
Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma

 , ,
, , 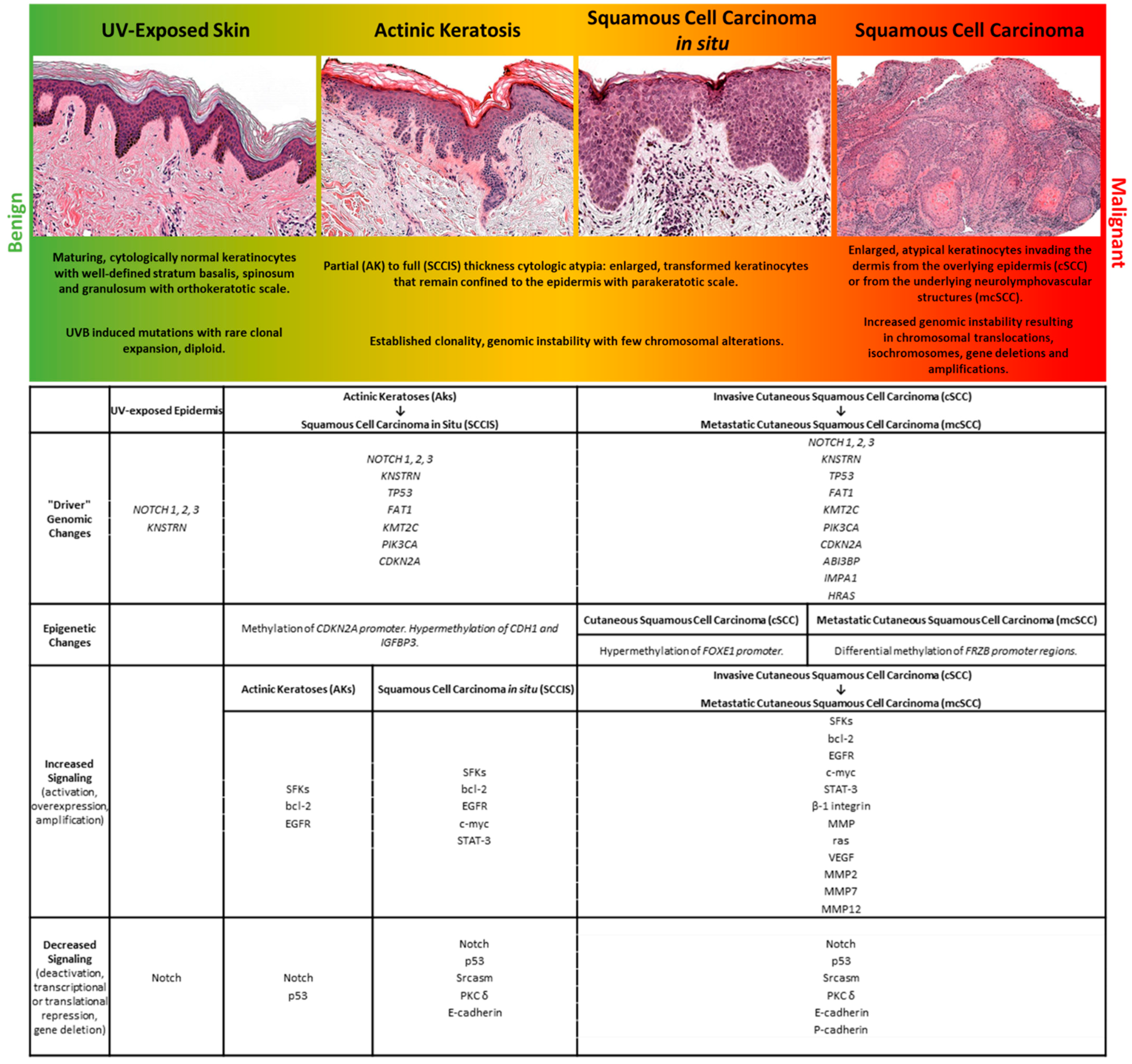{kind=link}
Abstract
1. Introduction
2. Genetic Alterations in cSCC
2.1. Mechanisms of Genetic Alterations in cSCC
2.2. Genomic Characterization of cSCC
2.2.1. TP53
2.2.2. NOTCH
2.2.3. RAS
2.2.4. CDKN2A
2.2.5. PIK3CA
2.2.6. KNSTRN
2.2.7. FAT1
2.2.8. CARD11
2.2.9. SRCASM
2.2.10. TP63
2.2.11. EGFR
2.3. Transcriptional Characterization of cSCC
3. Beyond the Genome: Epigenetic Regulations in cSCC
3.1. Aberrant DNA Methylation in cSCC
3.2. Histone Modifications in Cutaneous SCC
3.3. Long Noncoding RNA Dysregulation in cSCC
3.4. MicroRNA Dysregulation in cSCC
3.5. UV-Induced Epigenetic Regulation in cSCC
3.6. Epigenetic Regulation in cSCC Metastasis
3.7. Discovery of Novel Epigenetic Biomarkers in cSCC
3.8. CRISPR/Cas9 Tools of Epigenomic Editing
4. Metabolic Reprogramming in cSCC
5. Current Status of Molecular Targeted Therapies in cSCC
5.1. EGFR Inhibitors
5.2. Immune Checkpoint Inhibitors
5.3. SRC Family Kinase (SFK) Inhibitors
5.4. PI3K/mTOR Inhibitors
5.5. Epigenetic Modulators
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Schmults, C.D.; Karia, P.S.; Carter, J.B.; Han, J.; Qureshi, A.A. Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: A 10-year, single-institution cohort study. JAMA Dermatol. 2013, 149, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.R.; Zens, M.S.; Celaya, M.O.; Riddle, B.L.; Karagas, M.R.; Peacock, J.L. Survival after squamous cell and basal cell carcinoma of the skin: A retrospective cohort analysis. Int. J. Cancer 2015, 137, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Criscione, V.D.; Weinstock, M.A.; Naylor, M.F.; Luque, C.; Eide, M.J.; Bingham, S.F.; Department of Veteran Affairs Topical Tretinoin Chemoprevention Trial Group. Actinic keratoses: Natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer 2009, 115, 2523–2530. [Google Scholar] [CrossRef]
- Figueras Nart, I.; Cerio, R.; Dirschka, T.; Dreno, B.; Lear, J.T.; Pellacani, G.; Peris, K.; Ruiz de Casas, A. Defining the actinic keratosis field: A literature review and discussion. J. Eur. Acad. Dermatol. Venereol. JEADV 2018, 32, 544–563. [Google Scholar] [CrossRef]
- Sartini, D.; Campagna, R.; Lucarini, G.; Pompei, V.; Salvolini, E.; Mattioli-Belmonte, M.; Molinelli, E.; Brisigotti, V.; Campanati, A.; Bacchetti, T.; et al. Differential immunohistochemical expression of paraoxonase-2 in actinic keratosis and squamous cell carcinoma. Hum. Cell 2021, 34, 1929–1931. [Google Scholar] [CrossRef]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472. [Google Scholar] [CrossRef]
- Harwood, C.A.; Proby, C.M.; Arron, S.T. Genomics of SCC: Tumor Formation, Progression, and Future Therapeutic Implications for High-Risk Cutaneous Squamous Cell Carcinoma. In High-Risk Cutaneous Squamous Cell Carcinoma: A Practical Guide for Patient Management; Schmults, C.D., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 67–102. [Google Scholar] [CrossRef]
- Cornejo, C.M.; Jambusaria-Pahlajani, A.; Willenbrink, T.J.; Schmults, C.D.; Arron, S.T.; Ruiz, E.S. Field cancerization: Treatment. J. Am. Acad. Dermatol. 2020, 83, 719–730. [Google Scholar] [CrossRef]
- Ishitsuka, Y.; Hanaoka, Y.; Tanemura, A.; Fujimoto, M. Cutaneous Squamous Cell Carcinoma in the Age of Immunotherapy. Cancers 2021, 13, 1148. [Google Scholar] [CrossRef]
- Kim, C.; Cheng, J.; Colegio, O.R. Cutaneous squamous cell carcinomas in solid organ transplant recipients: Emerging strategies for surveillance, staging, and treatment. Semin. Oncol. 2016, 43, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Schierbeck, J.; Vestergaard, T.; Bygum, A. Skin Cancer Associated Genodermatoses: A Literature Review. Acta Derm. Venereol. 2019, 99, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Errol, C.F.; Graham, C.W.; Wolfram, S.; Richard, D.W.; Roger, A.S.; Tom, E. DNA Repair and Mutagenesis, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2006. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef]
- Leibeling, D.; Laspe, P.; Emmert, S. Nucleotide excision repair and cancer. J. Mol. Histol. 2006, 37, 225–238. [Google Scholar] [CrossRef]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef]
- Kraemer, K.H.; DiGiovanna, J.J. Xeroderma Pigmentosum. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Brash, D.E.; Rudolph, J.A.; Simon, J.A.; Lin, A.; McKenna, G.J.; Baden, H.P.; Halperin, A.J.; Ponten, J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 1991, 88, 10124–10128. [Google Scholar] [CrossRef]
- Molho-Pessach, V.; Lotem, M. Ultraviolet radiation and cutaneous carcinogenesis. Curr. Probl. Derm. 2007, 35, 14–27. [Google Scholar]
- Campagna, R.; Valentina, p.; Davide, S. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef]
- Setlow, R.B.; Carrier, W.L. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J. Mol. Biol. 1966, 17, 237–254. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Nairn, R.S. The biology of the (6-4) photoproduct. Photochem. Photobiol. 1989, 49, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV signature mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N.; et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 2016, 7, 12601. [Google Scholar] [CrossRef]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.A.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Thomson, J.; Bewicke-Copley, F.; Anene, C.A.; Gulati, A.; Nagano, A.; Purdie, K.; Inman, G.J.; Proby, C.M.; Leigh, I.M.; Harwood, C.A.; et al. The Genomic Landscape of Actinic Keratosis. J. Investig. Dermatol. 2021, 141, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Capell, B.C.; Parekh, V.; O’Day, C.; Atillasoy, C.; Bashir, H.M.; Yeh, C.; Shim, E.H.; Prouty, S.M.; Dentchev, T.; et al. Whole-Exome and Transcriptome Analysis of UV-Exposed Epidermis and Carcinoma In Situ Reveals Early Drivers of Carcinogenesis. J. Investig. Derm. 2021, 141, 295–307.e13. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bhaduri, A.; Mah, A.; Johnson, W.L.; Ungewickell, A.; Aros, C.J.; Nguyen, C.B.; Rios, E.J.; Siprashvili, Z.; Straight, A.; et al. Recurrent point mutations in the kinetochore gene KNSTRN in cutaneous squamous cell carcinoma. Nat. Genet. 2014, 46, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Kanellou, P.; Zaravinos, A.; Zioga, M.; Stratigos, A.; Baritaki, S.; Soufla, G.; Zoras, O.; Spandidos, D.A. Genomic instability, mutations and expression analysis of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in actinic keratosis. Cancer Lett. 2008, 264, 145–161. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef]
