
Marginal Zone B-Cell Populations and Their Regulatory Potential in the Context of HIV and Other Chronic Inflammatory Conditions

Abstract
:1. Introduction
2. Ontogeny of MZ B-Cells
3. MZ B-Cells and Their Antibody Responses
4. MZ B-Cell Populations and Their Regulatory Potential
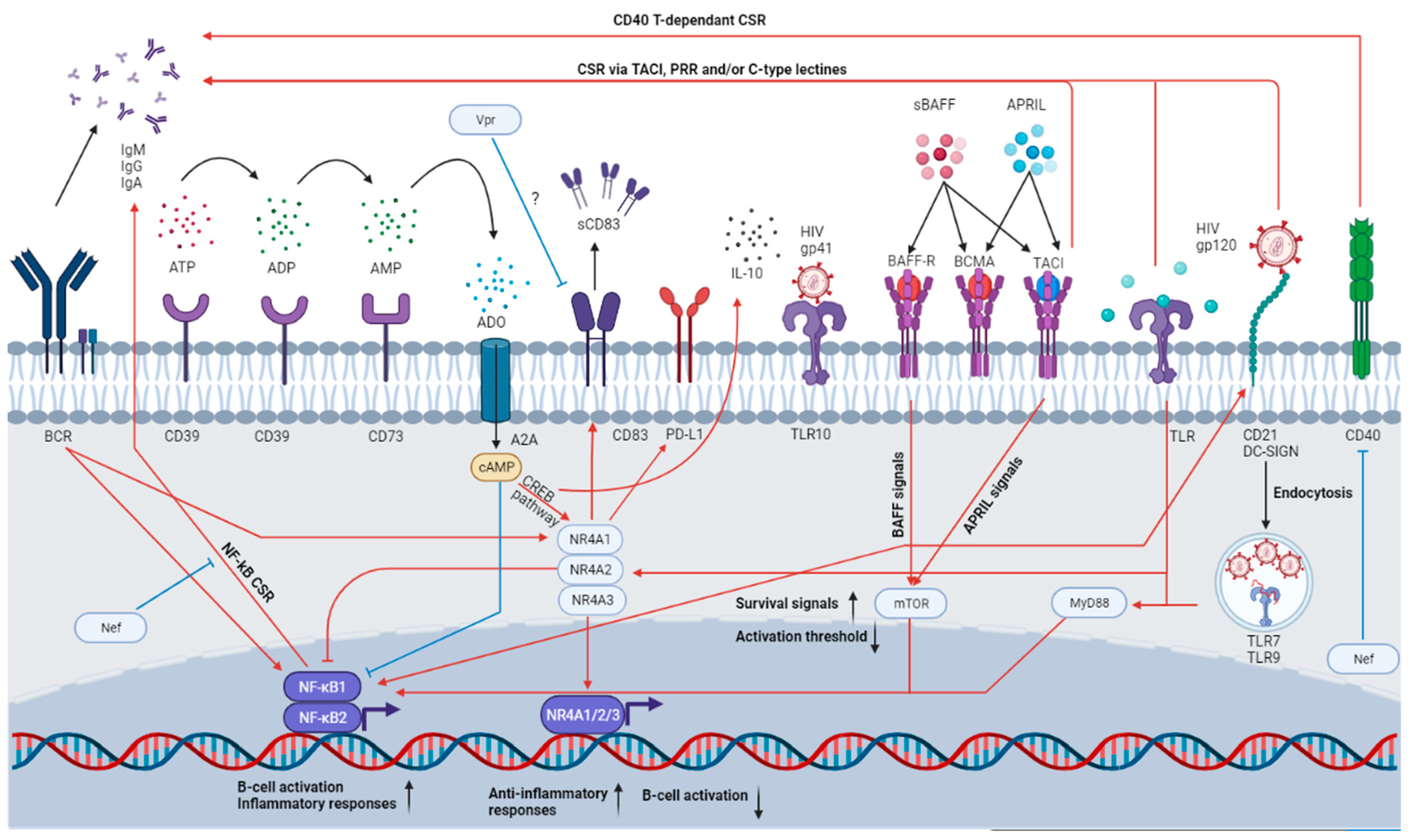
4.1. Importance of NR4As
4.2. Importance of CD39 and CD73
5. The BAFF/APRIL System
6. HIV Infection and the Dysregulation of the B-Cell Compartment
7. MZp in the Context of HIV
8. MZps and Similar Populations in Other Diseases
8.1. Autoimmune Diseases
8.2. Atherosclerosis
8.3. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2) and Other Viral Infections
8.4. Malignancies Associated with MZ Deregulations
9. Possible Therapeutic Avenues
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef] [Green Version]
- Palm, E.A.-K.; Kleinau, S. Marginal zone B cells: From housekeeping function to autoimmunity? J. Autoimmun. 2021, 119, 102627. [Google Scholar] [CrossRef] [PubMed]
- Weill, J.-C.; Weller, S.; Reynaud, C.-A. Human Marginal Zone B Cells. Annu. Rev. Immunol. 2009, 27, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Doyon-Laliberté, K.; Chagnon-Choquet, J.; Byrns, M.; Aranguren, M.; Memmi, M.; Chrobak, P.; Stagg, J.; Poudrier, J.; Roger, M. NR4A Expression by Human Marginal Zone B-Cells. Antibodies 2019, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, J.; Chagnon-Choquet, J.; Valcke, H.S.; Poudrier, J.; Roger, M.; the Montreal Primary HIV Infection and Long-Term Non-Progressor Study Groups. High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood 2011, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Chagnon-Choquet, J.; Fontaine, J.; Poudrier, J.; Roger, M.; for the Montreal Primary HIV Infection and Slow Progressor Study Groups. IL-10 and Lymphotoxin-α Expression Profiles within Marginal Zone-Like B-Cell Populations Are Associated with Control of HIV-1 Disease Progression. PLOS ONE 2014, 9, e101949. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Kearney, J.F. Marginal-zone B cells. Nat. Rev. Immunol. 2002, 2, 323–335. [Google Scholar] [CrossRef]
- Jackson, R.T.; Ling, R.E.; Roy, A. The Origin of B-cells: Human Fetal B Cell Development and Implications for the Patho-genesis of Childhood Acute Lymphoblastic Leukemia. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Hayakawa, K. B CELL DEVELOPMENT PATHWAYS. Annu. Rev. Immunol. 2001, 19, 595–621. [Google Scholar] [CrossRef] [PubMed]
- Nishana, M.; Raghavan, S.C. Role of recombination activating genes in the generation of antigen receptor diversity and beyond. Immunology 2012, 137, 271–281. [Google Scholar] [CrossRef]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Han, J.; Yang, M.; Zhu, J.; Jin, T. Transitional B cells involved in autoimmunity and their impact on neuroimmunological diseases. J. Transl. Med. 2020, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Cariappa, A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 2009, 9, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Tull, T.J.; Pitcher, M.J.; Guesdon, W.; Siu, J.H.; Lebrero-Fernández, C.; Zhao, Y.; Petrov, N.; Heck, S.; Ellis, R.; Dhami, P.; et al. Human marginal zone B cell development from early T2 progenitors. J. Exp. Med. 2021, 218, e20202001. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, S.L.; Russell, D.M.; Nemazee, D. Receptor editing in self-reactive bone marrow B cells. The Journal of Experimental Medicine. 1993. 177: 1009-1020. J. Immunol. 2011, 186, 1313–1324. [Google Scholar]
- Spencer, J.; Finn, T.; Pulford, K.A.; Mason, D.Y.; Isaacson, P.G. The human gut contains a novel population of B lymphocytes which resemble marginal zone cells. Clin. Exp. Immunol. 1985, 62, 607–612. [Google Scholar]
- Thomas, L.; Rothstein, D.O.; Griffin, N.E.; Holodick, T.D. Human B-1 cells take the stage. Ann. N. Y. Acad. Sci. 2013, 1285, 97–114. [Google Scholar]
- Baumgarth, N. A Hard(y) Look at B-1 Cell Development and Function. J. Immunol. 2017, 199, 3387–3394. [Google Scholar] [CrossRef]
- Lechner, M.; Engleitner, T.; Babushku, T.; Schmidt-Supprian, M.; Rad, R.; Strobl, L.J.; Zimber-Strobl, U. Notch2-mediated plasticity between marginal zone and follicular B cells. Nat. Commun. 2021, 12, 1111. [Google Scholar] [CrossRef]
- Cariappa, A.; Tang, M.; Parng, C.; Nebelitskiy, E.; Carroll, M.; Georgopoulos, K.; Pillai, S. The Follicular versus Marginal Zone B Lymphocyte Cell Fate Decision Is Regulated by Aiolos, Btk, and CD21. Immunity 2001, 14, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Hammad, H.; Vanderkerken, M.; Pouliot, P.; Deswarte, K.; Toussaint, W.; Vergote, K.; Vandersarren, L.; Janssens, S.; Ramou, I.; Savvides, S.N.; et al. Transitional B cells commit to marginal zone B cell fate by Taok3-mediated surface expression of ADAM10. Nat. Immunol. 2017, 18, 313–320. [Google Scholar] [CrossRef]
