 706–719. [Google Scholar] [CrossRef] [Green Version]
- Taurog, J.D.; Richardson, J.A.; Croft, J.T.; Simmons, W.A.; Zhou, M.; Fernandez-Sueiro, J.L.; Balish, E.; Hammer, R.E. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 1994, 180, 2359–2364. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, J.; Bobe, A.M.; Miyoshi, S.; Huang, Y.; Hubert, N.; Delmont, T.O.; Eren, A.M.; Leone, V.; Chang, E.B. Peripartum Antibiotics Promote Gut Dysbiosis, Loss of Immune Tolerance, and Inflammatory Bowel Disease in Genetically Prone Offspring. Cell Rep. 2017, 20, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Packey, C.D.; Sartor, R.B. Interplay of commensal and pathogenic bacteria, genetic mutations, and immunoregulatory defects in the pathogenesis of inflammatory bowel diseases. J. Intern. Med. 2008, 263, 597–606. [Google Scholar] [CrossRef]
- Cohen, L.J.; Cho, J.H.; Gevers, D.; Chu, H. Genetic Factors and the Intestinal Microbiome Guide Development of Microbe-Based Therapies for Inflammatory Bowel Diseases. Gastroenterology 2019, 156, 2174–2189. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J. Gastroenterol. 2020, 55, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, W.; Goethel, A.; Bedrani, L.; Croitoru Mdcm, K. Determinants of IBD Heritability: Genes, Bugs, and More. Inflamm. Bowel. Dis. 2018, 24, 1133–1148. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas Lopez, R.; Grande Burgos, M.J.; Galvez, A.; Perez Pulido, R. The human gastrointestinal tract and oral microbiota in inflammatory bowel disease: A state of the science review. APMIS 2017, 125, 3–10. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vazquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Chang, E.B. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues. Gastroenterology 2021, 160, 524–537. [Google Scholar] [CrossRef]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef]
- Tlaskalova-Hogenova, H.; Tuckova, L.; Stepankova, R.; Hudcovic, T.; Palova-Jelinkova, L.; Kozakova, H.; Rossmann, P.; Sanchez, D.; Cinova, J.; Hrncir, T.; et al. Involvement of innate immunity in the development of inflammatory and autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1051, 787–798. [Google Scholar] [CrossRef]
- Sartor, R.B. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: Antibiotics, probiotics, and prebiotics. Gastroenterology 2004, 126, 1620–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eun, C.S.; Mishima, Y.; Wohlgemuth, S.; Liu, B.; Bower, M.; Carroll, I.M.; Sartor, R.B. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10-/- mice. Infect. Immun. 2014, 82, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORgammat(+) Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224.e4. [Google Scholar] [CrossRef] [Green Version]
- Clooney, A.G.; Eckenberger, J.; Laserna-Mendieta, E.; Sexton, K.A.; Bernstein, M.T.; Vagianos, K.; Sargent, M.; Ryan, F.J.; Moran, C.; Sheehan, D.; et al. Ranking microbiome variance in inflammatory bowel disease: A large longitudinal intercontinental study. Gut 2021, 70, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Martin, R.; Rossi, O.; Bermudez-Humaran, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef]
- Oyri, S.F.; Muzes, G.; Sipos, F. Dysbiotic gut microbiome: A key element of Crohn’s disease. Comp. Immunol. Microbiol. Infect. Dis. 2015, 43, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Lordan, C.; Ross, R.P.; Cotter, P.D. Gut microbes from the phylogenetically diverse genus Eubacterium and their various contributions to gut health. Gut Microbes 2020, 12, 1802866. [Google Scholar] [CrossRef]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.; Dore, J.; Leclerc, M.; McSweeney, C.S. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm. Bowel. Dis. 2010, 16, 2034–2042. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vazquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Azimi, T.; Nasiri, M.J.; Chirani, A.S.; Pouriran, R.; Dabiri, H. The role of bacteria in the inflammatory bowel disease development: A narrative review. APMIS 2018, 126, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, W.; Marya, N.; Fahed, J.; Leslie, G.; Patel, K.; Cave, D.R. Clostridium difficile in Inflammatory Bowel Disease: A Retrospective Study. Gastroenterol. Res. Pract. 2017, 2017, 4803262. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Bautista, J.; Padilla-Suarez, C.; Bouza, E.; Munoz, P.; Menchen, L.; Marin-Jimenez, I. Listeria monocytogenes infection in inflammatory bowel disease patients: Case series and review of the literature. Eur. J. Gastroenterol. Hepatol. 2014, 26, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, A.; Del Chierico, F.; Altomare, A.; Zorzi, F.; Cella, E.; Putignani, L.; Guarino, M.P.L.; Monteleone, G.; Cicala, M.; Angeletti, S.; et al. Exploring the genetic diversity of the 16S rRNA gene of Akkermansia muciniphila in IBD and IBS. Future Microbiol. 2019, 14, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.; Xue, X.; Zhang, L.; Yang, X.; Zhao, L.; Zhang, C. Strain-Specific Anti-inflammatory Properties of Two Akkermansia muciniphila Strains on Chronic Colitis in Mice. Front. Cell. Infect. Microbiol. 2019, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ji, X.; Lu, G.; Zhang, F. The potential of Akkermansia muciniphila in inflammatory bowel disease. Appl. Microbiol. Biotechnol. 2021, 105, 5785–5794. [Google Scholar] [CrossRef]