- Benjamin, C.L.; Ananthaswamy, H.N. p53 and the pathogenesis of skin cancer. Toxicol. Appl. Pharmacol. 2007, 224, 241–248. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001, 2, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Vousden, K.H. Complicating the complexity of p53. Carcinogenesis 2005, 26, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Tan, T.; Chu, G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol. Cell. Biol. 2002, 22, 3247–3254. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Kolev, V.; Mandinova, A.; Guinea-Viniegra, J.; Hu, B.; Lefort, K.; Lambertini, C.; Neel, V.; Dummer, R.; Wagner, E.F.; Dotto, G.P. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat. Cell Biol. 2008, 10, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Marshall, C.; Griffin, T.; Hanson, M.; Hick, R.; Dentchev, T.; Williams, E.; Werth, A.; Miller, C.; et al. Srcasm inhibits Fyn-induced cutaneous carcinogenesis with modulation of Notch1 and p53. Cancer Res. 2009, 69, 9439–9447. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar] [PubMed]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Suh, Y.A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintas-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef]
- Weisz, L.; Oren, M.; Rotter, V. Transcription regulation by mutant p53. Oncogene 2007, 26, 2202–2211. [Google Scholar] [CrossRef]
- Bougeard, G.; Sesboue, R.; Baert-Desurmont, S.; Vasseur, S.; Martin, C.; Tinat, J.; Brugieres, L.; Chompret, A.; de Paillerets, B.B.; Stoppa-Lyonnet, D.; et al. Molecular basis of the Li-Fraumeni syndrome: An update from the French LFS families. J. Med. Genet. 2008, 45, 535–538. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Mirza, S.A.; Xu, S.; Schulz-Heddergott, R.; Marchenko, N.D.; Moll, U.M. p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis. 2017, 8, e2661. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Hum. Mutat. 2003, 21, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef]
- Brash, D.E.; Ziegler, A.; Jonason, A.S.; Simon, J.A.; Kunala, S.; Leffell, D.J. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J. Investig. Derm. Symp. Proc. 1996, 1, 136–142. [Google Scholar]
- Durinck, S.; Ho, C.; Wang, N.J.; Liao, W.; Jakkula, L.R.; Collisson, E.A.; Pons, J.; Chan, S.W.; Lam, E.T.; Chu, C.; et al. Temporal dissection of tumorigenesis in primary cancers. Cancer Discov. 2011, 1, 137–143. [Google Scholar] [CrossRef]
- Tanaka, N.; Zhao, M.; Tang, L.; Patel, A.A.; Xi, Q.; Van, H.T.; Takahashi, H.; Osman, A.A.; Zhang, J.; Wang, J.; et al. Gain-of-function mutant p53 promotes the oncogenic potential of head and neck squamous cell carcinoma cells by targeting the transcription factors FOXO3a and FOXM1. Oncogene 2018, 37, 1279–1292. [Google Scholar] [CrossRef]
- Stahl, P.L.; Stranneheim, H.; Asplund, A.; Berglund, L.; Ponten, F.; Lundeberg, J. Sun-induced nonsynonymous p53 mutations are extensively accumulated and tolerated in normal appearing human skin. J. Investig. Dermatol. 2011, 131, 504–508. [Google Scholar] [CrossRef]
- Fernandez-Figueras, M.T.; Saenz-Sarda, X.; Vargas, P.; Thompson, C.T.; Carrato, C.; Puig, L.; Ferrandiz, C.; Ariza, A. The depth of follicular extension in actinic keratosis correlates with the depth of invasion in squamous cell carcinoma: Implication for clinical treatment. J. Eur. Acad. Dermatol. Venereol. JEADV 2018, 32, 1657–1661. [Google Scholar] [CrossRef]
- Hedberg, M.; Seykora, J.T. Clarifying Progress on the Genomic Landscape of Actinic Keratosis. J. Investig. Dermatol. 2021, 141, 1622–1624. [Google Scholar] [CrossRef] [PubMed]
- Marks, R.; Rennie, G.; Selwood, T.S. Malignant transformation of solar keratoses to squamous cell carcinoma. Lancet 1988, 1, 795–797. [Google Scholar] [CrossRef]
- Gilchrest, B.A. Actinic Keratoses: Reconciling the Biology of Field Cancerization with Treatment Paradigms. J. Investig. Dermatol. 2021, 141, 727–731. [Google Scholar] [CrossRef]
- Quintana, R.M.; Dupuy, A.J.; Bravo, A.; Casanova, M.L.; Alameda, J.P.; Page, A.; Sanchez-Viera, M.; Ramirez, A.; Navarro, M. A transposon-based analysis of gene mutations related to skin cancer development. J. Investig. Dermatol. 2013, 133, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P. Notch tumor suppressor function. Oncogene 2008, 27, 5115. [Google Scholar] [CrossRef]
- Nowell, C.; Radtke, F. Cutaneous Notch signaling in health and disease. Cold Spring Harb. Perspect. Med. 2013, 3, a017772. [Google Scholar] [CrossRef]
- Okuyama, R.; Tagami, H.; Aiba, S. Notch signaling: Its role in epidermal homeostasis and in the pathogenesis of skin diseases. J. Dermatol. Sci. 2008, 49, 187–194. [Google Scholar] [CrossRef]
- Moretti, F.; Marinari, B.; Lo Iacono, N.; Botti, E.; Giunta, A.; Spallone, G.; Garaffo, G.; Vernersson-Lindahl, E.; Merlo, G.; Mills, A.A.; et al. A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. J. Clin. Investig. 2010, 120, 1570–1577. [Google Scholar] [CrossRef]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Tommasi di Vignano, A.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef]