- Moran, S.T.; Cariappa, A.; Liu, H.; Muir, B.; Sgroi, D.; Boboila, C.; Pillai, S. Synergism between NF-κB1/p50 and Notch2 during the Development of Marginal Zone B Lymphocytes. J. Immunol. 2007, 179, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Poe, J.C.; Su, H.; Anand, S.; Matsushima, G.K.; Rathmell, J.C.; Maillard, I.; Radojcic, V.; Imai, K.; Reyes, N.J.; et al. BAFF promotes heightened BCR responsiveness and manifestations of chronic GVHD after allogeneic stem cell transplantation. Blood 2021, 137, 2544–2557. [Google Scholar] [CrossRef] [PubMed]
- Dono, M.; Zupo, S.; Leanza, N.; Melioli, G.; Fogli, M.; Melagrana, A.; Chiorazzi, N.; Ferrarini, M. Heterogeneity of Tonsillar Subepithelial B Lymphocytes, the Splenic Marginal Zone Equivalents. J. Immunol. 2000, 164, 5596–5604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, S. Human blood IgM memory B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 2004, 104, 3647–3654. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Hendricks, J.; Bos, N.A.; Kroese, F.G. Heterogeneity of Memory Marginal Zone B Cells. Crit. Rev. Immunol. 2018, 38, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Berkowska, M.A.; Driessen, G.J.A.; Bikos, V.; Grosserichter-Wagener, C.; Stamatopoulos, K.; Cerutti, A.; He, B.; Biermann, K.; Lange, J.F.; van der Burg, M.; et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood 2011, 118, 2150–2158. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Küppers, R. Human memory B cells. Leukemia 2016, 30, 2283–2292. [Google Scholar] [CrossRef]
- Janeway, C.A.Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Bendelac, A.; Bonneville, M.; Kearney, J.F. Autoreactivity by design: Innate B and T lymphocytes. Nat. Rev. Immunol. 2001, 1, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Okigaki, M.; Schlessinger, J.; Ravetch, J.V. Absence of marginal zone B cells in Pyk-2–deficient mice defines their role in the humoral response. Nat. Immunol. 2000, 1, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Oliver, A.M.; Kearney, J.F. Marginal Zone and B1 B Cells Unite in the Early Response against T-Independent Blood-Borne Particulate Antigens. Immunity 2001, 14, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Cerny, J. Functional heterogeneity of marginal zone B cells revealed by their ability to generate both early anti-body-forming cells and germinal centers with hypermutation and memory in response to a T-dependent antigen. J. Exp. Med. 2003, 198, 1923–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, C.P.; Draves, K.E.; Giltiay, N.V.; Clark, E.A. Extrafollicular B cell activation by marginal zone dendritic cells drives T cell-dependent antibody re-sponses. J. Exp. Med. 2012, 209, 1825–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinamon, G.; Zachariah, M.A.; Lam, O.M.; Foss, F.W.; Cyster, J.G. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008, 9, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biram, A.; Davidzohn, N.; Shulman, Z. T cell interactions with B cells during germinal center formation, a three-step model. Immunol. Rev. 2019, 288, 37–48. [Google Scholar] [CrossRef]
- He, B.; Qiao, X.; Klasse, P.J.; Chiu, A.; Chadburn, A.; Knowles, D.M.; Moore, J.P.; Cerutti, A. HIV-1 Envelope Triggers Polyclonal Ig Class Switch Recombination through a CD40-Independent Mechanism Involving BAFF and C-Type Lectin Receptors. J. Immunol. 2006, 176, 3931–3941. [Google Scholar] [CrossRef] [Green Version]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350.e7. [Google Scholar] [CrossRef]
- Gatto, D.; Brink, R. The germinal center reaction. J. Allergy Clin. Immunol. 2010, 126, 898–907. [Google Scholar] [CrossRef]
- Weller, S.; Mamani-Matsuda, M.; Picard, C.; Cordier, C.; Lecoeuche, D.; Gauthier, F.; Weill, J.C.; Reynaud, C.A. Somatic diversification in the absence of antigen-driven responses is the hallmark of the IgM+ IgD+ CD27+ B cell repertoire in infants. J. Exp. Med. 2008, 205, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Scheeren, F.A. T cell-independent development and induction of somatic hypermutation in human IgM+ IgD+ CD27+ B cells. J. Exp. Med. 2008, 205, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell–helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Santamaria, R.; Xu, W.; Cols, M.; Chen, K.; Puga, I.; Shan, M.; Xiong, H.; Bussel, J.B.; Chiu, A.; et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat. Immunol. 2010, 11, 836–845. [Google Scholar] [CrossRef] [Green Version]
- Treml, L.S.; Carlesso, G.; Hoek, K.L.; Stadanlick, J.E.; Kambayashi, T.; Bram, R.J.; Cancro, M.P.; Khan, W.N. TLR stimulation modifies BLyS receptor expression in follicular and marginal zone B cells. J. Immunol. 2007, 178, 7531–7539. [Google Scholar] [CrossRef] [Green Version]
- Attanavanich, K.; Kearney, J.F. Marginal Zone, but Not Follicular B Cells, Are Potent Activators of Naive CD4 T Cells. J. Immunol. 2004, 172, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Marinkovic, D.; Marinkovic, T. Putative role of marginal zone B cells in pathophysiological processes. Scand. J. Immunol. 2020, 92, e12920. [Google Scholar] [CrossRef]
- Bialecki, E.; Paget, C.; Fontaine, J.; Capron, M.; Trottein, F.; Faveeuw, C. Role of Marginal Zone B Lymphocytes in Invariant NKT Cell Activation. J. Immunol. 2009, 182, 6105–6113. [Google Scholar] [CrossRef]