- Zocco, M.A.; dal Verme, L.Z.; Cremonini, F.; Piscaglia, A.C.; Nista, E.C.; Candelli, M.; Novi, M.; Rigante, D.; Cazzato, I.A.; Ojetti, V.; et al. Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment. Pharm. 2006, 23, 1567–1574. [Google Scholar] [CrossRef]
- Turroni, F.; Duranti, S.; Milani, C.; Lugli, G.A.; van Sinderen, D.; Ventura, M. Bifidobacterium bifidum: A Key Member of the Early Human Gut Microbiota. Microorganisms 2019, 7, 544. [Google Scholar] [CrossRef] [Green Version]
- Elguezabal, N.; Chamorro, S.; Molina, E.; Garrido, J.M.; Izeta, A.; Rodrigo, L.; Juste, R.A. Lactase persistence, NOD2 status and Mycobacterium avium subsp. paratuberculosis infection associations to Inflammatory Bowel Disease. Gut Pathog. 2012, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Sibartie, S.; Scully, P.; Keohane, J.; O’Neill, S.; O’Mahony, J.; O’Hanlon, D.; Kirwan, W.O.; O’Mahony, L.; Shanahan, F. Mycobacterium avium subsp. Paratuberculosis (MAP) as a modifying factor in Crohn’s disease. Inflamm. Bowel. Dis. 2010, 16, 296–304. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Mirsepasi-Lauridsen, H.C.; Vallance, B.A.; Krogfelt, K.A.; Petersen, A.M. Escherichia coli Pathobionts Associated with Inflammatory Bowel Disease. Clin. Microbiol. Rev. 2019, 32, e00060-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.A.; Garrett, W.S. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu. Rev. Microbiol. 2016, 70, 395–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neut, C.; Bulois, P.; Desreumaux, P.; Membre, J.M.; Lederman, E.; Gambiez, L.; Cortot, A.; Quandalle, P.; van Kruiningen, H.; Colombel, J.F. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 939–946. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [Green Version]
- Zafar, H.; Saier, M.H., Jr. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef]
- Goethel, A.; Croitoru, K.; Philpott, D.J. The interplay between microbes and the immune response in inflammatory bowel disease. J. Physiol. 2018, 596, 3869–3882. [Google Scholar] [CrossRef] [Green Version]
- Kolho, K.L.; Korpela, K.; Jaakkola, T.; Pichai, M.V.; Zoetendal, E.G.; Salonen, A.; de Vos, W.M. Fecal Microbiota in Pediatric Inflammatory Bowel Disease and Its Relation to Inflammation. Am. J. Gastroenterol. 2015, 110, 921–930. [Google Scholar] [CrossRef]
- Zhuang, X.; Liu, C.; Zhan, S.; Tian, Z.; Li, N.; Mao, R.; Zeng, Z.; Chen, M. Gut Microbiota Profile in Pediatric Patients With Inflammatory Bowel Disease: A Systematic Review. Front. Pediatr. 2021, 9, 626232. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Ribaldone, D.G.; Caviglia, G.P.; Abdulle, A.; Pellicano, R.; Ditto, M.C.; Morino, M.; Fusaro, E.; Saracco, G.M.; Bugianesi, E.; Astegiano, M. Adalimumab Therapy Improves Intestinal Dysbiosis in Crohn’s Disease. J. Clin. Med. 2019, 8, 1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, N.; MacDonald, T.T. Recent advances in gut immunology. Parasite Immunol. 2017, 39, e12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nkamga, V.D.; Lotte, R.; Chirio, D.; Lonjon, M.; Roger, P.M.; Drancourt, M.; Ruimy, R. Methanobrevibacter oralis detected along with Aggregatibacter actinomycetemcomitans in a series of community-acquired brain abscesses. Clin. Microbiol. Infect. 2018, 24, 207–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matijasic, M.; Mestrovic, T.; Paljetak, H.C.; Peric, M.; Baresic, A.; Verbanac, D. Gut Microbiota beyond Bacteria-Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.B.; Rostami, E.; Sephay, A.A.; Shahrokh, S.; Balaii, H.; Aghdaei, H.A.; Zali, M.R. Alterations of the human gut Methanobrevibacter smithii as a biomarker for inflammatory bowel diseases. Microb. Pathog. 2018, 117, 285–289. [Google Scholar] [CrossRef]
- Blais Lecours, P.; Marsolais, D.; Cormier, Y.; Berberi, M.; Hache, C.; Bourdages, R.; Duchaine, C. Increased prevalence of Methanosphaera stadtmanae in inflammatory bowel diseases. PLoS ONE 2014, 9, e87734. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Perez-Brocal, V.; Garcia-Lopez, R.; Nos, P.; Beltran, B.; Moret, I.; Moya, A. Metagenomic Analysis of Crohn’s Disease Patients Identifies Changes in the Virome and Microbiome Related to Disease Status and Therapy, and Detects Potential Interactions and Biomarkers. Inflamm. Bowel. Dis. 2015, 21, 2515–2532. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef] [Green Version]
- Beheshti-Maal, A.; Shahrokh, S.; Ansari, S.; Mirsamadi, E.S.; Yadegar, A.; Mirjalali, H.; Zali, M.R. Gut mycobiome: The probable determinative role of fungi in IBD patients. Mycoses 2021, 64, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.L.; Erb Downward, J.R.; Mason, K.D.; Falkowski, N.R.; Eaton, K.A.; Kao, J.Y.; Young, V.B.; Huffnagle, G.B. Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect. Immun. 2012, 80, 3371–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tilburg Bernardes, E.; Pettersen, V.K.; Gutierrez, M.W.; Laforest-Lapointe, I.; Jendzjowsky, N.G.; Cavin, J.B.; Vicentini, F.A.; Keenan, C.M.; Ramay, H.R.; Samara, J.; et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat. Commun. 2020, 11, 2577. [Google Scholar] [CrossRef] [PubMed]
- de Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Ng, S.C.; Tang, W.; Leong, R.W.; Chen, M.; Ko, Y.; Studd, C.; Niewiadomski, O.; Bell, S.; Kamm, M.A.; de Silva, H.J.; et al. Environmental risk factors in inflammatory bowel disease: A population-based case-control study in Asia-Pacific. Gut 2015, 64, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Liu, H.; Xu, G.; Laugsand, J.B.; Pang, Z. Exploring Links Between Industrialization, Urbanization, and Chinese Inflammatory Bowel Disease. Front. Med. 2021, 8, 757025. [Google Scholar] [CrossRef]