- Okuyama, R.; Nguyen, B.C.; Talora, C.; Ogawa, E.; Tommasi di Vignano, A.; Lioumi, M.; Chiorino, G.; Tagami, H.; Woo, M.; Dotto, G.P. High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev. Cell 2004, 6, 551–562. [Google Scholar] [CrossRef]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef]
- Mandinova, A.; Lefort, K.; Tommasi di Vignano, A.; Stonely, W.; Ostano, P.; Chiorino, G.; Iwaki, H.; Nakanishi, J.; Dotto, G.P. The FoxO3a gene is a key negative target of canonical Notch signalling in the keratinocyte UVB response. EMBO J 2008, 27, 1243–1254. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- van der Schroeff, J.G.; Evers, L.M.; Boot, A.J.; Bos, J.L. Ras oncogene mutations in basal cell carcinomas and squamous cell carcinomas of human skin. J. Investig. Dermatol. 1990, 94, 423–425. [Google Scholar] [CrossRef]
- Spencer, J.M.; Kahn, S.M.; Jiang, W.; DeLeo, V.A.; Weinstein, I.B. Activated ras genes occur in human actinic keratoses, premalignant precursors to squamous cell carcinomas. Arch. Dermatol. 1995, 131, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Pierceall, W.E.; Goldberg, L.H.; Tainsky, M.A.; Mukhopadhyay, T.; Ananthaswamy, H.N. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol. Carcinog. 1991, 4, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Ashford, B.G.; Clark, J.; Gupta, R.; Iyer, N.G.; Yu, B.; Ranson, M. Reviewing the genetic alterations in high-risk cutaneous squamous cell carcinoma: A search for prognostic markers and therapeutic targets. Head Neck 2017, 39, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P. Non-melanoma skin cancer: What drives tumor development and progression? Carcinogenesis 2005, 26, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef]
- Huang, P.Y.; Balmain, A. Modeling cutaneous squamous carcinoma development in the mouse. Cold Spring Harb. Perspect. Med. 2014, 4, a013623. [Google Scholar] [CrossRef]
- Serrano, M.; Lee, H.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef]
- Honda, R.; Yasuda, H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J 1999, 18, 22–27. [Google Scholar] [CrossRef]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Soufir, N.; Moles, J.P.; Vilmer, C.; Moch, C.; Verola, O.; Rivet, J.; Tesniere, A.; Dubertret, L.; Basset-Seguin, N. P16 UV mutations in human skin epithelial tumors. Oncogene 1999, 18, 5477–5481. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, Z.; Liloglou, T.; Zafiropoulos, A.; Koumantaki, E.; Zoras, O.; Spandidos, D.A. Mutational analysis of CDKN2A genes in patients with squamous cell carcinoma of the skin. Br. J. Dermatol. 2003, 148, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Blokx, W.A.; Ruiter, D.J.; Verdijk, M.A.; de Wilde, P.C.; Willems, R.W.; de Jong, E.M.; Ligtenberg, M.J. INK4-ARF and p53 mutations in metastatic cutaneous squamous cell carcinoma: Case report and archival study on the use of Ink4a-ARF and p53 mutation analysis in identification of the corresponding primary tumor. Am. J. Surg. Pathol. 2005, 29, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Mortier, L.; Marchetti, P.; Delaporte, E.; Martin de Lassalle, E.; Thomas, P.; Piette, F.; Formstecher, P.; Polakowska, R.; Danze, P.M. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16(INK4a) tumor suppressor. Cancer Lett. 2002, 176, 205–214. [Google Scholar] [CrossRef]
- Hedberg, M.L.; Peyser, N.D.; Bauman, J.E.; Gooding, W.E.; Li, H.; Bhola, N.E.; Zhu, T.R.; Zeng, Y.; Brand, T.M.; Kim, M.O.; et al. Use of nonsteroidal anti-inflammatory drugs predicts improved patient survival for PIK3CA-altered head and neck cancer. J. Exp. Med. 2019, 216, 419–427. [Google Scholar] [CrossRef]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Cai, Y.; Yousef, A.; Grandis, J.R.; Johnson, D.E. NSAID therapy for PIK3CA-Altered colorectal, breast, and head and neck cancer. Adv. Biol. Regul. 2020, 75, 100653. [Google Scholar] [CrossRef]
- Fang, L.; Seki, A.; Fang, G. SKAP associates with kinetochores and promotes the metaphase-to-anaphase transition. Cell Cycle 2009, 8, 2819–2827. [Google Scholar] [CrossRef]
- Schmitz, L.; Grinblat, B.; Novak, B.; Hoeh, A.K.; Handschke, K.; von Dobbeler, C.; Bierhoff, E.; Szeimies, R.M.; Gambichler, T.; Torezan, L.; et al. Somatic mutations in kinetochore gene KNSTRN are associated with basal proliferating actinic keratoses and cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. JEADV 2019, 33, 1535–1540. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Yilmaz, A.S.; Ozer, H.G.; Gillespie, J.L.; Allain, D.C.; Bernhardt, M.N.; Furlan, K.C.; Castro, L.T.; Peters, S.B.; Nagarajan, P.; Kang, S.Y.; et al. Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors. Cancer 2017, 123, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, S.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8. [Google Scholar] [CrossRef] [PubMed]
- Bertin, J.; Wang, L.; Guo, Y.; Jacobson, M.D.; Poyet, J.L.; Srinivasula, S.M.; Merriam, S.; DiStefano, P.S.; Alnemri, E.S. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J. Biol. Chem. 2001, 276, 11877–11882. [Google Scholar] [CrossRef]
- Watt, S.A.; Purdie, K.J.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; McHugh, A.T.; Xue, D.J.; Dayal, J.H.; Proby, C.M.; Harwood, C.A.; et al. Novel CARD11 Mutations in Human Cutaneous Squamous Cell Carcinoma Lead to Aberrant NF-kappaB Regulation. Am. J. Pathol. 2015, 185, 2354–2363. [Google Scholar] [CrossRef]
- Sur, I.; Ulvmar, M.; Toftgard, R. The two-faced NF-kappaB in the skin. Int. Rev. Immunol. 2008, 27, 205–223. [Google Scholar] [CrossRef]
- van Hogerlinden, M.; Rozell, B.L.; Ahrlund-Richter, L.; Toftgard, R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999, 59, 3299–3303. [Google Scholar]
- Zhang, J.Y.; Tao, S.; Kimmel, R.; Khavari, P.A. CDK4 regulation by TNFR1 and JNK is required for NF-kappaB-mediated epidermal growth control. J. Cell Biol. 2005, 168, 561–566. [Google Scholar] [CrossRef]