- Mauri, C.; Menon, M. The expanding family of regulatory B cells. Int. Immunol. 2015, 27, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Mauri, C.; Bosma, A. Immune Regulatory Function of B Cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Iizuka-Koga, M.; Nakatsukasa, H.; Ito, M.; Akanuma, T.; Lu, Q.; Yoshimura, A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J. Autoimmun. 2017, 83, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K.; Bouaziz, J.-D.; Matsushita, T.; Tsubata, T.; Tedder, T.F. The Development and Function of Regulatory B Cells Expressing IL-10 (B10 Cells) Requires Antigen Receptor Diversity and TLR Signals. J. Immunol. 2009, 182, 7459–7472. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K.; Bouaziz, J.-D.; Haas, K.M.; Poe, J.C.; Fujimoto, M.; Tedder, T.F. A Regulatory B Cell Subset with a Unique CD1dhiCD5+ Phenotype Controls T Cell-Dependent Inflammatory Responses. Immunity 2008, 28, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, K.; Sármay, G.; Kövesdi, D. MZ B cells migrate in a T-bet dependent manner and might contribute to the remission of collagen-induced arthritis by the secretion of IL-10. Eur. J. Immunol. 2016, 46, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.G.; Chavez-Rueda, K.A.; Eddaoudi, A.; Meyer-Bahlburg, A.; Rawlings, D.J.; Ehrenstein, M.; Mauri, C. Novel Suppressive Function of Transitional 2 B Cells in Experimental Arthritis. J. Immunol. 2007, 178, 7868–7878. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-H.; Li, K.-P.; Chu, K.-H.; Chiang, B.-L. A B-1a cell subset induces Foxp3− T cells with regulatory activity through an IL-10-independent pathway. Cell Mol. Immunol. 2015, 12, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillatreau, S. Natural regulatory plasma cells. Curr. Opin. Immunol. 2018, 55, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Baba, A.; Yokota, T.; Nishikawa, H.; Ohkawa, Y.; Kayama, H.; Kallies, A.; Nutt, S.L.; Sakaguchi, S.; Takeda, K.; et al. Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity 2014, 41, 1040–1051. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Yeung, M.; Camirand, G.; Zeng, Q.; Akiba, H.; Yagita, H.; Chalasani, G.; Sayegh, M.H.; Najafian, N.; Rothstein, D.M. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J. Clin. Investig. 2011, 121, 3645–3656. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, J.; Zhou, N.; Zhang, Y.; Wu, M.; Xu, J.; Shen, C.; An, X.; Shen, G.; Yang, M.; et al. The Unknown Aspect of BAFF: Inducing IL-35 Production by a CD5(+)CD1d(hi)FcγRIIb(hi) Regulatory B-Cell Subset in Lupus. J. Invest. Derm. 2017, 137, 2532–2543. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Basu, S.; Williams, C.B.; Salzman, N.; Dittel, B.N. A Novel IL-10–Independent Regulatory Role for B Cells in Suppressing Autoimmunity by Maintenance of Regulatory T Cells via GITR Ligand. J. Immunol. 2012, 188, 3188–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundy, S.K. Killer B lymphocytes: The evidence and the potential. Agents Actions 2009, 58, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Zekzer, D.; Hanssen, L.; Lu, Y.; Olcott, A.; Kaufman, D.L. Lipopolysaccharide-Activated B Cells Down-Regulate Th1 Immunity and Prevent Autoimmune Diabetes in Nonobese Diabetic Mice. J. Immunol. 2001, 167, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.R.; Hams, E.; Floudas, A.; Sparwasser, T.; Weaver, C.T.; Fallon, P. PD-L1hi B cells are critical regulators of humoral immunity. Nat. Commun. 2015, 6, 5997. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. Exosomes Exercise Inhibition of Anti-Tumor Immunity during Chemotherapy. Immunity 2019, 50, 547–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaku, H. A novel mechanism of B cell-mediated immune suppression through CD73 expression and adenosine pro-duction. J. Immunol. 2014, 193, 5904–5913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafei, M.; Hsieh, J.; Zehntner, S.; Li, M.; Forner, K.; Birman, E.; Boivin, M.-N.; Young, Y.K.; Perreault, C.; Galipeau, J. A granulocyte-macrophage colony–stimulating factor and interleukin-15 fusokine induces a regulatory B cell population with immune suppressive properties. Nat. Med. 2009, 15, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.A.; Yassin-Noreña, L.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.; Ehrenstein, M.; Mauri, C. CD19+CD24hiCD38hi B Cells Exhibit Regulatory Capacity in Healthy Individuals but Are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Weiqian, C.; Lihuan, Y. Peripheral CD24 hi CD27 + CD19 + B cells subset as a potential biomarker in naïve systemic lupus erythematosus. Int. J. Rheum. Dis. 2013, 16, 698–708. [Google Scholar] [CrossRef]
- van de Veen, W.; Stanic, B.; Yaman, G.; Wawrzyniak, M.; Söllner, S.; Akdis, D.G. IgG4 production is confined to human IL-10-producing regulatory B cells that suppress antigen-specific immune responses. J. Allergy Clin. Immunol. 2013, 131, 1204–1212. [Google Scholar] [CrossRef]