- Zuo, T.; Kamm, M.A.; Colombel, J.F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef]
- Nielsen, C.C.; Gascon, M.; Osornio-Vargas, A.R.; Shier, C.; Guttman, D.S.; Becker, A.B.; Azad, M.B.; Sears, M.R.; Lefebvre, D.L.; Moraes, T.J.; et al. Natural environments in the urban context and gut microbiota in infants. Environ. Int. 2020, 142, 105881. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Vavricka, S. Exposome in IBD: Recent insights in environmental factors that influence the onset and course of IBD. Inflamm. Bowel. Dis. 2015, 21, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Saidel-Odes, L.; Odes, S. Hygiene hypothesis in inflammatory bowel disease. Ann. Gastroenterol. 2014, 27, 189–190. [Google Scholar] [PubMed]
- Probert, C.S.; Jayanthi, V.; Pinder, D.; Wicks, A.C.; Mayberry, J.F. Epidemiological study of ulcerative proctocolitis in Indian migrants and the indigenous population of Leicestershire. Gut 1992, 33, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Carr, I.; Mayberry, J.F. The effects of migration on ulcerative colitis: A three-year prospective study among Europeans and first- and second- generation South Asians in Leicester (1991–1994). Am. J. Gastroenterol. 1999, 94, 2918–2922. [Google Scholar] [CrossRef]
- Benchimol, E.I.; Manuel, D.G.; To, T.; Mack, D.R.; Nguyen, G.C.; Gommerman, J.L.; Croitoru, K.; Mojaverian, N.; Wang, X.; Quach, P.; et al. Asthma, type 1 and type 2 diabetes mellitus, and inflammatory bowel disease amongst South Asian immigrants to Canada and their children: A population-based cohort study. PLoS ONE 2015, 10, e0123599. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.; Kariyawasam, V.; Karnib, M.; Butcher, R.; Samuel, D.; Alrubaie, A.; Rahme, N.; McDonald, C.; Cowlishaw, J.; Katelaris, P.; et al. Inflammatory Bowel Disease Environmental Risk Factors: A Population-Based Case-Control Study of Middle Eastern Migration to Australia. Clin. Gastroenterol. Hepatol. 2015, 13, 1453–1463.e1. [Google Scholar] [CrossRef]
- Carr, E.J.; Dooley, J.; Garcia-Perez, J.E.; Lagou, V.; Lee, J.C.; Wouters, C.; Meyts, I.; Goris, A.; Boeckxstaens, G.; Linterman, M.A.; et al. The cellular composition of the human immune system is shaped by age and cohabitation. Nat. Immunol. 2016, 17, 461–468. [Google Scholar] [CrossRef]
- Valles-Colomer, M.; Bacigalupe, R.; Vieira-Silva, S.; Suzuki, S.; Darzi, Y.; Tito, R.Y.; Yamada, T.; Segata, N.; Raes, J.; Falony, G. Variation and transmission of the human gut microbiota across multiple familial generations. Nat. Microbiol. 2022, 7, 87–96. [Google Scholar] [CrossRef]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e620. [Google Scholar] [CrossRef] [Green Version]
- Hanski, I.; von Hertzen, L.; Fyhrquist, N.; Koskinen, K.; Torppa, K.; Laatikainen, T.; Karisola, P.; Auvinen, P.; Paulin, L.; Makela, M.J.; et al. Environmental biodiversity, human microbiota, and allergy are interrelated. Proc. Natl. Acad. Sci. USA 2012, 109, 8334–8339. [Google Scholar] [CrossRef] [Green Version]
- Gascon, M.; Vrijheid, M.; Nieuwenhuijsen, M.J. The Built Environment and Child Health: An Overview of Current Evidence. Curr. Environ. Health Rep. 2016, 3, 250–257. [Google Scholar] [CrossRef]
- Genuneit, J.; Standl, M. Epidemiology of Allergy: Natural Course and Risk Factors of Allergic Diseases. Handb. Exp. Pharm. 2022, 268, 21–27. [Google Scholar] [CrossRef]
- Kuenzig, M.E.; Bishay, K.; Leigh, R.; Kaplan, G.G.; Benchimol, E.I.; Crowdscreen, S.R.R.T. Co-occurrence of Asthma and the Inflammatory Bowel Diseases: A Systematic Review and Meta-analysis. Clin. Transl. Gastroenterol. 2018, 9, 188. [Google Scholar] [CrossRef]
- da Luz Moreira, A.; de Campos Lobato, L.F.; de Lima Moreira, J.P.; Luiz, R.R.; Elia, C.; Fiocchi, C.; de Souza, H.S.P. Geosocial Features and Loss of Biodiversity Underlie Variable Rates of Inflammatory Bowel Disease in a Large Developing Country: A Population-Based Study. Inflamm. Bowel. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Bernstein, C.N.; Gearry, R.; Hviid, A.; Kolho, K.L.; Kronman, M.P.; Shaw, S.; Van Kruiningen, H.; Colombel, J.F.; Atreja, A. Antibiotics associated with increased risk of new-onset Crohn’s disease but not ulcerative colitis: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Aniwan, S.; Tremaine, W.J.; Raffals, L.E.; Kane, S.V.; Loftus, E.V., Jr. Antibiotic Use and New-Onset Inflammatory Bowel Disease in Olmsted County, Minnesota: A Population-Based Case-Control Study. J. Crohns Colitis 2018, 12, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.Y.; Blanchard, J.F.; Bernstein, C.N. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Am. J. Gastroenterol. 2010, 105, 2687–2692. [Google Scholar] [CrossRef]
- Hviid, A.; Svanstrom, H.; Frisch, M. Antibiotic use and inflammatory bowel diseases in childhood. Gut 2011, 60, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Ortqvist, A.K.; Lundholm, C.; Halfvarson, J.; Ludvigsson, J.F.; Almqvist, C. Fetal and early life antibiotics exposure and very early onset inflammatory bowel disease: A population-based study. Gut 2019, 68, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, H.; Malmborg, P.; Askling, J.; Ekbom, A.; Montgomery, S.M. Early-life exposures associated with antibiotic use and risk of subsequent Crohn’s disease. Scand. J. Gastroenterol. 2008, 43, 961–966. [Google Scholar] [CrossRef]
