
Toward a Treatment of Cancer: Design and In Vitro/In Vivo Evaluation of Uncharged Pyrazoline Derivatives as a Series of Novel SHP2 Inhibitors

 ,
, 

Abstract
:
1. Introduction
2. Results
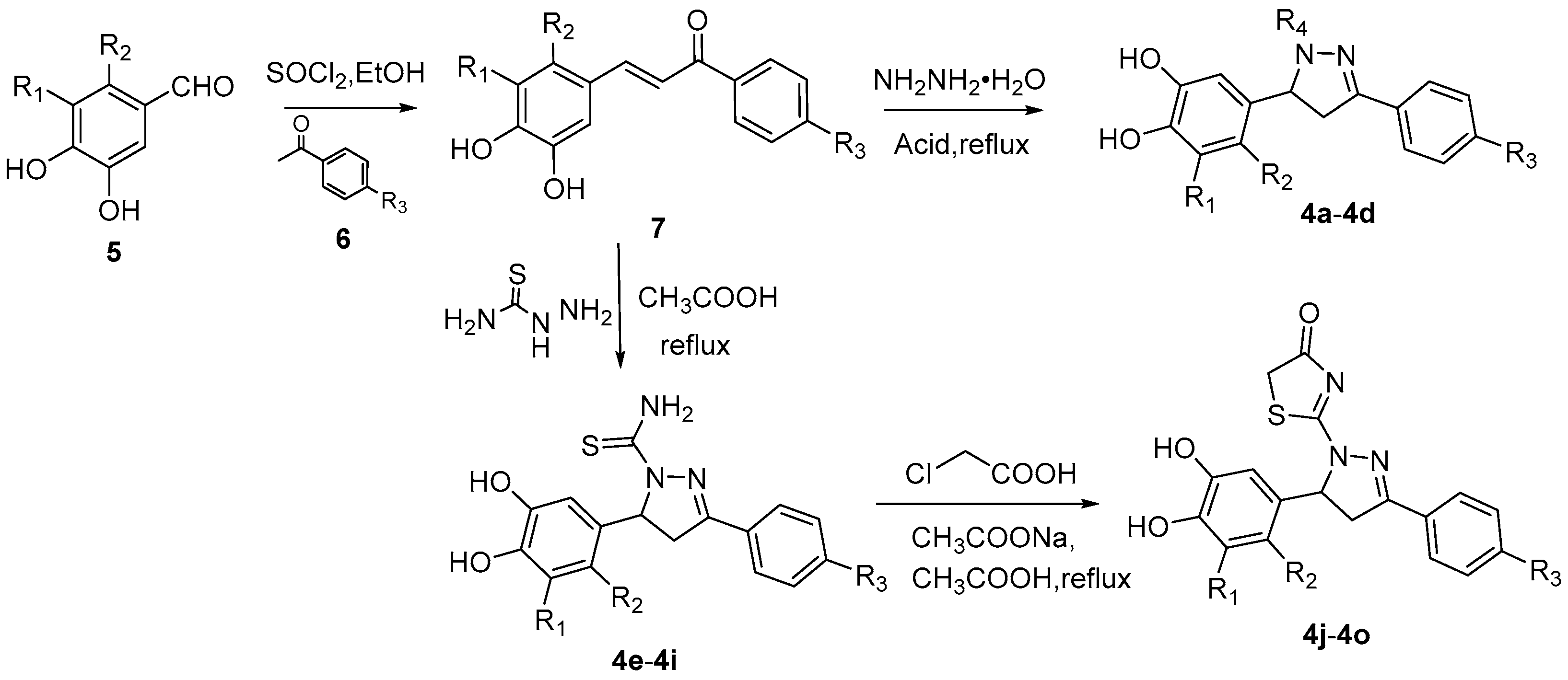
2.1. Design and Synthesis of SHP2 Inhibitors
2.2. Inhibition of SHP2
2.3. Compound 4o Is a Competitive SHP2 Inhibitor
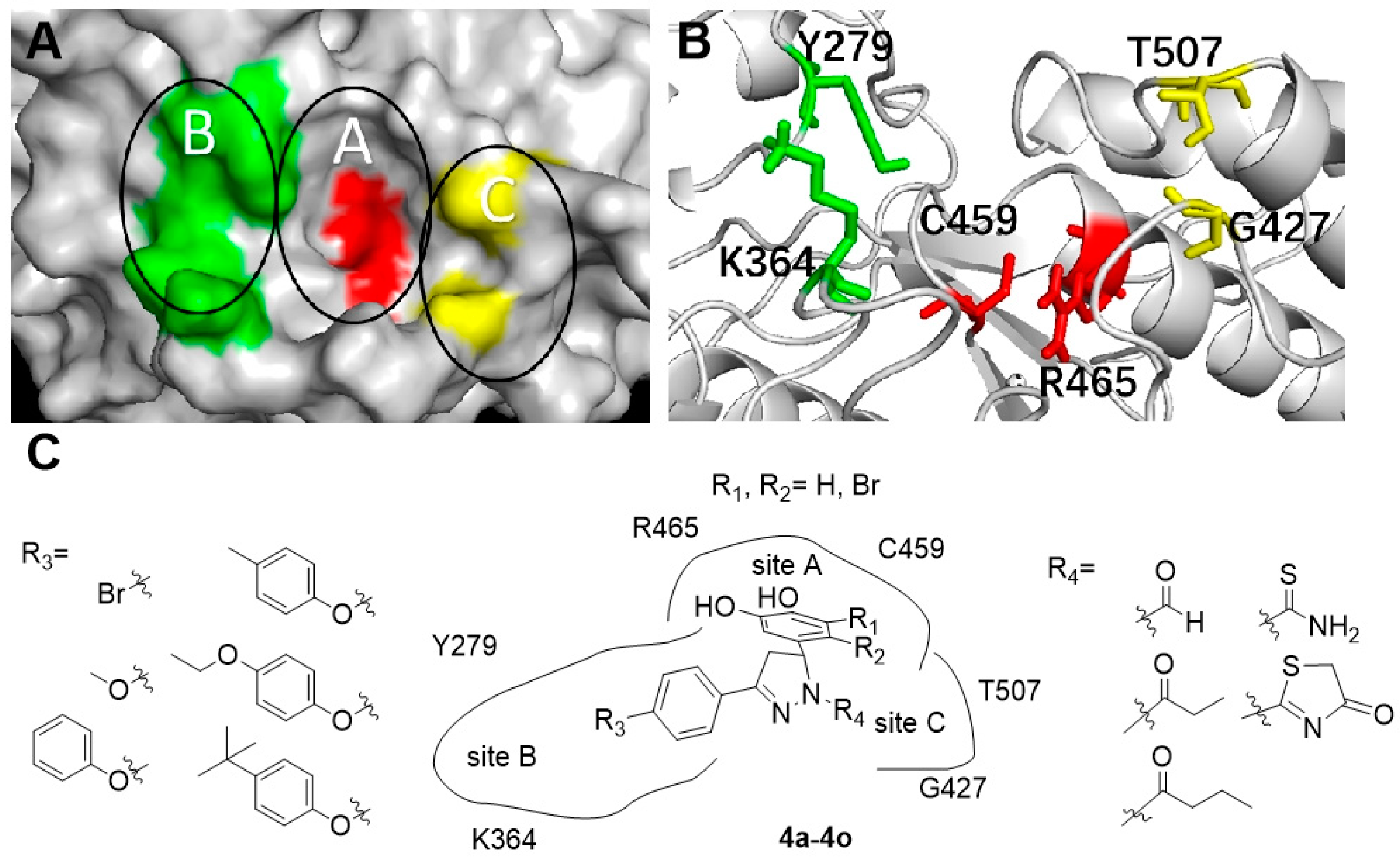
2.4. Molecular Docking
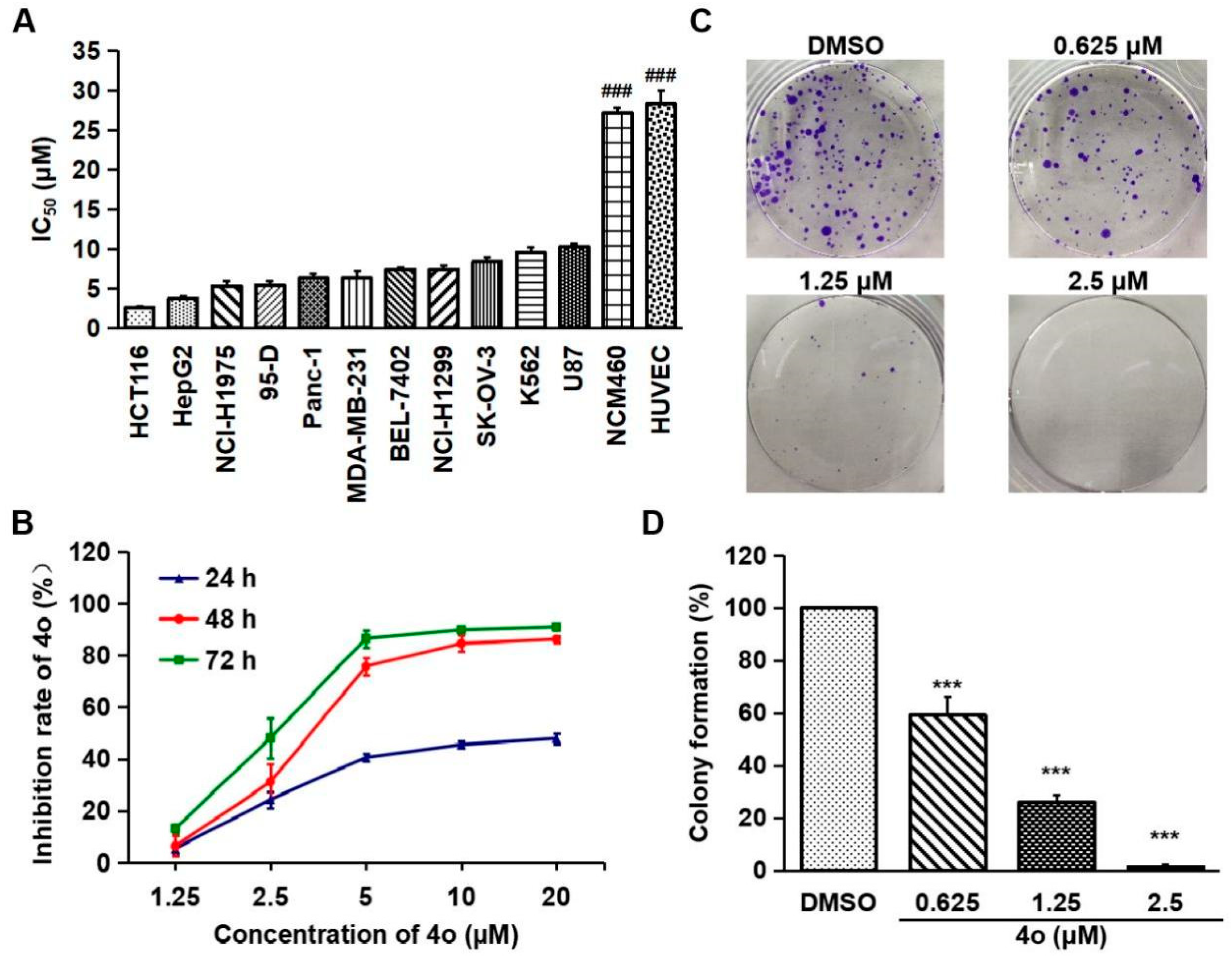
2.5. Compound 4o Inhibits the Proliferation of Various Cancer Cells
2.6. Compound 4o Induces Apoptosis in HCT116 Cells
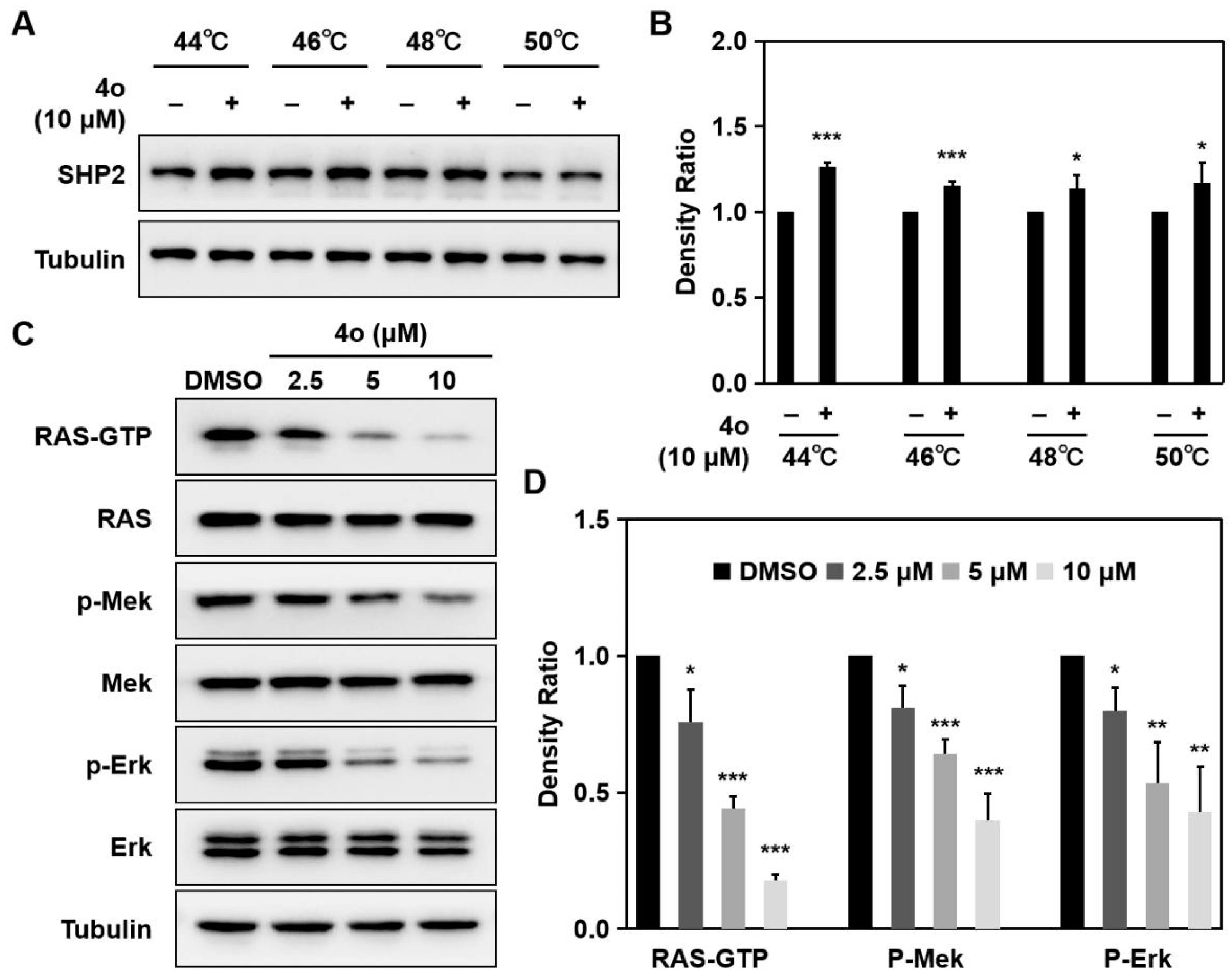
2.7. Compound 4o Interacts with SHP2 and Suppresses RAS/MAPK Signaling Pathway in HCT116 Cells
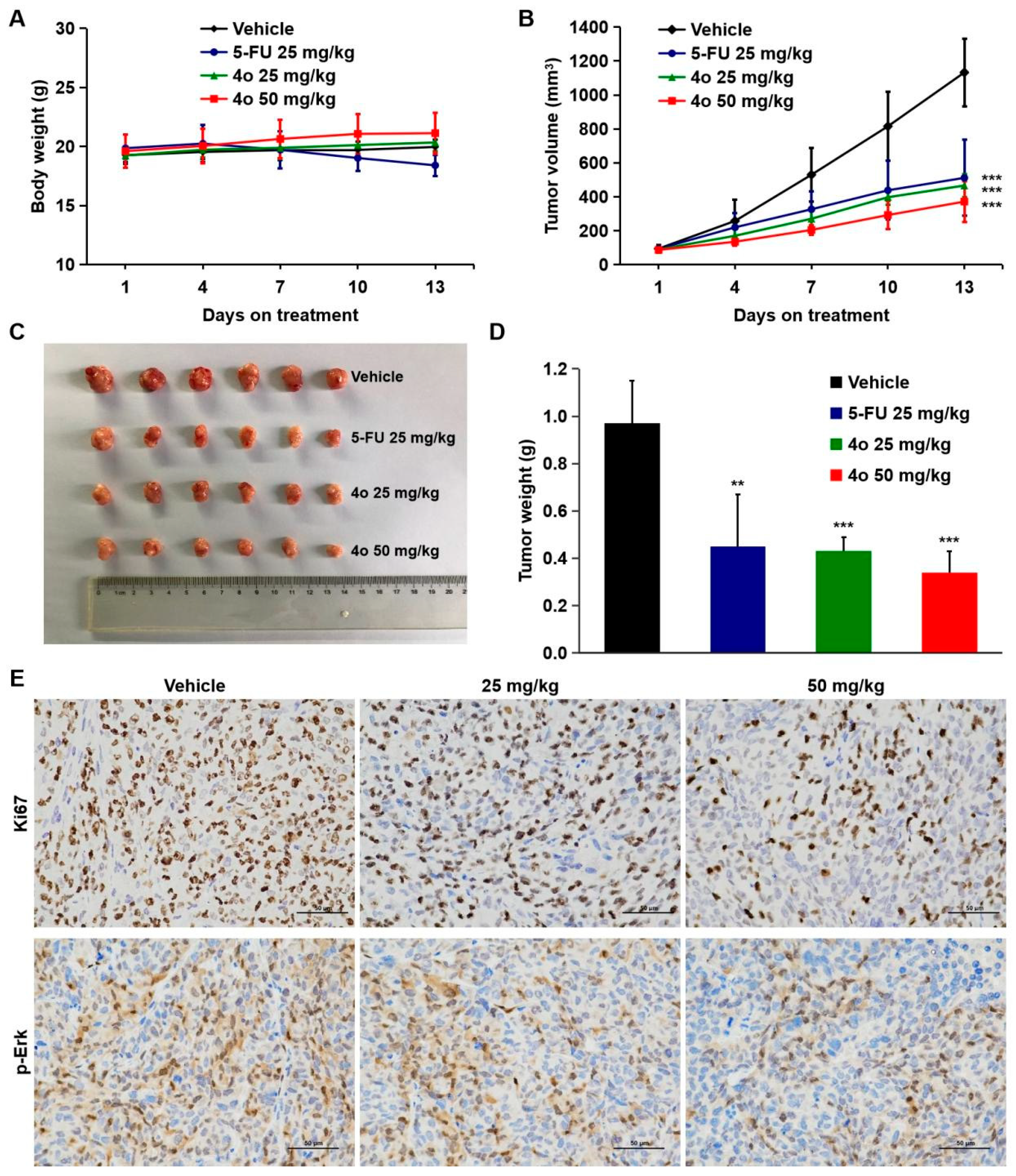
2.8. Compound 4o Induces Potent Anti-Tumor Activities In Vivo
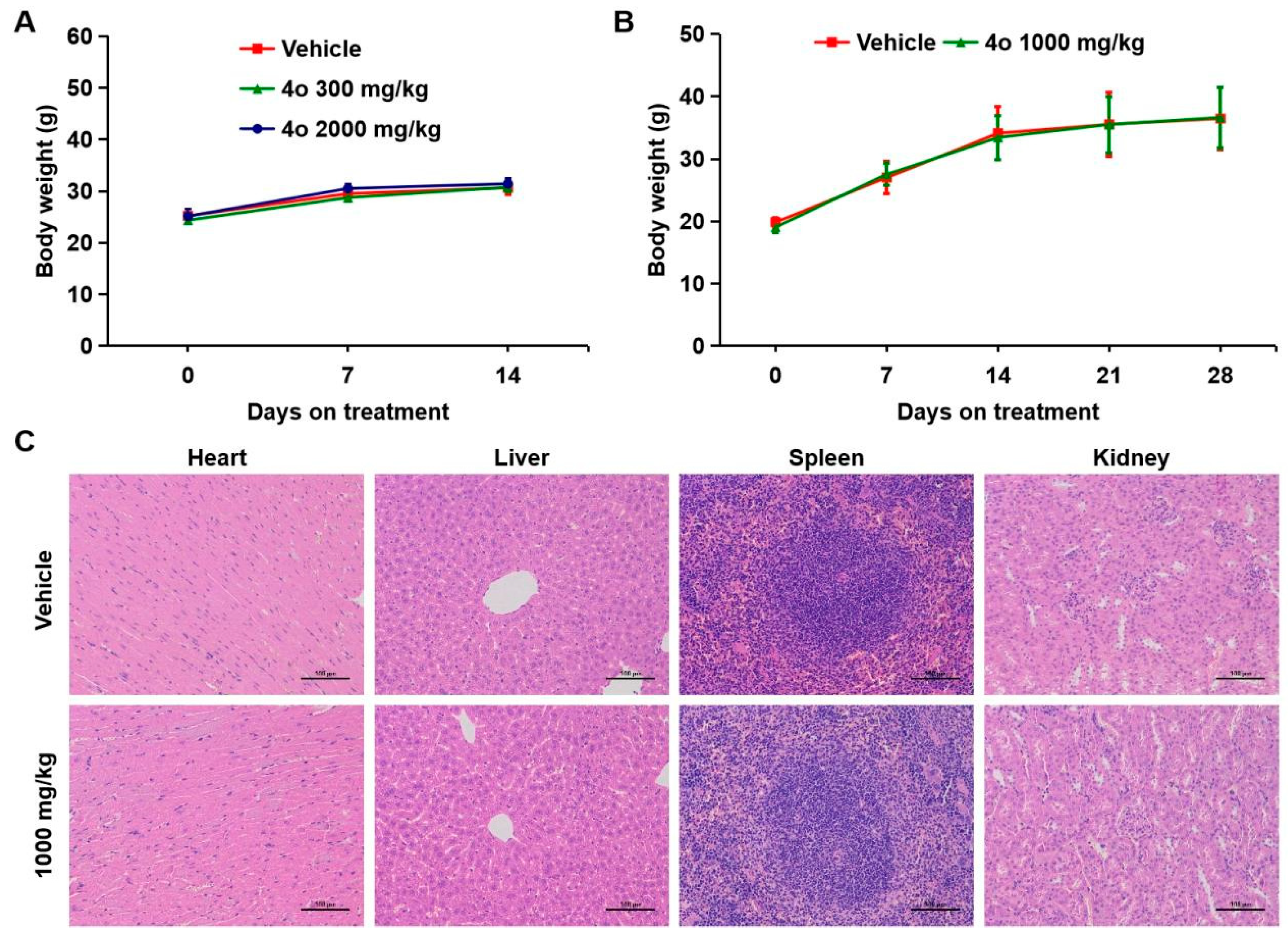
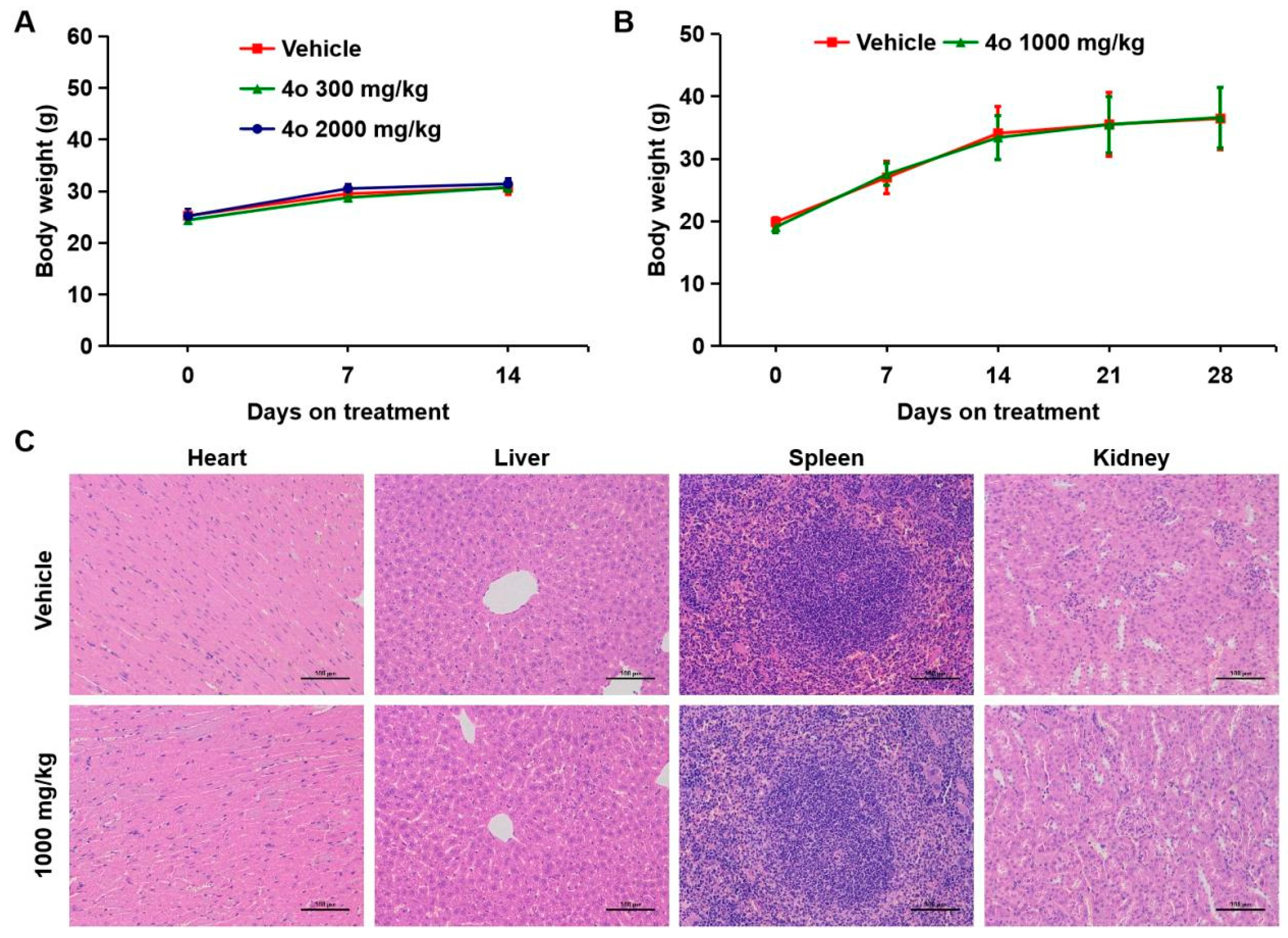
2.9. Toxicological Safety Evaluation of Compound 4o In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. Synthesis
4.3. Protein Expression and Purification
4.4. SHP2 Inhibition Assay
4.5. Inhibition Type of SHP2
4.6. Molecular Docking
4.7. Cell Culture
4.8. Cell Viability Assay
4.9. Colony Formation Assay
4.10. Western Blotting
4.11. Hoechst 33258 Staining
4.12. CETSA
4.13. RAS Activation Assay
4.14. Anti-Tumor Activity Studies In Vivo
4.15. Toxicological Safety Evaluation In Vivo
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, R.J.; Feng, G.-S. PTPN11 Is the First Identified Proto-Oncogene That Encodes a Tyrosine Phosphatase. Blood 2006, 109, 862–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.-X.; Wang, H.; Liu, C.; Kovats, S.; Velazquez, R.; Lu, H.; Pant, B.; Shirley, M.; Meyer, M.J.; Pu, M.; et al. Tumor Intrinsic Efficacy by SHP2 and RTK Inhibitors in KRAS-Mutant Cancers. Mol. Cancer Ther. 2019, 18, 2368–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.O.N.; Tong, M.; Chung, K.P.S.; Zhou, L.; Che, N.; Tang, K.H.; Ding, J.; Lau, E.Y.T.; Ng, I.O.L.; Ma, S.; et al. Overriding Adaptive Resistance to Sorafenib through Combination Therapy with Src Homology 2 Domain–Containing Phosphatase 2 Blockade in Hepatocellular Carcinoma. Hepatology 2020, 72, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wen, L.; Wang, S. SHP2 Inhibition Benefits Epidermal Growth Factor Receptor-Mutated Non-Small Cell Lung Cancer Therapy. Mini-Rev. Med. Chem. 2020, 21, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohseni, M.; Grauel, A.; Diez, J.E.; Guan, W.; Liang, S.; Choi, J.E.; Pu, M.; Chen, D.; Laszewski, T.; et al. SHP2 Blockade Enhances Anti-Tumor Immunity via Tumor Cell Intrinsic and Extrinsic Mechanisms. Sci. Rep. 2021, 11, 1399. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Kalaitzidis, D.; Neel, B.G. The Tyrosine Phosphatase Shp2 (PTPN11) in Cancer. Cancer Metastasis Rev. 2008, 27, 179–192. [Google Scholar] [CrossRef]
- Lawrence, H.R.; Pireddu, R.; Chen, L.; Luo, Y.; Sung, S.-S.; Szymanski, A.M.; Yip, M.L.R.; Guida, W.C.; Sebti, S.M.; Wu, J.; et al. Inhibitors of Src Homology-2 Domain Containing Protein Tyrosine Phosphatase-2 (Shp2) Based on Oxindole Scaffolds. J. Med. Chem. 2008, 51, 4948–4956. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhao, M.; Zhang, H.; Yu, B. Double-Edged Roles of Protein Tyrosine Phosphatase SHP2 in Cancer and Its Inhibitors in Clinical Trials. Pharmacol. Ther. 2022, 230, 107966. [Google Scholar] [CrossRef]
- Ding, Y.; Ouyang, Z.; Zhang, C.; Zhu, Y.; Xu, Q.; Sun, H.; Qu, J.; Sun, Y. Tyrosine Phosphatase SHP2 Exacerbates Psoriasis-like Skin Inflammation in Mice via ERK5-dependent NETosis. MedComm 2022, 3, e120. [Google Scholar] [CrossRef] [PubMed]
- Maroun, C.R.; Naujokas, M.A.; Holgado-Madruga, M.; Wong, A.J.; Park, M. The Tyrosine Phosphatase SHP-2 Is Required for Sustained Activation of Extracellular Signal-Regulated Kinase and Epithelial Morphogenesis Downstream from the Met Receptor Tyrosine Kinase. Mol. Cell. Biol. 2000, 20, 8513–8525. [Google Scholar] [CrossRef] [Green Version]
- Matozaki, T.; Murata, Y.; Saito, Y.; Okazawa, H.; Ohnishi, H. Protein Tyrosine Phosphatase SHP-2: A Proto-Oncogene Product That Promotes Ras Activation. Cancer Sci. 2009, 100, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 Is Required for Growth of KRAS-Mutant Non-Small-Cell Lung Cancer In Vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Sarver, P.; Acker, M.; Bagdanoff, J.T.; Chen, Z.; Chen, Y.-N.; Chan, H.; Firestone, B.; Fodor, M.; Fortanet, J.; Hao, H.; et al. 6-Amino-3-Methylpyrimidinones as Potent, Selective, and Orally Efficacious SHP2 Inhibitors. J. Med. Chem. 2019, 62, 1793–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.; Chen, W.; Zhu, J.; Wu, G.; Shen, R.; Xi, M.; Sun, H. Therapeutic Potential of Targeting SHP2 in Human Developmental Disorders and Cancers. Eur. J. Med. Chem. 2020, 190, 112117. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Heynen, G.J.J.E.; Germano, G.; Willems, S.M.; Evers, B.; Vecchione, L.; Gambino, V.; Lieftink, C.; Beijersbergen, R.L.; Di Nicolantonio, F.; et al. PTPN11 Is a Central Node in Intrinsic and Acquired Resistance to Targeted Cancer Drugs. Cell Rep. 2015, 12, 1978–1985. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Coad, J.; Ducatman, B.; Agazie, Y.M. SHP2 Is Up-Regulated in Breast Cancer Cells and in Infiltrating Ductal Carcinoma of the Breast, Implying Its Involvement in Breast Oncogenesis. Histopathology 2008, 53, 389–402. [Google Scholar] [CrossRef]
- Aceto, N.; Sausgruber, N.; Brinkhaus, H.; Gaidatzis, D.; Martiny-Baron, G.; Mazzarol, G.; Confalonieri, S.; Quarto, M.; Hu, G.; Balwierz, P.J.; et al. Tyrosine Phosphatase SHP2 Promotes Breast Cancer Progression and Maintains Tumor-Initiating Cells via Activation of Key Transcription Factors and a Positive Feedback Signaling Loop. Nat. Med. 2012, 18, 529–537. [Google Scholar] [CrossRef]
- Miyamoto, D.; Miyamoto, M.; Takahashi, A.; Yomogita, Y.; Higashi, H.; Kondo, S.; Hatakeyama, M. Isolation of a Distinct Class of Gain-of-Function SHP-2 Mutants with Oncogenic RAS-like Transforming Activity from Solid Tumors. Oncogene 2008, 27, 3508–3515. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Cardona, A.F.; Bracht, J.W.P.; Aldeguer, E.; Drozdowskyj, A.; Fernandez-Bruno, M.; Chaib, I.; Berenguer, J.; Santarpia, M.; Ito, M.; et al. Integrin-Linked Kinase (ILK) and Src Homology 2 Domain-Containing Phosphatase 2 (SHP2): Novel Targets in EGFR-Mutation Positive Non-Small Cell Lung Cancer (NSCLC). EBioMedicine 2019, 39, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Lu, J.; Wang, M.; Yang, C.-Y.; Wang, S. Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J. Med. Chem. 2020, 63, 7510–7528. [Google Scholar] [CrossRef]
- Nagamura, Y.; Miyazaki, M.; Nagano, Y.; Tomiyama, A.; Ohki, R.; Yanagihara, K.; Sakai, R.; Yamaguchi, H. SHP2 as a Potential Therapeutic Target in Diffuse-Type Gastric Carcinoma Addicted to Receptor Tyrosine Kinase Signaling. Cancers 2021, 13, 4309. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, S.; Overduin, M.; Barr, A.J. Targeting Protein Tyrosine Phosphatase SHP2 for Therapeutic Intervention. Future Med. Chem. 2014, 6, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sung, S.