- Gu, X.L.; He, H.; Lin, L.; Luo, G.X.; Wen, Y.F.; Xiang, D.C.; Qiu, J. Tim-1(+) B cells suppress T cell interferon-gamma production and promote Foxp3 expression, but have impaired regulatory function in coronary artery disease. Apmis 2017, 125, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Pan, F.; Wu, Z.; Wang, Z.; Zhou, Y.; Zhang, P.; Gou, M.; Dai, G. CD19loCD27hi Plasmablasts Suppress Harmful Th17 Inflammation Through Interleukin 10 Pathway in Colorectal Cancer. DNA Cell Biol. 2017, 36, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Rybojad, M.; Hau, E.; Monfort, B.J.; Branchtein, M.; Michonneau, D.; Dessirier, V.; Socié, G. CD24(hi)CD27+ and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood 2015, 125, 1830–1839. [Google Scholar]
- Fehres, C.M.; Van Uden, N.O.; Yeremenko, N.G.; Fernandez, L.; Salinas, G.F.; Van Duivenvoorde, L.M.; Huard, B.; Morel, J.; Spits, H.; Hahne, M.; et al. APRIL Induces a Novel Subset of IgA+ Regulatory B Cells That Suppress Inflammation via Expression of IL-10 and PD-L1. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, Y.; Zhang, M.; Wang, H.; Jiang, Y.; Gao, P. Ulcerative Colitis Is Characterized by a Decrease in Regulatory B Cells. J. Crohn's Colitis 2016, 10, 1212–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rensburg, I.C.; Loxton, A.G. B-cells with a FasL expressing regulatory phenotype are induced following successful anti-tuberculosis treatment. Tuberculosis 2018, 108, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Figueiro, F.; Muller, L.; Funk, S.; Jackson, E.K.; Battastini, A.M.O.; Whiteside, T.L. Phenotypic and functional characteristics of CD39high human regulatory B cells (Breg). OncoImmunology 2016, 5, e1082703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouël, A.; Pochard, P.; Simon, Q.; Ségalen, I.; Le Meur, Y.; Pers, J.; Hillion, S. B-Cells induce regulatory T cells through TGF-β/IDO production in A CTLA-4 dependent manner. J. Autoimmun. 2015, 59, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Sabourin-Poirier, C.; Fourcade, L.; Chagnon-Choquet, J.; Labbé, A.C.; Alary, M.; Guédou, F.; Roger, M. Blood B Lymphocyte Stimulator (BLyS)/BAFF levels may reflect natural immunity to HIV in highly exposed uninfected Beninese Commercial Sex Workers. Sci. Rep. 2016, 6, 32318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safe, S.; Jin, U.-H.; Morpurgo, B.; Abudayyeh, A.; Singh, M.; Tjalkens, R.B. Nuclear receptor 4A (NR4A) family—orphans no more. J. Steroid Biochem. Mol. Biol. 2015, 157, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, T.; Hibino, S.; Saeki, K.; Kanamori, M.; Takaki, S.; Yoshimura, A. Nr4a Receptors Regulate Development and Death of Labile Treg Precursors to Prevent Generation of Path-ogenic Self-Reactive Cells. Cell Rep. 2018, 24, 1627–1638.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandukwala, H.S.; Rao, A. Nurrishing Treg cells: Nr4a transcription factors control Foxp3 expression. Nat. Immunol. 2013, 14, 201–203. [Google Scholar] [CrossRef]
- Ashouri, J.; Weiss, A. Endogenous Nur77 Is a Specific Indicator of Antigen Receptor Signaling in Human T and B Cells. J. Immunol. 2016, 198, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Crean, D.; Cummins, E.P.; Bahar, B.; Mohan, H.; McMorrow, J.P.; Murphy, E.P. Adenosine Modulates NR4A Orphan Nuclear Receptors To Attenuate Hyperinflammatory Responses in Monocytic Cells. J. Immunol. 2015, 195, 1436–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, P.K.-T.; Siu, M.K.-Y.; Leung, T.H.-Y.; Mo, X.-T.; Chan, K.K.-L.; Ngan, H.Y.-S. Role of Nurr1 in Carcinogenesis and Tumor Immunology: A State of the Art Review. Cancers 2020, 12, 3044. [Google Scholar] [CrossRef]
- Hibino, S.; Chikuma, S.; Kondo, T.; Ito, M.; Nakatsukasa, H.; Omata-Mise, S.; Yoshimura, A. Inhibition of Nr4a Receptors Enhances Antitumor Immunity by Breaking Treg-Mediated Immune Tolerance. Cancer Res. 2018, 78, 3027–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulet, S.; Daudelin, J.-F.; Odagiu, L.; Pelletier, A.-N.; Yun, T.J.; Lesage, S.; Cheong, C.; Labrecque, N. The orphan nuclear receptor NR4A3 controls the differentiation of monocyte-derived dendritic cells following microbial stimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 15150–15159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mildner, A.; Schönheit, J.; Giladi, A.; David, E.; Lara-Astiaso, D.; Vivas, E.L.; Paul, F.; Chappell-Maor, L.; Priller, J.; Leutz, A.; et al. Genomic Characterization of Murine Monocytes Reveals C/EBPβ Transcription Factor Dependence of Ly6C—Cells. Immunity 2017, 46, 849–862.e7. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C− monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Tel-Karthaus, N.; Kers-Rebel, E.D.; Looman, M.W.; Ichinose, H.; De Vries, C.J.; Ansems, M. Nuclear Receptor Nur77 Deficiency Alters Dendritic Cell Function. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Karki, K.; Wright, G.A.; Mohankumar, K.; Jin, U.-H.; Zhang, X.