- Vu Thi Ngoc, B.; Ho Bich, H.; Galazzo, G.; Vu Tien Viet, D.; Oomen, M.; Nghiem Nguyen Minh, T.; Tran Huy, H.; van Doorn, H.R.; Wertheim, H.F.L.; Penders, J. Cross-Sectional Analysis of the Microbiota of Human Gut and Its Direct Environment in a Household Cohort with High Background of Antibiotic Use. Microorganisms 2021, 9, 2115. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Sartorio, M.U.A.; Pendezza, E.; Coppola, S.; Paparo, L.; D’Auria, E.; Zuccotti, G.V.; Berni Canani, R. Potential Role of Omega-3 Polyunsaturated Fatty Acids in Pediatric Food Allergy. Nutrients 2021, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, Y.; Gao, H.; Li, D.; Jiang, R.; Ge, L.; Tong, C.; Xu, K. Associations among Dietary Omega-3 Polyunsaturated Fatty Acids, the Gut Microbiota, and Intestinal Immunity. Mediat. Inflamm. 2021, 2021, 8879227. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Bolick, D.T.; Leng, J.; Medlock, G.L.; Kolling, G.L.; Papin, J.A.; Swann, J.R.; Guerrant, R.L. Protein- and zinc-deficient diets modulate the murine microbiome and metabolic phenotype. Am. J. Clin. Nutr. 2016, 104, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, S.M. Dietary factors in the development of type 1 diabetes. Pediatr. Diabetes 2016, 17 (Suppl. S22), 49–55. [Google Scholar] [CrossRef] [PubMed]
- Del Pinto, R.; Pietropaoli, D.; Chandar, A.K.; Ferri, C.; Cominelli, F. Association Between Inflammatory Bowel Disease and Vitamin D Deficiency: A Systematic Review and Meta-analysis. Inflamm. Bowel. Dis. 2015, 21, 2708–2717. [Google Scholar] [CrossRef] [Green Version]
- de Bruyn, J.R.; van Heeckeren, R.; Ponsioen, C.Y.; van den Brink, G.R.; Lowenberg, M.; Bredenoord, A.J.; Frijstein, G.; D’Haens, G.R. Vitamin D deficiency in Crohn’s disease and healthy controls: A prospective case-control study in the Netherlands. J. Crohns Colitis 2014, 8, 1267–1273. [Google Scholar] [CrossRef] [Green Version]
- Kearney, J. Food consumption trends and drivers. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2793–2807. [Google Scholar] [CrossRef]
- Cho, J.H.; Brant, S.R. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011, 140, 1704–1712. [Google Scholar] [CrossRef] [Green Version]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo. Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lange, K.M.; Barrett, J.C. Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun. 2015, 64, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Oshima, T.; Tomita, T.; Fukui, H.; Miwa, H. Butyrate Alleviates Cytokine-Induced Barrier Dysfunction by Modifying Claudin-2 Levels. Biology 2021, 10, 205. [Google Scholar] [CrossRef]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campoy, E.; Colombo, M.I. Autophagy in intracellular bacterial infection. Biochim. Biophys. Acta 2009, 1793, 1465–1477. [Google Scholar] [CrossRef] [Green Version]
- Elliott, T.R.; Hudspith, B.N.; Rayment, N.B.; Prescott, N.J.; Petrovska, L.; Hermon-Taylor, J.; Brostoff, J.; Boussioutas, A.; Mathew, C.G.; Sanderson, J.D. Defective macrophage handling of Escherichia coli in Crohn’s disease. J. Gastroenterol. Hepatol. 2015, 30, 1265–1274. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Messer, J.S.; Murphy, S.F.; Logsdon, M.F.; Lodolce, J.P.; Grimm, W.A.; Bartulis, S.J.; Vogel, T.P.; Burn, M.; Boone, D.L. The Crohn’s disease: Associated ATG16L1 variant and Salmonella invasion. BMJ Open 2013, 3, e002790. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.L.; Kuballa, P.; Song, J.H.; Patel, K.K.; Castoreno, A.B.; Yilmaz, O.H.; Jijon, H.B.; Zhang, M.; Aldrich, L.N.; Villablanca, E.J.; et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 2013, 145, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharl, M.; Rogler, G. Inflammatory bowel disease: Dysfunction of autophagy? Dig. Dis. 2012, 30 (Suppl. S3), 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, J.D.; Schlossmacher, M.G.; Philpott, D.J. LRRK2 and Nod2 promote lysozyme sorting in Paneth cells. Nat. Immunol. 2015, 16, 898–900. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Komatsu, M.; Virgin, H.W.t.; Stappenbeck, T.S. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009, 5, 250–252. [Google Scholar] [CrossRef] [Green Version]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Baxt, L.A.; Xavier, R.J. Role of Autophagy in the Maintenance of Intestinal Homeostasis. Gastroenterology 2015, 149, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Negroni, A.; Pierdomenico, M.; Cucchiara, S.; Stronati, L. NOD2 and inflammation: Current insights. J. Inflamm. Res. 2018, 11, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naser, S.A.; Arce, M.; Khaja, A.; Fernandez, M.; Naser, N.; Elwasila, S.; Thanigachalam, S. Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J. Gastroenterol. 2012, 18, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Lassen, K.G.; Xavier, R.J. Advances in inflammatory bowel disease pathogenesis: Linking host genetics and the microbiome. Gut 2013, 62, 1505–1510. [Google Scholar] [CrossRef]
- Wehkamp, J.; Salzman, N.H.; Porter, E.; Nuding, S.; Weichenthal, M.; Petras, R.E.; Shen, B.; Schaeffeler, E.; Schwab, M.; Linzmeier, R.; et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 18129–18134. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, H.H.; McKenzie, B.S.; Hue, S.; Thompson, C.; Joyce-Shaikh, B.; Stepankova, R.; Robinson, N.; Buonocore, S.; Tlaskalova-Hogenova, H.; Cua, D.J.; et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 2006, 25, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Cong, Y.; Weaver, C.T.; Schoeb, T.R.; McClanahan, T.K.; Fick, R.B.; Kastelein, R.A. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 2007, 132, 2359–2370. [Google Scholar] [CrossRef]