-S.; Yip, M.L.R.; Lawrence, H.R.; Ren, Y.; Guida, W.C.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Discovery of a Novel Shp2 Protein Tyrosine Phosphatase Inhibitor. Mol. Pharm. 2006, 70, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmuth, K.; Grosskopf, S.; Lum, C.T.; Würtele, M.; Röder, N.; von Kries, J.P.; Rosario, M.; Rademann, J.; Birchmeier, W. Specific Inhibitors of the Protein Tyrosine Phosphatase Shp2 Identified by High-Throughput Docking. Proc. Natl. Acad. Sci. USA 2008, 105, 7275–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Yu, Z.-H.; Zhang, R.-Y.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Shim, J.S.; Zeng, L.-F.; He, Y.; Chen, L.; et al. Exploring the Existing Drug Space for Novel PTyr Mimetic and SHP2 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.-Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, 63, 11368–11396. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Zhao, M.; Yang, X.; Yu, B. Strategies Targeting Protein Tyrosine Phosphatase SHP2 for Cancer Therapy. J. Med. Chem. 2022, 65, 3066–3079. [Google Scholar] [CrossRef]
- Zhang, X.; He, Y.; Liu, S.; Yu, Z.; Jiang, Z.-X.; Yang, Z.; Dong, Y.; Nabinger, S.C.; Wu, L.; Gunawan, A.M.; et al. Salicylic Acid Based Small Molecule Inhibitor for the Oncogenic Src Homology-2 Domain Containing Protein Tyrosine Phosphatase-2 (SHP2). J. Med. Chem. 2010, 53, 2482–2493. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-Y. Protein Tyrosine Phosphatases: Structure and Function, Substrate Specificity, and Inhibitor Development. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 209–234. [Google Scholar] [CrossRef]
- Mitra, R.; Ayyannan, S.R. Small-Molecule Inhibitors of Shp2 Phosphatase as Potential Chemotherapeutic Agents for Glioblastoma: A Minireview. ChemMedChem 2021, 16, 777–787. [Google Scholar] [CrossRef]
- Scott, L.M.; Lawrence, H.R.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Targeting Protein Tyrosine Phosphatases for Anticancer Drug Discovery. Curr. Pharm. Des. 2010, 16, 1843–1862. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xu, Q.; Jiang, B.; Zhang, R.; Jia, X.; Li, X.; Wang, L.; Guo, C.; Wu, N.; Shi, D. Selectivity, Cell Permeability and Oral Availability Studies of Novel Bromophenol Derivative HPN as Protein Tyrosine Phosphatase 1B Inhibitor. Br. J. Pharmacol. 2018, 175, 140–153. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zeng, L.