- Seykora, J.T.; Mei, L.; Dotto, G.P.; Stein, P.L. ‘Srcasm: A novel Src activating and signaling molecule. J. Biol. Chem. 2002, 277, 2812–2822. [Google Scholar] [CrossRef]
- Li, W.; Marshall, C.; Mei, L.; Gelfand, J.; Seykora, J.T. Srcasm corrects Fyn-induced epidermal hyperplasia by kinase down-regulation. J. Biol. Chem. 2007, 282, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Marshall, C.; Mei, L.; Dzubow, L.; Schmults, C.; Dans, M.; Seykora, J. Srcasm modulates EGF and Src-kinase signaling in keratinocytes. J. Biol. Chem. 2005, 280, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [CrossRef] [PubMed]
- Gatti, V.; Fierro, C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Peschiaroli, A. ΔNp63 in squamous cell carcinoma: Defining the oncogenic routes affecting epigenetic landscape and tumour microenvironment. Mol. Oncol. 2019, 13, 981–1001. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Vanbokhoven, H.; Melino, G.; Candi, E.; Declercq, W. p63, a story of mice and men. J. Investig. Dermatol. 2011, 131, 1196–1207. [Google Scholar] [CrossRef]
- Candi, E.; Cipollone, R.; Rivetti di Val Cervo, P.; Gonfloni, S.; Melino, G.; Knight, R. p63 in epithelial development. Cell. Mol. Life Sci. CMLS 2008, 65, 3126–3133. [Google Scholar] [CrossRef]
- Petitjean, A.; Ruptier, C.; Tribollet, V.; Hautefeuille, A.; Chardon, F.; Cavard, C.; Puisieux, A.; Hainaut, P.; Caron de Fromentel, C. Properties of the six isoforms of p63: p53-like regulation in response to genotoxic stress and cross talk with DeltaNp73. Carcinogenesis 2008, 29, 273–281. [Google Scholar] [CrossRef]
- Rokudai, S.; Li, Y.; Otaka, Y.; Fujieda, M.; Owens, D.M.; Christiano, A.M.; Nishiyama, M.; Prives, C. STXBP4 regulates APC/C-mediated p63 turnover and drives squamous cell carcinogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E4806–E4814. [Google Scholar] [CrossRef]
- Dohn, M.; Zhang, S.; Chen, X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 2001, 20, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; Solovei, I.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Liu, B.; Rapisarda, V.; Poterlowicz, K.; Malashchuk, I.; Rudolf, J.; Sharov, A.A.; Jahoda, C.A.; Fessing, M.Y.; Benitah, S.A.; et al. Cbx4 maintains the epithelial lineage identity and cell proliferation in the developing stratified epithelium. J. Cell Biol. 2016, 212, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cao, Y.; Gong, X.; Li, H. Long noncoding RNAs in head and neck cancer. Oncotarget 2017, 8, 10726–10740. [Google Scholar] [CrossRef]
- Yugawa, T.; Narisawa-Saito, M.; Yoshimatsu, Y.; Haga, K.; Ohno, S.-i.; Egawa, N.; Fujita, M.; Kiyono, T. ΔNp63α Repression of the Notch1 Gene Supports the Proliferative Capacity of Normal Human Keratinocytes and Cervical Cancer Cells. Cancer Res. 2010, 70, 4034–4044. [Google Scholar] [CrossRef]
- Westfall, M.D.; Mays, D.J.; Sniezek, J.C.; Pietenpol, J.A. The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol. Cell. Biol. 2003, 23, 2264–2276. [Google Scholar] [CrossRef]
- Devos, M.; Gilbert, B.; Denecker, G.; Leurs, K.; Mc Guire, C.; Lemeire, K.; Hochepied, T.; Vuylsteke, M.; Lambert, J.; Van Den Broecke, C.; et al. Elevated DeltaNp63alpha Levels Facilitate Epidermal and Biliary Oncogenic Transformation. J. Investig. Dermatol. 2017, 137, 494–505. [Google Scholar] [CrossRef]
- Ramsey, M.R.; Wilson, C.; Ory, B.; Rothenberg, S.M.; Faquin, W.; Mills, A.A.; Ellisen, L.W. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Investig. 2013, 123, 3525–3538. [Google Scholar] [CrossRef]
- Abraham, C.G.; Ludwig, M.P.; Andrysik, Z.; Pandey, A.; Joshi, M.; Galbraith, M.D.; Sullivan, K.D.; Espinosa, J.M. ΔNp63α Suppresses TGFB2 Expression and RHOA Activity to Drive Cell Proliferation in Squamous Cell Carcinomas. Cell Rep. 2018, 24, 3224–3236. [Google Scholar] [CrossRef]
- Hazawa, M.; Lin, D.C.; Kobayashi, A.; Jiang, Y.Y.; Xu, L.; Dewi, F.R.P.; Mohamed, M.S.; Nakada, M.; Meguro-Horike, M.; Horike, S.I.; et al. ROCK-dependent phosphorylation of NUP62 regulates p63 nuclear transport and squamous cell carcinoma proliferation. EMBO Rep. 2018, 19, 73–88. [Google Scholar] [CrossRef]
- Li, Y.; Peart, M.J.; Prives, C. Stxbp4 regulates DeltaNp63 stability by suppression of RACK1-dependent degradation. Mol. Cell. Biol. 2009, 29, 3953–3963. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.J.; Patel, A.; Purdie, K.J.; Wang, J.; Rizvi, H.; Hufbauer, M.; Ostano, P.; Akgul, B.; Chiorino, G.; Harwood, C.A.; et al. Epigenetic Regulation of iASPP-p63 Feedback Loop in Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 1658–1671.e8. [Google Scholar] [CrossRef] [PubMed]
- de Lima, P.O.; Joseph, S.; Panizza, B.; Simpson, F. Epidermal Growth Factor Receptor’s Function in Cutaneous Squamous Cell Carcinoma and Its Role as a Therapeutic Target in the Age of Immunotherapies. Curr. Treat. Options Oncol. 2020, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Jost, M.; Kari, C.; Rodeck, U. The EGF receptor—An essential regulator of multiple epidermal functions. Eur. J. Dermatol. EJD 2000, 10, 505–510. [Google Scholar] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- Toll, A.; Salgado, R.; Yebenes, M.; Martin-Ezquerra, G.; Gilaberte, M.; Baro, T.; Sole, F.; Alameda, F.; Espinet, B.; Pujol, R.M. Epidermal growth factor receptor gene numerical aberrations are frequent events in actinic keratoses and invasive cutaneous squamous cell carcinomas. Exp. Dermatol. 2010, 19, 151–153. [Google Scholar] [CrossRef]
- Canueto, J.; Cardenoso, E.; Garcia, J.L.; Santos-Briz, A.; Castellanos-Martin, A.; Fernandez-Lopez, E.; Blanco Gomez, A.; Perez-Losada, J.; Roman-Curto, C. Epidermal growth factor receptor expression is associated with poor outcome in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 176, 1279–1287. [Google Scholar] [CrossRef]