- Yu, R.Y.; Gallagher, G. A naturally occurring, soluble antagonist of human IL-23 inhibits the development and in vitro function of human Th17 cells. J. Immunol. 2010, 185, 7302–7308. [Google Scholar] [CrossRef]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [Green Version]
- Spalinger, M.R.; McCole, D.F.; Rogler, G.; Scharl, M. Protein tyrosine phosphatase non-receptor type 2 and inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1034–1044. [Google Scholar] [CrossRef] [Green Version]
- Spalinger, M.R.; Manzini, R.; Hering, L.; Riggs, J.B.; Gottier, C.; Lang, S.; Atrott, K.; Fettelschoss, A.; Olomski, F.; Kundig, T.M.; et al. PTPN2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer. Cell Rep. 2018, 22, 1835–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharl, M.; Mwinyi, J.; Fischbeck, A.; Leucht, K.; Eloranta, J.J.; Arikkat, J.; Pesch, T.; Kellermeier, S.; Mair, A.; Kullak-Ublick, G.A.; et al. Crohn’s disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm. Bowel. Dis. 2012, 18, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Spalinger, M.R.; Kasper, S.; Chassard, C.; Raselli, T.; Frey-Wagner, I.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Mair, F.; et al. PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal. Immunol. 2015, 8, 918–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagawa, T.; Kitani, A.; Fuss, I.; Levine, B.; Brant, S.R.; Peter, I.; Tajima, M.; Nakamura, S.; Strober, W. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci. Transl. Med. 2018, 10, eaan8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellermayer, R. Challenges for epigenetic research in inflammatory bowel diseases. Epigenomics 2017, 9, 527–538. [Google Scholar] [CrossRef]
- Benchimol, E.I.; Mack, D.R.; Guttmann, A.; Nguyen, G.C.; To, T.; Mojaverian, N.; Quach, P.; Manuel, D.G. Inflammatory bowel disease in immigrants to Canada and their children: A population-based cohort study. Am. J. Gastroenterol. 2015, 110, 553–563. [Google Scholar] [CrossRef]
- Dabritz, J.; Menheniott, T.R. Linking immunity, epigenetics, and cancer in inflammatory bowel disease. Inflamm. Bowel. Dis. 2014, 20, 1638–1654. [Google Scholar] [CrossRef] [Green Version]
- Amatullah, H.; Jeffrey, K.L. Epigenome-metabolome-microbiome axis in health and IBD. Curr. Opin. Microbiol. 2020, 56, 97–108. [Google Scholar] [CrossRef]
- Woo, V.; Alenghat, T. Epigenetic regulation by gut microbiota. Gut Microbes 2022, 14, 2022407. [Google Scholar] [CrossRef]
- Wawrzyniak, M.; Scharl, M. Genetics and epigenetics of inflammatory bowel disease. Swiss Med. Wkly 2018, 148, w14671. [Google Scholar] [CrossRef]
- Ray, G.; Longworth, M.S. Epigenetics, DNA Organization, and Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2019, 25, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A. MicroRNAs: New players in inflammatory bowel disease. Gut 2015, 64, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Chen, L.; Xu, L.; Bilotta, A.J.; Yao, S.; Liu, Z.; Cong, Y. MicroRNA-10a Negatively Regulates CD4(+) T Cell IL-10 Production through Suppression of Blimp1. J. Immunol. 2021, 207, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.G.; Pekow, J. The emerging role of miRNAs in inflammatory bowel disease: A review. Ther. Adv. Gastroenterol. 2015, 8, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Ceppi, M.; Pereira, P.M.; Dunand-Sauthier, I.; Barras, E.; Reith, W.; Santos, M.A.; Pierre, P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2735–2740. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrova, K.; Romero-Mosquera, B.; Hernandez, V. Diet, Gut Microbiome and Epigenetics: Emerging Links with Inflammatory Bowel Diseases and Prospects for Management and Prevention. Nutrients 2017, 9, 962. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Alim, I.; Bultman, S.J.; Ratan, R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016, 625, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [Green Version]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediat. Inflamm. 2014, 2014, 162021. [Google Scholar] [CrossRef]
- Paul, B.; Barnes, S.; Demark-Wahnefried, W.; Morrow, C.; Salvador, C.; Skibola, C.; Tollefsbol, T.O. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin. Epigenetics 2015, 7, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miro-Blanch, J.; Yanes, O. Epigenetic Regulation at the Interplay Between Gut Microbiota and Host Metabolism. Front. Genet. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, A. Microbial pathogenesis in inflammatory bowel diseases. Microb. Pathog. 2022, 163, 105383. [Google Scholar] [CrossRef]
- Lecuyer, E.; Rakotobe, S.; Lengline-Garnier, H.; Lebreton, C.; Picard, M.; Juste, C.; Fritzen, R.; Eberl, G.; McCoy, K.D.; Macpherson, A.J.; et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 2014, 40, 608–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Chen, H.; Shu, X.; Yin, Y.; Li, J.; Qin, J.; Chen, L.; Peng, K.; Xu, F.; Gu, W.; et al. Presence of Segmented Filamentous Bacteria in Human Children and Its Potential Role in the Modulation of Human Gut Immunity. Front. Microbiol. 2018, 9, 1403. [Google Scholar] [CrossRef]