-F.; He, Y.; Zhang, S.; Zhang, Z.-Y. Small Molecule Tools for Functional Interrogation of Protein Tyrosine Phosphatases. FEBS J. 2013, 280, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Q.; Li, C.; Luo, J.; Li, X.; Wang, L.; Jiang, B.; Shi, D. Toward a Treatment of Diabesity: In Vitro and in Vivo Evaluation of Uncharged Bromophenol Derivatives as a New Series of PTP1B Inhibitors. Eur. J. Med. Chem. 2019, 166, 178–185. [Google Scholar] [CrossRef]
- Zeng, L.-F.; Zhang, R.-Y.; Yu, Z.-H.; Li, S.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Mali, R.S.; Li, X.; Chan, R.J.; et al. Therapeutic Potential of Targeting the Oncogenic SHP2 Phosphatase. J. Med. Chem. 2014, 57, 6594–6609. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.; Ito, E.; Lau, K.S.; Mocanu, J.D.; Bastianutto, C.; Schimmer, A.D.; Liu, F.-F. Increased Efficiency for Performing Colony Formation Assays in 96-Well Plates: Novel Applications to Combination Therapies and High-Throughput Screening. BioTechniques 2008, 44, ix–xiv. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell Death beyond Apoptosis. Leukemia 2000, 14, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Diamantopoulos, P.T.; Sofotasiou, M.; Papadopoulou, V.; Polonyfi, K.; Iliakis, T.; Viniou, N.-A. PARP1-Driven Apoptosis in Chronic Lymphocytic Leukemia. BioMed Res. Int. 2014, 2014, e106713. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Jacobson, K.N.; McDermott, K.M.; Reddy, S.P.; Cress, A.E.; Tang, H.; Dudek, S.M.; Black, S.M.; Garcia, J.G.N.; Makino, A.; et al. ATP Promotes Cell Survival via Regulation of Cytosolic [Ca2+] and Bcl-2/Bax Ratio in Lung Cancer Cells. Am. J. Physiol.-Cell Physiol. 2016, 310, C99–C114. [Google Scholar] [CrossRef] [Green Version]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Chen, A.; Zhu, M.; Qi, X.; Tang, W.; Liu, M.; Li, D.; Gu, Q.; Li, J. Penicisulfuranol A, a Novel C-Terminal Inhibitor Disrupting Molecular Chaperone Function of Hsp90 Independent of ATP Binding Domain. Biochem. Pharmacol. 2019, 163, 404–415. [Google Scholar] [CrossRef]