- Grandis, J.R.; Zeng, Q.; Drenning, S.D.; Tweardy, D.J. Normalization of EGFR mRNA levels following restoration of wild-type p53 in a head and neck squamous cell carcinoma cell line. Int. J. Oncol. 1998, 13, 375–378. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, L.; Ma, S.; Ma, J.; Wang, Y.; Li, S.; Hu, X.; Han, S.; Zhou, M.; Zhou, L.; et al. MALAT1-KTN1-EGFR regulatory axis promotes the development of cutaneous squamous cell carcinoma. Cell Death Differ. 2019, 26, 2061–2073. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Dai, Y.; Niu, X.; Zhou, M. miR-27a Downregulation Promotes Cutaneous Squamous Cell Carcinoma Progression via Targeting EGFR. Front. Oncol. 2019, 9, 1565. [Google Scholar] [CrossRef]
- Nindl, I.; Dang, C.; Forschner, T.; Kuban, R.J.; Meyer, T.; Sterry, W.; Stockfleth, E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol. Cancer 2006, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Dooley, T.P.; Reddy, S.P.; Wilborn, T.W.; Davis, R.L. Biomarkers of human cutaneous squamous cell carcinoma from tissues and cell lines identified by DNA microarrays and qRT-PCR. Biochem. Biophys. Res. Commun. 2003, 306, 1026–1036. [Google Scholar] [CrossRef]
- Hameetman, L.; Commandeur, S.; Bavinck, J.N.B.; Wisgerhof, H.C.; de Gruijl, F.R.; Willemze, R.; Mullenders, L.; Tensen, C.P.; Vrieling, H. Molecular profiling of cutaneous squamous cell carcinomas and actinic keratoses from organ transplant recipients. BMC Cancer 2013, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diez, I.; Hernandez-Munoz, I.; Hernandez-Ruiz, E.; Nonell, L.; Puigdecanet, E.; Bodalo-Torruella, M.; Andrades, E.; Pujol, R.M.; Toll, A. Transcriptome and cytogenetic profiling analysis of matched in situ/invasive cutaneous squamous cell carcinomas from immunocompetent patients. Genes Chromosomes Cancer 2019, 58, 164–174. [Google Scholar] [CrossRef]
- Mitsui, H.; Suarez-Farinas, M.; Gulati, N.; Shah, K.R.; Cannizzaro, M.V.; Coats, I.; Felsen, D.; Krueger, J.G.; Carucci, J.A. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J. Investig. Dermatol. 2014, 134, 1418–1427. [Google Scholar] [CrossRef]
- Padilla, R.S.; Sebastian, S.; Jiang, Z.; Nindl, I.; Larson, R. Gene expression patterns of normal human skin, actinic keratosis, and squamous cell carcinoma: A spectrum of disease progression. Arch. Dermatol. 2010, 146, 288–293. [Google Scholar] [CrossRef]
- Kathpalia, V.P.; Mussak, E.N.; Chow, S.S.; Lam, P.H.; Skelley, N.; Time, M.; Markelewicz, R.J., Jr.; Kanduc, D.; Lomas, L.; Xiang, Z.; et al. Genome-wide transcriptional profiling in human squamous cell carcinoma of the skin identifies unique tumor-associated signatures. J. Dermatol. 2006, 33, 309–318. [Google Scholar] [CrossRef]
- Ra, S.H.; Li, X.; Binder, S. Molecular discrimination of cutaneous squamous cell carcinoma from actinic keratosis and normal skin. Mod. Pathol. 2011, 24, 963–973. [Google Scholar] [CrossRef]
- Hudson, L.G.; Gale, J.M.; Padilla, R.S.; Pickett, G.; Alexander, B.E.; Wang, J.; Kusewitt, D.F. Microarray analysis of cutaneous squamous cell carcinomas reveals enhanced expression of epidermal differentiation complex genes. Mol. Carcinog. 2010, 49, 619–629. [Google Scholar] [CrossRef]
- Kar, A.; Gutierrez-Hartmann, A. Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 522–543. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Epimutation and cancer: A new carcinogenic mechanism of Lynch syndrome (Review). Int. J. Oncol. 2012, 41, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.P.; Proby, C.M. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Venza, I.; Visalli, M.; Tripodo, B.; De Grazia, G.; Loddo, S.; Teti, D.; Venza, M. FOXE1 is a target for aberrant methylation in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2010, 162, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Darr, O.A.; Colacino, J.A.; Tang, A.L.; McHugh, J.B.; Bellile, E.L.; Bradford, C.R.; Prince, M.P.; Chepeha, D.B.; Rozek, L.S.; Moyer, J.S. Epigenetic alterations in metastatic cutaneous carcinoma. Head Neck 2015, 37, 994–1001. [Google Scholar] [CrossRef]
- Chiles, M.C.; Ai, L.; Zuo, C.; Fan, C.Y.; Smoller, B.R. E-cadherin promoter hypermethylation in preneoplastic and neoplastic skin lesions. Mod. Pathol. 2003, 16, 1014–1018. [Google Scholar] [CrossRef]
- Fraga, M.F.; Herranz, M.; Espada, J.; Ballestar, E.; Paz, M.F.; Ropero, S.; Erkek, E.; Bozdogan, O.; Peinado, H.; Niveleau, A.; et al. A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Res. 2004, 64, 5527–5534. [Google Scholar] [CrossRef]
- Nessa, A.; Rashid, M.H.U.; E-Ferdous, N.; Chowdhury, A. Screening for and management of high-grade cervical intraepithelial neoplasia in Bangladesh: A cross-sectional study comparing two protocols. J. Obstet. Gynaecol. Res. 2013, 39, 564–571. [Google Scholar] [CrossRef]
- Missero, C.; Calautti, E.; Eckner, R.; Chin, J.; Tsai, L.H.; Livingston, D.M.; Dotto, G.P. Involvement of the cell-cycle inhibitor Cip1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc. Natl. Acad. Sci. USA 1995, 92, 5451–5455. [Google Scholar] [CrossRef]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell. Mol. Life Sci. CMLS 2013, 70, 3989–4008. [Google Scholar] [CrossRef]
- McCabe, M.T.; Creasy, C.L. EZH2 as a potential target in cancer therapy. Epigenomics 2014, 6, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hung, M.C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2014, 46, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Balasubramanian, S.; Kerr, C.; Huang, J.M.; Eckert, R.L. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis 2015, 36, 800–810. [Google Scholar] [CrossRef]