-H.; Safe, S. A Bis-Indole–Derived NR4A1 Antagonist Induces PD-L1 Degradation and Enhances Antitumor Immunity. Cancer Res. 2020, 80, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridi, M.S.; Hawk, J.D.; Chatterjee, S.; Safe, S.; Abel, T. Pharmacological Activators of the NR4A Nuclear Receptors Enhance LTP in a CREB/CBP-Dependent Manner. Neuropsychopharmacology 2016, 42, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duren, R.P.; Boudreaux, S.P.; Conneely, O.M. Genome Wide Mapping of NR4A Binding Reveals Cooperativity with ETS Factors to Promote Epigenetic Activation of Distal Enhancers in Acute Myeloid Leukemia Cells. PLoS ONE 2016, 11, e0150450. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Arp, J.; Lian, D.; Liu, W.; Baroja, M.L.; Jiang, J.; Ramcharran, S.; Eldeen, F.Z.; Zinser, E.; Steinkasserer, A.; et al. Immunosuppression Involving Soluble CD83 Induces Tolerogenic Dendritic Cells That Prevent Cardiac Allograft Rejection. Transplantation 2010, 90, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Lan, Z.; Ge, W.; Arp, J.; Jiang, J.; Liu, W.; Gordon, D.; Healey, D.; Debenedette, M.; Nicolette, C.; Garcia, B.; et al. Induction of Kidney Allograft Tolerance by Soluble CD83 Associated With Prevalence of Tolerogenic Dendritic Cells and Indoleamine 2,3-Dioxygenase. Transplantation 2010, 90, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liang, S.; Zhong, Z.; Wen, J.; Li, W.; Wang, L.; Xu, J.; Zhong, F.; Li, X. Soluble CD83 inhibits human monocyte differentiation into dendritic cells in vitro. Cell. Immunol. 2014, 292, 25–31. [Google Scholar] [CrossRef]
- Bates, J.M.; Flanagan, K.; Mo, L.; Ota, N.; Ding, J.; Ho, S.; Liu, S.; Roose-Girma, M.; Warming, S.; Diehl, L. Dendritic cell CD83 homotypic interactions regulate inflammation and promote mucosal homeostasis. Mucosal Immunol. 2014, 8, 414–428. [Google Scholar] [CrossRef]
- Li, Z.; Ju, X.; Silveira, P.A.; Abadir, E.; Hsu, W.-H.; Hart, D.N.J.; Clark, G. CD83: Activation Marker for Antigen Presenting Cells and Its Therapeutic Potential. Front. Immunol. 2019, 10, 1312. [Google Scholar] [CrossRef] [Green Version]
- Wild, A.B.; Krzyzak, L.; Peckert, K.; Stich, L.; Kuhnt, C.; Butterhof, A.; Seitz, C.; Mattner, J.; Grüner, N.; Gänsbauer, M.; et al. CD83 orchestrates immunity toward self and non-self in dendritic cells. JCI Insight 2019, 4, e126246. [Google Scholar] [CrossRef] [Green Version]
- Sénéchal, B.; Boruchov, A.M.; Reagan, J.L.; Hart, D.N.J.; Young, J.W. Infection of mature monocyte-derived dendritic cells with human cytomegalovirus inhibits stimulation of T-cell proliferation via the release of soluble CD83. Blood 2004, 103, 4207–4215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, F.; Rössner, S.; Onderka, J.; Lechmann, M.; Pallotta, M.T.; Fallarino, F.; Boon, L.; Nicolette, C.; Debenedette, M.A.; Tcherepanova, I.Y.; et al. Topical Application of Soluble CD83 Induces IDO-Mediated Immune Modulation, Increases Foxp3+ T Cells, and Prolongs Allogeneic Corneal Graft Survival. J. Immunol. 2013, 191, 1965–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebbeler, M.; Koenig, C.; Krzyzak, L.; Seitz, C.; Wild, A.; Ulas, T.; Baßler, K.; Kopelyanskiy, D.; Butterhof, A.; Kuhnt, C.; et al. CD83 expression is essential for Treg cell differentiation and stability. JCI Insight 2018, 3, e99712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saze, Z.; Schuler, P.J.; Hong, C.-S.; Cheng, D.; Jackson, E.K.; Whiteside, T.L. Adenosine production by human B cells and B cell–mediated suppression of activated T cells. Blood 2013, 122, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minguet, S.; Huber, M.; Rosenkranz, L.; Schamel, W.W.; Reth, M.; Brummer, T. Adenosine and cAMP are potent inhibitors of the NF-kappa B pathway downstream of immunoreceptors. Eur. J. Immunol. 2005, 35, 31–41. [Google Scholar] [CrossRef]
- Dwyer, K.M.; Deaglio, S.; Gao, W.; Friedman, D.; Strom, T.B.; Robson, S.C. CD39 and control of cellular immune responses. Purinergic Signal. 2007, 3, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Rheumatol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Vincent, F.; Morand, E.; Schneider, P.; Mackay, F. The BAFF/APRIL system in SLE pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, L.; Opalka, N.; Kappler, J.; Shu, H.-B.; Zhang, G. Crystal Structure of sTALL-1 Reveals a Virus-like Assembly of TNF Family Ligands. Cell 2002, 108, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Cachero, T.G.; Schwartz, I.M.; Qian, F.; Day, E.S.; Bossen, C.; Ingold, K.; Tardivel, A.; Krushinskie, D.; Eldredge, J.; Silvian, L.; et al. Formation of Virus-like Clusters Is an Intrinsic Property of the Tumor Necrosis Factor Family Member BAFF (B Cell Activating Factor). Biochemistry 2006, 45, 2006–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossen, C.; Cachero, T.G.; Tardivel, A.; Ingold, K.; Willen, L.; Dobles, M.; Scott, M.L.; Maquelin, A.; Belnoue, E.; Siegrist, C.-A.; et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 2008, 111, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Dillon, S.R.; Gross, J.A.; Ansell, S.M.; Novak, A.J. An APRIL to remember: Novel TNF ligands as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Roschke, V.; Sosnovtseva, S.; Ward, C.D.; Hong, J.S.; Smith, R.; Albert, V.; Stohl, W.; Baker, K.P.; Ullrich, S.; Nardelli, B.; et al. BLyS and APRIL Form Biologically Active Heterotrimers That Are Expressed in Patients with Systemic Immune-Based Rheumatic Diseases. J. Immunol. 2002, 169, 4314–4321. [Google Scholar] [CrossRef] [Green Version]