- Grossmann, K.S.; Rosário, M.; Birchmeier, C.; Birchmeier, W. The Tyrosine Phosphatase Shp2 in Development and Cancer. Adv. Cancer Res. 2010, 106, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS Nucleotide Cycling Underlies the SHP2 Phosphatase Dependence of Mutant BRAF-, NF1- and RAS-Driven Cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-Fluorouracil and Other Fluoropyrimidines in Colorectal Cancer: Past, Present and Future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef] [PubMed]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 Is a Promising Molecular Target in the Diagnosis of Cancer (Review). Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [Green Version]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating Mutations of the Noonan Syndrome-Associated SHP2/PTPN11 Gene in Human Solid Tumors and Adult Acute Myelogenous Leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.T.; Meng, F.Y.; Li, Q.; Ho, C.Y.; Lam, T.S.; To, Y.; Lee, W.H.; Li, M.; Chu, K.H.; Toh, M. Cucurbitacin B Induces Apoptosis and S Phase Cell Cycle Arrest in BEL-7402 Human Hepatocellular Carcinoma Cells and Is Effective via Oral Administration. Cancer Lett. 2010, 294, 118–124. [Google Scholar] [CrossRef]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing News: SH2 Domain-Containing Tyrosine Phosphatases in Cell Signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein Tyrosine Phosphatases: From Genes, to Function, to Disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Yu, B.; Liu, W.; Yu, W.-M.; Loh, M.L.; Alter, S.; Guvench, O.; Mackerell, A.D.; Tang, L.-D.; Qu, C.-K. Targeting Protein Tyrosine Phosphatase SHP2 for the Treatment of PTPN11-Associated Malignancies. Mol. Cancer 2013, 12, 1738–1748. [Google Scholar] [CrossRef] [Green Version]
- Pathak, M.K.; Yi, T. Sodium Stibogluconate Is a Potent Inhibitor of Protein Tyrosine Phosphatases and Augments Cytokine Responses in Hemopoietic Cell Lines. J. Immunol. 2001, 167, 3391–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Borden, E.; Yi, T. Interferon-Gamma Is Induced in Human Peripheral Blood Immune Cells in Vitro by Sodium Stibogluconate/Interleukin-2 and Mediates Its Antitumor Activity In Vivo. J. Interferon Cytokine Res. 2009, 29, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, T.; Elson, P.; Mitsuhashi, M.; Jacobs, B.; Hollovary, E.; Budd, T.G.; Spiro, T.; Triozzi, P.; Borden, E.C. Phosphatase Inhibitor, Sodium Stibogluconate, in Combination with Interferon (IFN) Alpha 2b: Phase I Trials to Identify Pharmacodynamic and Clinical Effects. Oncotarget 2011, 2, 1155–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yu, Z.; Yu, X.; Huang, S.