- Behrens, C.; Solis, L.M.; Lin, H.; Yuan, P.; Tang, X.; Kadara, H.; Riquelme, E.; Galindo, H.; Moran, C.A.; Kalhor, N.; et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6556–6565. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, H.; Heilman, E.R.; Walsh, M.G.; Haseeb, M.A.; Gupta, R. Increased expression of enhancer of Zeste Homolog 2 (EZH2) differentiates squamous cell carcinoma from normal skin and actinic keratosis. Eur. J. Dermatol. EJD 2014, 24, 41–45. [Google Scholar] [CrossRef]
- Wu, C.; Chen, Z.; Kuchroo, V.K. Ezh2 lines up the chromatin in T regulatory cells. Immunity 2015, 42, 201–203. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, B.; Wen, X.; Hao, D.; Du, D.; He, G.; Jiang, X. The Roles of lncRNA in Cutaneous Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 158. [Google Scholar] [CrossRef]
- Kretz, M. TINCR, staufen1, and cellular differentiation. RNA Biol. 2013, 10, 1597–1601. [Google Scholar] [CrossRef]
- Piipponen, M.; Nissinen, L.; Farshchian, M.; Riihila, P.; Kivisaari, A.; Kallajoki, M.; Peltonen, J.; Peltonen, S.; Kahari, V.M. Long Noncoding RNA PICSAR Promotes Growth of Cutaneous Squamous Cell Carcinoma by Regulating ERK1/2 Activity. J. Investig. Dermatol. 2016, 136, 1701–1710. [Google Scholar] [CrossRef]
- Mei, X.L.; Zhong, S. Long noncoding RNA LINC00520 prevents the progression of cutaneous squamous cell carcinoma through the inactivation of the PI3K/Akt signaling pathway by downregulating EGFR. Chin. Med. J. 2019, 132, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liao, J.; Duan, X.; He, Y.; Liao, Y. Upregulation of LINC00319 indicates a poor prognosis and promotes cell proliferation and invasion in cutaneous squamous cell carcinoma. J. Cell. Biochem. 2018, 119, 10393–10405. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Q.; Kuang, S.; Zhao, C.; Yang, L.; Zhang, Y.; Zhu, H.; Yang, R. USF1-induced upregulation of LINC01048 promotes cell proliferation and apoptosis in cutaneous squamous cell carcinoma by binding to TAF15 to transcriptionally activate YAP1. Cell Death Dis. 2019, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Li, C.; Luo, S.; Liu-Smith, F.; Yang, J.; Wang, X.; Wang, N.; Lai, B.; Lei, T.; Wang, Q.; et al. Yes-Associated Protein Contributes to the Development of Human Cutaneous Squamous Cell Carcinoma via Activation of RAS. J. Investig. Dermatol. 2016, 136, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Chan, C.W.; Fang, J.Y.; Shih, Y.M.; Liu, Y.W.; Wang, T.V.; Chen, C.Y. 2-O-Methylmagnolol upregulates the long non-coding RNA, GAS5, and enhances apoptosis in skin cancer cells. Cell Death Dis. 2017, 8, e2638. [Google Scholar] [CrossRef]
- Yu, G.J.; Sun, Y.; Zhang, D.W.; Zhang, P. Long non-coding RNA HOTAIR functions as a competitive endogenous RNA to regulate PRAF2 expression by sponging miR-326 in cutaneous squamous cell carcinoma. Cancer Cell Int. 2019, 19, 270. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- García-Sancha, N.; Corchado-Cobos, R.; Pérez-Losada, J.; Cañueto, J. MicroRNA Dysregulation in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar] [CrossRef]
- Benaich, N.; Woodhouse, S.; Goldie, S.J.; Mishra, A.; Quist, S.R.; Watt, F.M. Rewiring of an epithelial differentiation factor, miR-203, to inhibit human squamous cell carcinoma metastasis. Cell Rep. 2014, 9, 104–117. [Google Scholar] [CrossRef]
- Yamane, K.; Jinnin, M.; Etoh, T.; Kobayashi, Y.; Shimozono, N.; Fukushima, S.; Masuguchi, S.; Maruo, K.; Inoue, Y.; Ishihara, T.; et al. Down-regulation of miR-124/-214 in cutaneous squamous cell carcinoma mediates abnormal cell proliferation via the induction of ERK. J. Mol. Med. 2013, 91, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, W.; Ma, S.; Cao, H.; Peng, X.; Guo, L.; Zhou, X.; Zheng, L.; Guo, L.; Wan, M.; et al. A novel onco-miR-365 induces cutaneous squamous cell carcinoma. Carcinogenesis 2013, 34, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, Y.; Zhou, M.; Zhang, Y.; Wang, P.; Li, X.; Yang, J.; Wang, H.; Ding, Z. HOXA9 inhibits HIF-1alpha-mediated glycolysis through interacting with CRIP2 to repress cutaneous squamous cell carcinoma development. Nat. Commun. 2018, 9, 1480. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhang, L.; Meisgen, F.; Harada, M.; Heilborn, J.; Homey, B.; Grander, D.; Stahle, M.; Sonkoly, E.; Pivarcsi, A. MicroRNA-125b down-regulates matrix metallopeptidase 13 and inhibits cutaneous squamous cell carcinoma cell proliferation, migration, and invasion. J. Biol. Chem. 2012, 287, 29899–29908. [Google Scholar] [CrossRef]
- Olasz, E.B.; Seline, L.N.; Schock, A.M.; Duncan, N.E.; Lopez, A.; Lazar, J.; Flister, M.J.; Lu, Y.; Liu, P.; Sokumbi, O.; et al. MicroRNA-135b Regulates Leucine Zipper Tumor Suppressor 1 in Cutaneous Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0125412. [Google Scholar] [CrossRef]
- Davis, A.J.; Tsinkevich, M.; Rodencal, J.; Abbas, H.A.; Su, X.H.; Gi, Y.J.; Fang, B.; Rajapakshe, K.; Coarfa, C.; Gunaratne, P.H.; et al. TAp63-regulated microRNAs suppress cutaneous squamous cell carcinoma through inhibition of a network of cell cycle genes. Cancer Res. 2020, 80, 2484–2497. [Google Scholar] [CrossRef]
- Chen, I.P.; Henning, S.; Faust, A.; Boukamp, P.; Volkmer, B.; Greinert, R. UVA-induced epigenetic regulation of P16(INK4a) in human epidermal keratinocytes and skin tumor derived cells. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2012, 11, 180–190. [Google Scholar] [CrossRef]
- Yang, H.; Schramek, D.; Adam, R.C.; Keyes, B.E.; Wang, P.; Zheng, D.; Fuchs, E. ETS family transcriptional regulators drive chromatin dynamics and malignancy in squamous cell carcinomas. eLife 2015, 4, e10870. [Google Scholar] [CrossRef]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. SnapShot: Histone modifications. Cell 2014, 159, 458. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Kasraian, Z.; Rezvani, H.R. Energy metabolism in skin cancers: A therapeutic perspective. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 712–722. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hosseini, M.; Dousset, L.; Mahfouf, W.; Serrano-Sanchez, M.; Redonnet-Vernhet, I.; Mesli, S.; Kasraian, Z.; Obre, E.; Bonneu, M.; Claverol, S.; et al. Energy Metabolism Rewiring Precedes UVB-Induced Primary Skin Tumor Formation. Cell Rep. 2018, 23, 3621–3634. [Google Scholar] [CrossRef]