- Hansen, J.J.; Sartor, R.B. Therapeutic Manipulation of the Microbiome in IBD: Current Results and Future Approaches. Curr. Treat. Options Gastroenterol. 2015, 13, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Richard, M.L.; Sokol, H. The gut mycobiota: Insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 331–345. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Sanders, D.J.; Inniss, S.; Sebepos-Rogers, G.; Rahman, F.Z.; Smith, A.M. The role of the microbiome in gastrointestinal inflammation. Biosci. Rep. 2021, 41, BSR20203850. [Google Scholar] [CrossRef]
- Nanini, H.F.; Bernardazzi, C.; Castro, F.; de Souza, H.S.P. Damage-associated molecular patterns in inflammatory bowel disease: From biomarkers to therapeutic targets. World J. Gastroenterol. 2018, 24, 4622–4634. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.H.; Liu, L.; Hou, Y.Y.; Shen, S.N.; Wang, T.T. C-type lectin receptor-mediated immune recognition and response of the microbiota in the gut. Gastroenterol. Rep. 2019, 7, 312–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Okabe, Y.; Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014, 157, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Ebihara, T. Dichotomous Regulation of Acquired Immunity by Innate Lymphoid Cells. Cells 2020, 9, 1193. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Modulation of inflammasome pathways by bacterial and viral pathogens. J. Immunol. 2011, 187, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Vanhove, W.; Peeters, P.M.; Staelens, D.; Schraenen, A.; Van der Goten, J.; Cleynen, I.; De Schepper, S.; Van Lommel, L.; Reynaert, N.L.; Schuit, F.; et al. Strong Upregulation of AIM2 and IFI16 Inflammasomes in the Mucosa of Patients with Active Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2015, 21, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Dong, Y.; Ye, M.; Jin, S.; Yang, J.; Joosse, M.E.; Sun, Y.; Zhang, J.; Lazarev, M.; Brant, S.R.; et al. The Pathogenic Role of NLRP3 Inflammasome Activation in Inflammatory Bowel Diseases of Both Mice and Humans. J. Crohns Colitis 2017, 11, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Geremia, A.; Arancibia-Carcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef]
- Shepherd, F.R.; McLaren, J.E. T Cell Immunity to Bacterial Pathogens: Mechanisms of Immune Control and Bacterial Evasion. Int. J. Mol. Sci. 2020, 21, 6144. [Google Scholar] [CrossRef]
- Silberger, D.J.; Zindl, C.L.; Weaver, C.T. Citrobacter rodentium: A model enteropathogen for understanding the interplay of innate and adaptive components of type 3 immunity. Mucosal. Immunol. 2017, 10, 1108–1117. [Google Scholar] [CrossRef] [Green Version]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef]
- Ledder, O.; Turner, D. Antibiotics in IBD: Still a Role in the Biological Era? Inflamm. Bowel. Dis. 2018, 24, 1676–1688. [Google Scholar] [CrossRef]
- Selby, W.; Pavli, P.; Crotty, B.; Florin, T.; Radford-Smith, G.; Gibson, P.; Mitchell, B.; Connell, W.; Read, R.; Merrett, M.; et al. Two-year combination antibiotic therapy with clarithromycin, rifabutin, and clofazimine for Crohn’s disease. Gastroenterology 2007, 132, 2313–2319. [Google Scholar] [CrossRef]
- Thia, K.T.; Mahadevan, U.; Feagan, B.G.; Wong, C.; Cockeram, A.; Bitton, A.; Bernstein, C.N.; Sandborn, W.J. Ciprofloxacin or metronidazole for the treatment of perianal fistulas in patients with Crohn’s disease: A randomized, double-blind, placebo-controlled pilot study. Inflamm. Bowel. Dis. 2009, 15, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthes, H.; Krummenerl, T.; Giensch, M.; Wolff, C.; Schulze, J. Clinical trial: Probiotic treatment of acute distal ulcerative colitis with rectally administered Escherichia coli Nissle 1917 (EcN). BMC Complement. Altern. Med. 2010, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorak, R.N. Probiotics in the management of ulcerative colitis. Gastroenterol. Hepatol. 2010, 6, 688–690. [Google Scholar]
- Bibiloni, R.; Fedorak, R.N.; Tannock, G.W.; Madsen, K.L.; Gionchetti, P.; Campieri, M.; De Simone, C.; Sartor, R.B. VSL#3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am. J. Gastroenterol. 2005, 100, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Weingarden, A.R.; Vaughn, B.P. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 2017, 8, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasilewski, A.; Zielinska, M.; Storr, M.; Fichna, J. Beneficial Effects of Probiotics, Prebiotics, Synbiotics, and Psychobiotics in Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2015, 21, 1674–1682. [Google Scholar] [CrossRef]
- Motta, J.P.; Bermudez-Humaran, L.G.; Deraison, C.; Martin, L.; Rolland, C.; Rousset, P.; Boue, J.; Dietrich, G.; Chapman, K.; Kharrat, P.; et al. Food-grade bacteria expressing elafin protect against inflammation and restore colon homeostasis. Sci. Transl. Med. 2012, 4, 158ra144. [Google Scholar] [CrossRef]
- Hanson, M.L.; Hixon, J.A.; Li, W.; Felber, B.K.; Anver, M.R.; Stewart, C.A.; Janelsins, B.M.; Datta, S.K.; Shen, W.; McLean, M.H.; et al. Oral delivery of IL-27 recombinant bacteria attenuates immune colitis in mice. Gastroenterology 2014, 146, 210–221.e213. [Google Scholar] [CrossRef] [Green Version]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 2000, 289, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, J.G.; del Carmen, S.; Miyoshi, A.; Azevedo, V.; Sesma, F.; Langella, P.; Bermudez-Humaran, L.G.; Watterlot, L.; Perdigon, G.; de Moreno de LeBlanc, A. Use of superoxide dismutase and catalase producing lactic acid bacteria in TNBS induced Crohn’s disease in mice. J. Biotechnol. 2011, 151, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen, S.; de Moreno de LeBlanc, A.; Martin, R.; Chain, F.; Langella, P.; Bermudez-Humaran, L.G.; LeBlanc, J.G. Genetically engineered immunomodulatory Streptococcus thermophilus strains producing antioxidant enzymes exhibit enhanced anti-inflammatory activities. Appl. Environ. Microbiol. 2014, 80, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109.e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, A.; Mahajan, R.; Singh, A.; Midha, V.; Mehta, V.; Narang, V.; Singh, T.; Singh Pannu, A. Role of Faecal Microbiota Transplantation for Maintenance of Remission in Patients With Ulcerative Colitis: A Pilot Study. J. Crohns Colitis 2019, 13, 1311–1317. [Google Scholar] [CrossRef]