- Woodland, R.T.; Fox, C.J.; Schmidt, M.R.; Hammerman, P.S.; Opferman, J.T.; Korsmeyer, S.J.; Hilbert, D.M.; Thompson, C.B. Multiple signaling pathways promote B lymphocyte stimulator–dependent B-cell growth and survival. Blood 2008, 111, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Mambetsariev, N.; Lin, W.W.; Stunz, L.L.; Hanson, B.M.; Hildebrand, J.M.; Bishop, G.A. Nuclear TRAF3 is a negative regulator of CREB in B cells. Proc. Natl. Acad. Sci. USA 2016, 113, 1032–1037. [Google Scholar] [CrossRef] [Green Version]
- Moir, S.; Fauci, A.S. B cells in HIV infection and disease. Nat. Rev. Immunol. 2009, 9, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Stohl, W.; Cheema, G.S.; Briggs, W.S.; Xua, D.; Sosnovtsevac, S.; Roschkec, V.; Ferrara, D.E.; Labatd, K.; Sattler, F.R.; Pierangeli, S.S.; et al. B Lymphocyte Stimulator Protein-Associated Increase in Circulating Autoantibody Levels May Require CD4+ T Cells: Lessons from HIV-Infected Patients. Clin. Immunol. 2002, 104, 115–122. [Google Scholar] [CrossRef]
- Chagnon-Choquet, J.; Gauvin, J.; Roger, J.; Fontaine, J.; Poudrier, J.; Vassal, A.; Legault, M.; Routy, J.P.; Tremblay, C.; Thomas, R.; et al. HIV Nef Promotes Expression of B-Lymphocyte Stimulator by Blood Dendritic Cells During HIV Infection in Humans. J. Infect. Dis. 2014, 211, 1229–1240. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, B.; Valdez, H.; Freimuth, W.; Butler, T.; Asaad, R.; Lederman, M.M. Plasma levels of B-lymphocyte stimulator increase with HIV disease progression. AIDS 2003, 17, 1983–1985. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, L.; Sabourin-Poirier, C.; Perraud, V.; Faucher, M.-C.; Chagnon-Choquet, J.; Labbé, A.-C.; Alary, M.; Guédou, F.; Poudrier, J.; Roger, M. Natural Immunity to HIV is associated with Low BLyS/BAFF levels and low frequencies of innate marginal zone like CD1c+ B-cells in the genital tract. PLoS Pathog. 2019, 15, e1007840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poudrier, J.; Soulas, C.; Chagnon-Choquet, J.; Burdo, T.; Autissier, P.; Oskar, K.; Williams, K.C.; Roger, M. High Expression Levels of BLyS/BAFF by Blood Dendritic Cells and Granulocytes Are Associated with B-cell dysregulation in SIV-Infected Rhesus Macaques. PLoS ONE 2015, 10, e0131513. [Google Scholar] [CrossRef] [PubMed]
- Hannah, Z.; Kay, D.; Poudrier, J.; Jolicoeur, P. Division of Experimental Medicine; McGill University: Montreal, QC, Canada, 2022; Unpublished work. [Google Scholar]
- Gomez, A.M.; Ouellet, M.; Tremblay, M.J. HIV-1–Triggered Release of Type I IFN by Plasmacytoid Dendritic Cells Induces BAFF Production in Monocytes. J. Immunol. 2015, 194, 2300–2308. [Google Scholar] [CrossRef] [Green Version]
- Chu, V.T.; Enghard, P.; Riemekasten, G.; Berek, C. In Vitro and In Vivo Activation Induces BAFF and APRIL Expression in B Cells. J. Immunol. 2007, 179, 5947–5957. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Liu, Y.; Han, J.; Yang, Z.; Sheng, W.; Dai, H.; Wang, Y.; Xia, T.; Hou, M. TLR7 regulates dendritic cell-dependent B-cell responses through BlyS in immune thrombocytopenic purpura. Eur. J. Haematol. 2010, 86, 67–74. [Google Scholar] [CrossRef]
- Lane, H.C.; Masur, H.; Edgar, L.C.; Whalen, G.; Rook, A.H.; Fauci, A.S. Abnormalities of B-Cell Activation and Immunoregulation in Patients with the Acquired Immunodeficiency Syndrome. N. Engl. J. Med. 1983, 309, 453–458. [Google Scholar] [CrossRef]
- Mandl, J.N. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and non-pathogenic AIDS virus infections. Nat. Med. 2008, 14, 1077–1087. [Google Scholar] [CrossRef]
- Sandler, N.G.; Douek, D.C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Rev. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef]
- Jiang, W. Microbial Translocation and B Cell Dysfunction in Human Immunodeficiency Virus Disease. Am. J. Immunol. 2012, 8, 44–51. [Google Scholar]
- Pers, J.-O.; Daridon, C.; Devauchelle, V.; Jousse, S.; Saraux, A.; Jamin, C.; Youinou, P. BAFF Overexpression Is Associated with Autoantibody Production in Autoimmune Diseases. Ann. N. Y. Acad. Sci. 2005, 1050, 34–39. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Mörch, C.; Sönnerborg, A.; Chiodi, F. Loss of memory (CD27) B lymphocytes in HIV-1 infection. AIDS 2001, 15, 957–964. [Google Scholar] [CrossRef]