-X.; Luo, Y.; Wu, L.; Shen, W.; Yang, Z.; Wang, L.; Gunawan, A.M.; et al. SHP2 Is a Target of the Immunosuppressant Tautomycetin. Chem. Biol. 2011, 18, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-L.; Chen, X.-Y.; Gao, Y.; Gao, L.-X.; Sheng, L.; Zhu, J.; Xu, L.; Ding, Z.-Z.; Zhang, C.; Li, J.-Y.; et al. Benzo[c](1,2,5)Thiadiazole Derivatives: A New Class of Potent Src Homology-2 Domain Containing Protein Tyrosine Phosphatase-2 (SHP2) Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5154–5157. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qu, J.; Zhao, M.; Xu, Q.; Sun, Y. Targeting SHP2 as a Promising Strategy for Cancer Immunotherapy. Pharmacol. Res. 2020, 152, 104595. [Google Scholar] [CrossRef]
- Dai, J.; Zhu, M.; Qi, X.; Wang, Y.; Li, H.; Tang, S.; Wang, Q.; Chen, A.; Liu, M.; Gu, Q.; et al. Fungal Mycotoxin Penisuloxazin A, a Novel C-Terminal Hsp90 Inhibitor and Characteristics of Its Analogues on Hsp90 Function Related to Binding Sites. Biochem. Pharmacol. 2020, 182, 114218. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 (μM) |
| 4a | H | H | 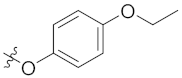 | -CHO | 13.33 ± 2.11 |
| 4b | H | H | 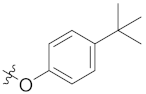 | -CHO | 13.74 ± 1.98 |
| 4c | H | H | 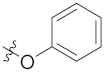 | 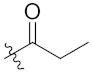 | 34.17 ± 5.90 |
| 4d | H | H |  | 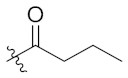 | 9.51 ± 2.01 |
| 4e | H | H |  | 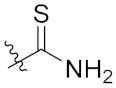 | 5.96 ± 0.92 |
| 4f | H | H | -OCH3 | 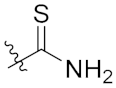 | 13.20 ± 1.89 |
| 4g | H | H | 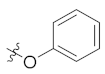 | 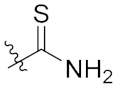 | 5.97 ± 0.88 |
| 4h | H | H | 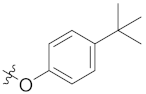 | 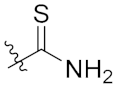 | 8.44 ± 2.03 |
| 4i | H | H | Br | 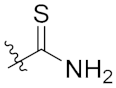 | 20.40 ± 2.45 |
| 4j | H | H | -OCH3 | 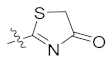 | 11.38 ± 1.56 |
| 4k | H | H | 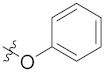 | 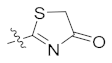 | 4.98 ± 0.77 |
| 4l | H | H | 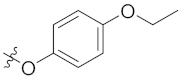 |  | 4.36 ± 0.65 |
| 4m | H | H | 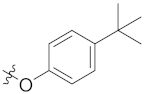 |  | 4.94 ± 0.79 |
| 4n | H | H | Br | 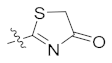 | 18.53 ± 2.32 |
| 4o | Br | Br | 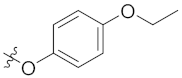 | 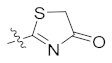 | 1.56 ± 0.25 |
| Na3VO4 | 10.68 ± 2.08 | ||||