- Dang, X.; Xu, M.; Liu, D.; Zhou, D.; Yang, W. Assessing the efficacy and safety of fecal microbiota transplantation and probiotic VSL#3 for active ulcerative colitis: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0228846. [Google Scholar] [CrossRef]
- Vich Vila, A.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef] [Green Version]
- Blachier, F.; Davila, A.M.; Mimoun, S.; Benetti, P.H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Bouillaud, F.; Tome, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2010, 39, 335–347. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef]
- Lewis, J.D.; Chen, E.Z.; Baldassano, R.N.; Otley, A.R.; Griffiths, A.M.; Lee, D.; Bittinger, K.; Bailey, A.; Friedman, E.S.; Hoffmann, C.; et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 2015, 18, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Gomez, G.; Mayorga, L.; Oyarzun, I.; Roca, J.; Borruel, N.; Casellas, F.; Varela, E.; Pozuelo, M.; Machiels, K.; Guarner, F.; et al. Dysbiosis and relapse-related microbiome in inflammatory bowel disease: A shotgun metagenomic approach. Comput. Struct. Biotechnol. J. 2021, 19, 6481–6489. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiser, M.; Simon, J.M.; Kochar, B.; Tovar, A.; Israel, J.W.; Robinson, A.; Gipson, G.R.; Schaner, M.S.; Herfarth, H.H.; Sartor, R.B.; et al. Molecular classification of Crohn’s disease reveals two clinically relevant subtypes. Gut 2018, 67, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.O.; Marigorta, U.M.; Schirmer, M.; Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, K.; Catesson, A.; Griffin, J.L.; Holmes, E.; Williams, H.R.T. Metabolomic Analysis in Inflammatory Bowel Disease: A Systematic Review. J. Crohns Colitis 2021, 15, 813–826. [Google Scholar] [CrossRef]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orru, S.; Blois, S.; et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Desbonnet, L.; Clarke, G.; Traplin, A.; O’Sullivan, O.; Crispie, F.; Moloney, R.D.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F. Gut microbiota depletion from early adolescence in mice: Implications for brain and behaviour. Brain Behav. Immun. 2015, 48, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248.e1. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Bennike, T.B.; Carlsen, T.G.; Ellingsen, T.; Bonderup, O.K.; Glerup, H.; Bogsted, M.; Christiansen, G.; Birkelund, S.; Stensballe, A.; Andersen, V. Neutrophil Extracellular Traps in Ulcerative Colitis: A Proteome Analysis of Intestinal Biopsies. Inflamm. Bowel. Dis. 2015, 21, 2052–2067. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Riaz, T.; Sollid, L.M.; Olsen, I.; de Souza, G.A. Quantitative Proteomics of Gut-Derived Th1 and Th1/Th17 Clones Reveal the Presence of CD28+ NKG2D- Th1 Cytotoxic CD4+ T cells. Mol. Cell Proteom. 2016, 15, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Duguet, F.; Locard-Paulet, M.; Marcellin, M.; Chaoui, K.; Bernard, I.; Andreoletti, O.; Lesourne, R.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; Saoudi, A. Proteomic Analysis of Regulatory T Cells Reveals the Importance of Themis1 in the Control of Their Suppressive Function. Mol. Cell Proteom. 2017, 16, 1416–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoodi, M.; Pearl, D.S.; Eiden, M.; Shute, J.K.; Brown, J.F.; Calder, P.C.; Trebble, T.M. Altered colonic mucosal Polyunsaturated Fatty Acid (PUFA) derived lipid mediators in ulcerative colitis: New insight into relationship with disease activity and pathophysiology. PLoS ONE 2013, 8, e76532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewell, G.W.; Hannun, Y.A.; Han, X.; Koster, G.; Bielawski, J.; Goss, V.; Smith, P.J.; Rahman, F.Z.; Vega, R.; Bloom, S.L.; et al. Lipidomic profiling in Crohn’s disease: Abnormalities in phosphatidylinositols, with preservation of ceramide, phosphatidylcholine and phosphatidylserine composition. Int. J. Biochem. Cell Biol. 2012, 44, 1839–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karner, M.; Kocjan, A.; Stein, J.; Schreiber, S.; von Boyen, G.; Uebel, P.; Schmidt, C.; Kupcinskas, L.; Dina, I.; Zuelch, F.; et al. First multicenter study of modified release phosphatidylcholine “LT-02” in ulcerative colitis: A randomized, placebo-controlled trial in mesalazine-refractory courses. Am. J. Gastroenterol. 2014, 109, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Microbiome Components | Presence in IBD | Possible Mechanisms | Evidence | References |
|---|---|---|---|---|
| Firmicutes | ||||