- Nagase, H.; Agematsu, K.; Kitano, K.; Takamoto, M.; Okubo, Y.; Komiyama, A.; Sugane, K. Mechanism of Hypergammaglobulinemia by HIV Infection: Circulating Memory B-Cell Reduction with Plasmacytosis. Clin. Immunol. 2001, 100, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Titanji, K.; Chiodi, F.; Bellocco, R.; Schepis, D.; Osorio, L.; Tassandin, C.; Tambussi, G.; Grutzmeier, S.; Lopalco, L.; De Milito, A. Primary HIV-1 infection sets the stage for important B lymphocyte dysfunctions. AIDS 2005, 19, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Moir, S.; Malaspina, A.; Pickeral, O.K.; Donoghue, E.T.; Vasquez, J.; Miller, N.J.; Krishnan, S.R.; Planta, M.A.; Turney, J.F.; Justement, J.S.; et al. Decreased Survival of B Cells of HIV-viremic Patients Mediated by Altered Expression of Receptors of the TNF Superfamily. J. Exp. Med. 2004, 200, 587–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darce, J.R.; Arendt, B.K.; Wu, X.; Jelinek, D.F. Regulated Expression of BAFF-Binding Receptors during Human B Cell Differentiation. J. Immunol. 2007, 179, 7276–7286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Grevenynghe, J.; Cubas, R.A.; Noto, A.; DaFonseca, S.; He, Z.; Peretz, Y.; Haddad, E.K. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J. Clin. Investig. 2011, 121, 3877–3888. [Google Scholar] [CrossRef] [PubMed]
- Amu, S.; Ruffin, N.; Rethi, B.; Chiodi, F. Impairment of B-cell functions during HIV-1 infection. AIDS 2013, 27, 2323–2334. [Google Scholar] [CrossRef]
- Cagigi, A.; Nilsson, A.; Pensieroso, S.; Chiodi, F. Dysfunctional B-cell responses during HIV-1 infection: Implication for influenza vaccination and highly active antiretroviral therapy. Lancet Infect. Dis. 2010, 10, 499–503. [Google Scholar] [CrossRef]
- Poudrier, J.; Weng, X.; Kay, D.G.; Paré, G.; Calvo, E.L.; Hanna, Z.; Kosco-Vilbois, M.H.; Jolicoeur, P. The AIDS Disease of CD4C/HIV Transgenic Mice Shows Impaired Germinal Centers and Autoantibodies and Develops in the Absence of IFN-γ and IL-6. Immunity 2001, 15, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Hanna, Z.; Kay, D.G.; Cool, M.; Jothy, S.; Rebai, N.; Jolicoeur, P. Transgenic Mice Expressing Human Immunodeficiency Virus Type 1 in Immune Cells Develop a Severe AIDS-Like Disease. J. Virol. 1998, 72, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Hanna, Z.; Kay, D.G.; Rebai, N.; Guimond, A.; Jothy, S.; Jolicoeur, P. Nef Harbors a Major Determinant of Pathogenicity for an AIDS-like Disease Induced by HIV-1 in Transgenic Mice. Cell 1998, 95, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J. Mice Transgenic for Baff Develop Lymphocytic Disorders along with Autoimmune Manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Levesque, M.C.; Moody, M.A.; Hwang, K.-K.; Marshall, D.J.; Whitesides, J.F.; Amos, J.D.; Gurley, T.C.; Allgood, S.; Haynes, B.B.; Vandergrift, N.A.; et al. Polyclonal B Cell Differentiation and Loss of Gastrointestinal Tract Germinal Centers in the Earliest Stages of HIV-1 Infection. PLoS Med. 2009, 6, e1000107. [Google Scholar] [CrossRef]
- Vanham, G.; Penne, L.; Devalck, J.; Kestens, L.; Colebunders, R.; Bosmans, E.; Thielemans, K.; Ceuppens, J.L. Decreased CD40 ligand induction in CD4 T cells and dysregulated IL-12 production during HIV infection. Clin. Exp. Immunol. 1999, 117, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Paiva, D.; Morais, J.; Pilotto, J.; Veloso, V.; Duarte, F.; Lenzi, H. Spectrum of Morphologic Changes of Lymph Nodes in HIV Infection. Mem. Inst. Oswaldo Cruz, 1996; 91, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Tenner-Rácz, K.; Rácz, P.; Gartner, S.; Ramsauer, J.; Dietrich, M.; Gluckman, J.C.; Popovic, M. Ultrastructural analysis of germinal centers in lymph nodes of patients with HIV-1-induced persistent generalized lymphadenopathy: Evidence for persistence of infection. Prog. AIDS Pathol. 1989, 1, 29–30. [Google Scholar] [PubMed]
- Gauvin, J.; Chagnon-Choquet, J.; Poudrier, J.; Roger, M.; Cohorts, M.P.H.I.A.S.P. Fluctuations in Blood Marginal Zone B-Cell Frequencies May Reflect Migratory Patterns Associated with HIV-1 Disease Progression Status. PLoS ONE 2016, 11, e0155868. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, E.; Struyf, S.; Van Damme, J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003, 14, 409–426. [Google Scholar] [CrossRef]
- Svensson, M. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J. Clin. Invest 2002, 110, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Zouali, M.; Richard, Y. Marginal Zone B-Cells, a Gatekeeper of Innate Immunity. Front. Immunol. 2011, 2, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sintes, J.; Gentile, M.; Zhang, S.; Garcia-Carmona, Y.; Magri, G.; Cassis, L.; Segura-Garzón, D.; Ciociola, A.; Grasset, E.K.; Bascones, S.; et al. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nat. Commun. 2017, 8, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrick, B.M.; Yao, X.-D.; Zahoor, M.; Abimiku, A.; Osawe, S.; Rosenthal, K.L. TLR10 Senses HIV-1 Proteins and Significantly Enhances HIV-1 Infection. Front. Immunol. 2019, 10, 482. [Google Scholar] [CrossRef] [PubMed]