- Flores, A.; Sandoval-Gonzalez, S.; Takahashi, R.; Krall, A.; Sathe, L.; Wei, L.; Radu, C.; Joly, J.H.; Graham, N.A.; Christofk, H.R.; et al. Increased lactate dehydrogenase activity is dispensable in squamous carcinoma cells of origin. Nat. Commun. 2019, 10, 91. [Google Scholar] [CrossRef]
- Goodwin, J.; Neugent, M.L.; Lee, S.Y.; Choe, J.H.; Choi, H.; Jenkins, D.M.R.; Ruthenborg, R.J.; Robinson, M.W.; Jeong, J.Y.; Wake, M.; et al. The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat. Commun. 2017, 8, 15503. [Google Scholar] [CrossRef]
- Zheng, W.; Tayyari, F.; Gowda, G.A.; Raftery, D.; McLamore, E.S.; Porterfield, D.M.; Donkin, S.S.; Bequette, B.; Teegarden, D. Altered glucose metabolism in Harvey-ras transformed MCF10A cells. Mol. Carcinog. 2015, 54, 111–120. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Bantubungi, K.; Hannou, S.A.; Caron-Houde, S.; Vallez, E.; Baron, M.; Lucas, A.; Bouchaert, E.; Paumelle, R.; Tailleux, A.; Staels, B. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1alpha pathway. Diabetes 2014, 63, 3199–3209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krishnan, K.J.; Birch-Machin, M.A. The incidence of both tandem duplications and the common deletion in mtDNA from three distinct categories of sun-exposed human skin and in prolonged culture of fibroblasts. J. Investig. Dermatol. 2006, 126, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Murphy, G.; Ralph, N.; O’Gorman, S.M.; Murphy, J.E. Mitochondrial DNA deletion percentage in sun exposed and non sun exposed skin. J. Photochem. Photobiol. B Biol. 2016, 165, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.J.; Turner, R.; Nikaido, O.; Rees, J.L.; Birch-Machin, M.A. The spectrum of mitochondrial DNA deletions is a ubiquitous marker of ultraviolet radiation exposure in human skin. J. Investig. Dermatol. 2000, 115, 674–679. [Google Scholar] [CrossRef]
- Yusoff, A.A.M.; Abdullah, W.S.W.; Khair, S.; Radzak, S.M.A. A comprehensive overview of mitochondrial DNA 4977-bp deletion in cancer studies. Oncol. Rev. 2019, 13, 409. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Kim, A.L.; Rossignol, R.; Ali, N.; Daly, M.; Mahfouf, W.; Bellance, N.; Taieb, A.; de Verneuil, H.; Mazurier, F.; et al. XPC silencing in normal human keratinocytes triggers metabolic alterations that drive the formation of squamous cell carcinomas. J. Clin. Investig. 2011, 121, 195–211. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Rossignol, R.; Ali, N.; Benard, G.; Tang, X.; Yang, H.S.; Jouary, T.; de Verneuil, H.; Taieb, A.; Kim, A.L.; et al. XPC silencing in normal human keratinocytes triggers metabolic alterations through NOX-1 activation-mediated reactive oxygen species. Biochim. Biophys. Acta 2011, 1807, 609–619. [Google Scholar] [CrossRef]
- Harbottle, A.; Birch-Machin, M.A. Real-time PCR analysis of a 3895 bp mitochondrial DNA deletion in nonmelanoma skin cancer and its use as a quantitative marker for sunlight exposure in human skin. Br. J. Cancer 2006, 94, 1887–1893. [Google Scholar] [CrossRef]
- Hosseini, M.; Dousset, L.; Michon, P.; Mahfouf, W.; Muzotte, E.; Bergeron, V.; Bortolotto, D.; Rossignol, R.; Moisan, F.; Taieb, A.; et al. UVB-induced DHODH upregulation, which is driven by STAT3, is a promising target for chemoprevention and combination therapy of photocarcinogenesis. Oncogenesis 2019, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Uribe, P.; Gonzalez, S. Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: Molecular bases for EGFR-targeted therapy. Pathol. Res. Pract. 2011, 207, 337–342. [Google Scholar] [CrossRef] [PubMed]
- William, W.N., Jr.; Feng, L.; Ferrarotto, R.; Ginsberg, L.; Kies, M.; Lippman, S.; Glisson, B.; Kim, E.S. Gefitinib for patients with incurable cutaneous squamous cell carcinoma: A single-arm phase II clinical trial. J. Am. Acad. Dermatol. 2017, 77, 1110–1113.e2. [Google Scholar] [CrossRef] [PubMed]
- Gold, K.A.; Kies, M.S.; William, W.N., Jr.; Johnson, F.M.; Lee, J.J.; Glisson, B.S. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: A single-arm phase 2 clinical trial. Cancer 2018, 124, 2169–2173. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef] [PubMed]
- Montaudie, H.; Viotti, J.; Combemale, P.; Dutriaux, C.; Dupin, N.; Robert, C.; Mortier, L.; Kaphan, R.; Duval-Modeste, A.B.; Dalle, S.; et al. Cetuximab is efficient and safe in patients with advanced cutaneous squamous cell carcinoma: A retrospective, multicentre study. Oncotarget 2020, 11, 378–385. [Google Scholar] [CrossRef]
- Foote, M.C.; McGrath, M.; Guminski, A.; Hughes, B.G.; Meakin, J.; Thomson, D.; Zarate, D.; Simpson, F.; Porceddu, S.V. Phase II study of single-agent panitumumab in patients with incurable cutaneous squamous cell carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 2047–2052. [Google Scholar] [CrossRef]
- Galbiati, D.; Cavalieri, S.; Alfieri, S.; Resteghini, C.; Bergamini, C.; Orlandi, E.; Platini, F.; Locati, L.; Giacomelli, L.; Licitra, L.; et al. Activity of platinum and cetuximab in cutaneous squamous cell cancer not amenable to curative treatment. Drugs Context 2019, 8, 212611. [Google Scholar] [CrossRef]
- Reigneau, M.; Robert, C.; Routier, E.; Mamelle, G.; Moya-Plana, A.; Tomasic, G.; Mateus, C. Efficacy of neoadjuvant cetuximab alone or with platinum salt for the treatment of unresectable advanced nonmetastatic cutaneous squamous cell carcinomas. Br. J. Dermatol. 2015, 173, 527–534. [Google Scholar] [CrossRef]