| Faecalibacterium prausnitzii |  | It is a highly active metabolic commensal bacterium involved in the production of butyrate. This metabolite plays a major role in gut physiology and has beneficial effects, including protection against pathogen invasion, modulation of the immune system, and promotion of anti-inflammatory activity | Presence of F. prausnitzii may serve as a biomarker of intestinal health in adults. Low levels of this bacteria could be predictive for CD. Its deficiency was shown in colonic CD. Low F. prausnitzii levels in patients with IBD undergoing surgery is associated with a higher risk of post-operative recurrence | [25,27] |
| Eubacterium spp. | | Involved in the production of SCFAs, especially butyrate. It is important in inflammation modulation and the promotion of epithelial barrier integrity | Found deficient in samples from patients with CD and UC | [24,28] |
| Ruminococcus albus | | Possibly involved in SCFA metabolism and its protective and anti-inflammatory roles | Found decreased in samples from patients with CD and UC | [24,29] |
| Ruminococcus gnavus |  | Involved in bile and amino acid biosynthesis pathways, including amino acid, energy, carbohydrate, and nucleotide metabolism Lack of supporting evidence of possible mechanisms involved | Found decreased in the stool of patients with treatment-naïve new-onset CD Found increased in samples from patients with CD compared to controls | [24,30] |
| Clostridioides difficile |  | A and B toxins produced by this bacterium may activate caspase-1 and secrete mature IL-1b and IL-18 (proinflammatory cytokines) that cause damage to the epithelial barrier and intestinal cells | High prevalence of infection by C. difficile has been demonstrated among patients with IBD | [31,32] |
| Listeria monocytogenes | | Possibly associated with invasive infection of epithelial cells | Listeria monocytogenes infection rates seem to be elevated in patients with IBD | [31,33] |
| Verrucomicrobia | ||||
| Akkermansia muciniphila | | Possibly involved with the production of SCFAs, which can activate the GPR43 and thereby increase the number of Foxp3+ regulatory T cells in the colon | Decreased in stools of both CD and UC patients. Human strain ATCC BAA-835T and murine strain 139 exerted anti-inflammatory effects on DSS-induced chronic colitis in mice | [34,35,36] |
| Actinobacteria | ||||
| Eggerthella lenta | | Lack of well-explored possible mechanisms | Found increased in samples from patients with CD compared to controls | [24] |
| Bifidobacterium bifidum | | Mucin metabolism performed by B. bifidum could activate enhanced production of mucin, thereby increasing the mucus layer depth and strengthening the epithelial barrier function | Found decreased in samples from patients with IBD. Some studies suggest that probiotics containing this bacterium could have positive responses in the treatment of IBD | [37,38] |
| Mycobacterium avium | | Associated with increased production of proinflammatory cytokines. Mutations in NOD2/CARD15 receptors may cause intracellular survival of the bacteria and ultimately cause infection | The abundance of this bacteria, especially the subspecies paratuberculosis, is higher in patients with IBD than in controls | [39,40] |
| Proteobacteria | ||||
| Escherichia coli | | Epithelium-associated invasive E. coli has frequently been isolated from ileal and colonic mucosa from patients with CD and can infect and damage intestinal epithelial cell monolayers, and synthesize α-hemolysin | Found increased numbers of E. coli strains with virulence properties isolated from samples of patients with IBD. Several studies indicate that there is a link between the prevalence of E. coli and IBD relapses | [41,42] |
| Haemophilus parainfluenzae | | Involved in glycerol-phospholipid and lipopolysaccharide metabolism, thereby promoting inflammation | Found increased in stool samples from patients with treatment-naïve new-onset CD | [30] |
| Campylobacter spp. | | Invasive strains of this bacteria in patients with IBD can survive in intracellular and anaerobic conditions | Increased in patients with IBD compared to controls | [43] |
| Eikenella corrodens | | Possibly involved in lipid and polysaccharide metabolism, resulting in proinflammatory responses | Increased in patients with IBD compared to controls | [30] |
| Fusobacteria | ||||
| Fusobacterium nucleatum | | Possibly involved in proinflammatory and tumorigenic responses | Increased in patients with IBD compared to controls. It is also associated with colorectal cancer | [43] |
| Bacteroidetes | ||||
| Bacteroides spp. | | Involved in mucin metabolism, possibly playing a role in damaging the protective mucus layer | Increased in CD samples. Prominent in patients with prior surgical resection | [24,44,45,46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana, P.T.; Rosas, S.L.B.; Ribeiro, B.E.; Marinho, Y.; de Souza, H.S.P. Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3464. https://doi.org/10.3390/ijms23073464
Santana PT, Rosas SLB, Ribeiro BE, Marinho Y, de Souza HSP. Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. International Journal of Molecular Sciences. 2022; 23(7):3464. https://doi.org/10.3390/ijms23073464
Chicago/Turabian StyleSantana, Patricia Teixeira, Siane Lopes Bittencourt Rosas, Beatriz Elias Ribeiro, Ygor Marinho, and Heitor S. P. de Souza. 2022. "Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets" International Journal of Molecular Sciences 23, no. 7: 3464. https://doi.org/10.3390/ijms23073464
APA StyleSantana, P. T., Rosas, S. L. B., Ribeiro, B. E., Marinho, Y., & de Souza, H. S. P. (2022). Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. International Journal of Molecular Sciences, 23(7), 3464. https://doi.org/10.3390/ijms23073464