| Cell Lines | IC50 (μM) |
|---|---|
| HCT116 | 2.64 ± 0.17 |
| HepG2 | 3.80 ± 0.22 |
| NCI-H1975 | 5.29 ± 0.60 |
| 95-D | 5.42 ± 0.44 |
| Panc-1 | 6.30 ± 0.50 |
| MDA-MB-231 | 6.34 ± 0.37 |
| BEL-7402 | 7.38 ± 0.31 |
| NCI-H1299 | 7.42 ± 0.43 |
| SK-OV-3 | 8.44 ± 0.45 |
| K562 | 9.60 ± 0.68 |
| U87 | 10.25 ± 0.39 |
| NCM460 | 27.10 ± 0.66 |
| HUVEC | 28.29 ± 1.63 |
| Groups | Mortality | Body Weight (g) | ||
|---|---|---|---|---|
| Day 0 | Day 7 | Day 14 | ||
| Vehicle | 0/6 | 25.25 ± 0.48 | 29.52 ± 1.06 | 30.67 ± 1.29 |
| 300 mg/kg | 0/6 | 24.41 ± 0.64 | 28.78 ± 0.78 | 30.75 ± 0.99 |
| 2000 mg/kg | 0/6 | 25.19 ± 1.45 | 30.51 ± 0.88 | 31.43 ± 1.09 |
| Groups | Mortality | Body Weight (g) | ||||
|---|---|---|---|---|---|---|
| Day 0 | Day 7 | Day 14 | Day 21 | Day 28 | ||
| Vehicle | 0/10 | 19.87 ± 0.75 | 26.99 ± 2.56 | 34.08 ± 4.24 | 35.51 ± 5.15 | 36.44 ± 4.93 |
| 1000 mg/kg | 0/10 | 19.08 ± 0.95 | 27.52 ± 1.77 | 33.41 ± 3.49 | 35.49 ± 4.52 | 36.63 ± 4.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, J.; Zhang, Y.; Gao, Y.; Bai, X.; Liu, F.; Li, S.; Yu, Y.; Hu, W.; Shi, T.; Shi, D.; et al. Toward a Treatment of Cancer: Design and In Vitro/In Vivo Evaluation of Uncharged Pyrazoline Derivatives as a Series of Novel SHP2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 3497. https://doi.org/10.3390/ijms23073497
Dai J, Zhang Y, Gao Y, Bai X, Liu F, Li S, Yu Y, Hu W, Shi T, Shi D, et al. Toward a Treatment of Cancer: Design and In Vitro/In Vivo Evaluation of Uncharged Pyrazoline Derivatives as a Series of Novel SHP2 Inhibitors. International Journal of Molecular Sciences. 2022; 23(7):3497. https://doi.org/10.3390/ijms23073497
Chicago/Turabian StyleDai, Jiajia, Yiting Zhang, Yanan Gao, Xiaoyi Bai, Fang Liu, Shuo Li, Yanyan Yu, Wenpeng Hu, Ting Shi, Dayong Shi, and et al. 2022. "Toward a Treatment of Cancer: Design and In Vitro/In Vivo Evaluation of Uncharged Pyrazoline Derivatives as a Series of Novel SHP2 Inhibitors" International Journal of Molecular Sciences 23, no. 7: 3497. https://doi.org/10.3390/ijms23073497
APA StyleDai, J., Zhang, Y., Gao, Y., Bai, X., Liu, F., Li, S., Yu, Y., Hu, W., Shi, T., Shi, D., & Li, X. (2022). Toward a Treatment of Cancer: Design and In Vitro/In Vivo Evaluation of Uncharged Pyrazoline Derivatives as a Series of Novel SHP2 Inhibitors. International Journal of Molecular Sciences, 23(7), 3497. https://doi.org/10.3390/ijms23073497