- Corchado-Cobos, R.; Garcia-Sancha, N.; Gonzalez-Sarmiento, R.; Perez-Losada, J.; Canueto, J. Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 2956. [Google Scholar] [CrossRef]
- Burova, E.; Hermann, A.; Waite, J.; Potocky, T.; Lai, V.; Hong, S.; Liu, M.; Allbritton, O.; Woodruff, A.; Wu, Q.; et al. Characterization of the Anti-PD-1 Antibody REGN2810 and Its Antitumor Activity in Human PD-1 Knock-In Mice. Mol. Cancer Ther. 2017, 16, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet. Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Grob, J.J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef]
- Maubec, E.; Boubaya, M.; Petrow, P.; Beylot-Barry, M.; Basset-Seguin, N.; Deschamps, L.; Grob, J.J.; Dreno, B.; Scheer-Senyarich, I.; Bloch-Queyrat, C.; et al. Phase II Study of Pembrolizumab As First-Line, Single-Drug Therapy for Patients With Unresectable Cutaneous Squamous Cell Carcinomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3051–3061. [Google Scholar] [CrossRef]
- Keeping, S.; Xu, Y.; Chen, C.I.; Cope, S.; Mojebi, A.; Kuznik, A.; Konidaris, G.; Ayers, D.; Sasane, M.; Allen, R.; et al. Comparative efficacy of cemiplimab versus other systemic treatments for advanced cutaneous squamous cell carcinoma. Future Oncol. 2021, 17, 611–627. [Google Scholar] [CrossRef]
- Chen, A.; Ali, N.; Boasberg, P.; Ho, A.S. Clinical Remission of Cutaneous Squamous Cell Carcinoma of the Auricle with Cetuximab and Nivolumab. J. Clin. Med. 2018, 7, 10. [Google Scholar] [CrossRef]
- Sernicola, A.; Lampitelli, S.; Marraffa, F.; Maddalena, P.; Grassi, S.; Richetta, A.G.; Calvieri, S. Case Report: Cetuximab use in advanced cutaneous squamous cell carcinoma resistant to chemotherapy. F1000Research 2019, 8, 933. [Google Scholar] [CrossRef]
- Yang, X.; Daifallah, A.E.M.; Shankar, S.; Beer, J.; Marshall, C.; Dentchev, T.; Seykora, F.; D’Armas, S.; Hahn, J.; Lee, V.; et al. Topical kinase inhibitors induce regression of cutaneous squamous cell carcinoma. Exp. Dermatol. 2019, 28, 609–613. [Google Scholar] [CrossRef]
- Naing, A.; Cohen, R.; Dy, G.K.; Hong, D.S.; Dyster, L.; Hangauer, D.G.; Kwan, R.; Fetterly, G.; Kurzrock, R.; Adjei, A.A. A phase I trial of KX2-391, a novel non-ATP competitive substrate-pocket- directed SRC inhibitor, in patients with advanced malignancies. Investig. New Drugs 2013, 31, 967–973. [Google Scholar] [CrossRef]
- Olivieri, A.; Manzione, L. Dasatinib: A new step in molecular target therapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2007, 18 (Suppl. 6), vi42–vi46. [Google Scholar] [CrossRef] [PubMed]
- Smolinski, M.P.; Bu, Y.; Clements, J.; Gelman, I.H.; Hegab, T.; Cutler, D.L.; Fang, J.W.S.; Fetterly, G.; Kwan, R.; Barnett, A.; et al. Discovery of Novel Dual Mechanism of Action Src Signaling and Tubulin Polymerization Inhibitors (KX2-391 and KX2-361). J. Med. Chem. 2018, 61, 4704–4719. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Kempers, S.; Lain, E.; Schlesinger, T.; Tyring, S.; Forman, S.; Ablon, G.; Martin, G.; Wang, H.; Cutler, D.L.; et al. Phase 3 Trials of Tirbanibulin Ointment for Actinic Keratosis. N. Engl. J. Med. 2021, 384, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Yanai, H.; Kaji, T.; Kubo, T.; Ennishi, D.; Hirasawa, A.; Yoshino, T. Secretory carcinoma of the skin with lymph node metastases and recurrence in both lungs: A case report. J. Cutan. Pathol. 2021, 48, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kang, H.; Mehra, R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC). Cancers Head Neck 2018, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Darido, C.; Georgy, S.R.; Cullinane, C.; Partridge, D.D.; Walker, R.; Srivastava, S.; Roslan, S.; Carpinelli, M.R.; Dworkin, S.; Pearson, R.B.; et al. Stage-dependent therapeutic efficacy in PI3K/mTOR-driven squamous cell carcinoma of the skin. Cell Death Differ. 2018, 25, 1146–1159. [Google Scholar] [CrossRef]
- Zou, Y.; Ge, M.; Wang, X. Targeting PI3K-AKT-mTOR by LY3023414 inhibits human skin squamous cell carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 490, 385–392. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Chung, C. Current targeted therapies in lymphomas. Am. J. Health Syst. Pharm. 2019, 76, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Kurundkar, D.; Srivastava, R.K.; Chaudhary, S.C.; Ballestas, M.E.; Kopelovich, L.; Elmets, C.A.; Athar, M. Vorinostat, an HDAC inhibitor attenuates epidermoid squamous cell carcinoma growth by dampening mTOR signaling pathway in a human xenograft murine model. Toxicol. Appl. Pharmacol. 2013, 266, 233–244. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Torres-Lockhart, K.; Forster, N.; Ramakrishnan, S.; Greninger, P.; Garnett, M.J.; McDermott, U.; Rothenberg, S.M.; Benes, C.H.; Ellisen, L.W. Mcl-1 and FBW7 control a dominant survival pathway underlying HDAC and Bcl-2 inhibitor synergy in squamous cell carcinoma. Cancer Discov. 2013, 3, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hedberg, M.L.; Berry, C.T.; Moshiri, A.S.; Xiang, Y.; Yeh, C.J.; Attilasoy, C.; Capell, B.C.; Seykora, J.T. Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3478. https://doi.org/10.3390/ijms23073478
Hedberg ML, Berry CT, Moshiri AS, Xiang Y, Yeh CJ, Attilasoy C, Capell BC, Seykora JT. Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2022; 23(7):3478. https://doi.org/10.3390/ijms23073478
Chicago/Turabian StyleHedberg, Matthew L., Corbett T. Berry, Ata S. Moshiri, Yan Xiang, Christopher J. Yeh, Cem Attilasoy, Brian C. Capell, and John T. Seykora. 2022. "Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma" International Journal of Molecular Sciences 23, no. 7: 3478. https://doi.org/10.3390/ijms23073478
APA StyleHedberg, M. L., Berry, C. T., Moshiri, A. S., Xiang, Y., Yeh, C. J., Attilasoy, C., Capell, B. C., & Seykora, J. T. (2022). Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences, 23(7), 3478. https://doi.org/10.3390/ijms23073478