- Moir, S.; Malaspina, A.; Li, Y.; Chun, T.-W.; Lowe, T.; Adelsberger, J.; Baseler, M.; Ehler, L.A.; Liu, S.Jr.; et al. B Cells of HIV-1–Infected Patients Bind Virions through Cd21–Complement Interactions and Transmit Infectious Virus to Activated T Cells. J. Exp. Med. 2000, 192, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X. Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in by-stander B cells. Nat. Immunol. 2006, 7, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Majumder, B. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-cell activation: Im-plications for viral immune escape. J. Virol. 2005, 79, 7990–8003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthumani, K.; Hwang, D.S.; Choo, A.Y.; Mayilvahanan, S.; Dayes, N.S.; Thieu, K.P.; Weiner, D.B. HIV-1 Vpr inhibits the maturation and activation of macrophages and dendritic cells in vitro. Int. Immunol. 2004, 17, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Jenks, S.A.; Cashman, K.S.; Woodruff, M.C.; Lee, F.E.; Sanz, I. Extrafollicular responses in humans andSLE. Immunol. Rev. 2019, 288, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; O’Neill, P.; Naradikian, M.S.; Scholz, J.L.; Cancro, M.P. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 2011, 118, 1294–1304. [Google Scholar] [CrossRef] [Green Version]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhou, S.; Qian, J.; Wang, Y.; Yu, X.; Dai, D.; Dai, M.; Wu, L.; Liao, Z.; Xue, Z.; et al. T-bet+CD11c+ B cells are critical for antichromatin immunoglobulin G production in the development of lupus. Arthritis Res. Ther. 2017, 19, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat. Commun. 2018, 9, 1758. [Google Scholar] [CrossRef] [PubMed]
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L.; Hussain, A. The Prevalence of Autoimmune Disorders in Women: A Narrative Review. Cureus 2020, 12, e8094. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Rui, K.; Wang, S.; Tian, J. Advances of Regulatory B Cells in Autoimmune Diseases. Front. Immunol. 2021, 12, 1131. [Google Scholar] [CrossRef] [PubMed]
- Möckel, T.; Basta, F.; Weinmann-Menke, J.; Schwarting, A. B cell activating factor (BAFF): Structure, functions, autoimmunity and clinical implications in Systemic Lupus Erythematosus (SLE). Autoimmun. Rev. 2020, 20, 102736. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. Immune dysfunction, inflammation, and accelerated aging in patients on antiretroviral therapy. Top. HIV Med. Publ. Int. AIDS Soc. USA 2009, 17, 118–123. [Google Scholar]
- Deeks, S.G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Kearns, A.; Gordon, J.; Burdo, T.H.; Qin, X. HIV-1-Associated Atherosclerosis: Unraveling the Missing Link. J. Am. Coll. Cardiol. 2017, 69, 3084–3098. [Google Scholar] [CrossRef]
- Triant, V.A. Cardiovascular Disease and HIV Infection. Curr. HIV/AIDS Rep. 2013, 10, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Sage, A.P.; Göderle, L.; Ozsvar-Kozma, M.; Murphy, D.; Porsch, F.; Binder, C.J. B Cell-Activating Factor Neutralization Aggravates Atherosclerosis. Circulation 2018, 138, 2263–2273. [Google Scholar] [CrossRef]
- Jackson, S.W.; Scharping, N.; Jacobs, H.M.; Wang, S.; Chait, A.; Rawlings, D.J. Cutting Edge: BAFF Overexpression Reduces Atherosclerosis via TACI-Dependent B Cell Activation. J. Immunol. 2016, 197, 4529–4534. [Google Scholar] [CrossRef] [Green Version]
- Nus, M.; Basatemur, G.; Galan, M.; Cros-Brunsó, L.; Zhao, T.X.; Masters, L.; Harrison, J.; Figg, N.; Tsiantoulas, D.; Geissmann, F.; et al. NR4A1 Deletion in Marginal Zone B Cells Exacerbates Atherosclerosis in Mice—Brief Report. Arter. Thromb. Vasc. Biol. 2020, 40, 2598–2604. [Google Scholar] [CrossRef] [PubMed]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Tay, C.; Liu, Y.-H.; Kanellakis, P.; Kallies, A.; Li, Y.; Cao, A.; Hosseini, H.; Tipping, P.; Toh, B.-H.; Bobik, A.; et al. Follicular B Cells Promote Atherosclerosis via T Cell–Mediated Differentiation Into Plasma Cells and Secreting Pathogenic Immunoglobulin G. Arter. Thromb. Vasc. Biol. 2018, 38, e71–e84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, E.; Nezos, A.; Antypa, E.; Ioakeimidis, D.; Koutsilieris, M.; Tektonidou, M.; Moutsopoulos, H.M.; Mavragani, C.P. B-cell activating factor and related genetic variants in lupus related atherosclerosis. J. Autoimmun. 2018, 92, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.C.; Sanguankeo, A.; Upala, S. Association between primary Sjogren’s syndrome, arterial stiffness, and subclinical atherosclerosis: A systematic review and meta-analysis. Clin. Rheumatol. 2018, 38, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Choi, B.-H.; Cheon, H.-G.; Do, M.-S. B cell activation factor (BAFF) is a novel adipokine that links obesity and inflammation. Exp. Mol. Med. 2009, 41, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Do, M.-S. BAFF knockout improves systemic inflammation via regulating adipose tissue distribution in high-fat diet-induced obesity. Exp. Mol. Med. 2015, 47, e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, N.; Kuo, H.-H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.e13. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020, 21, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Karbuz, A.; Gervais, A.; Tayoun, A.A.; Aiuti, A.; Belot, A.; Bolze, A.; Gaudet, A.; Bondarenko, A.; et al. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schultheiß, C.; Paschold, L.; Simnica, D.; Mohme, M.; Willscher, E.; von Wenserski, L.; Scholz, R.; Wieters, I.; Dahlke, C.; Tolosa, E.; et al. Next-Generation Sequencing of T and B Cell Receptor Repertoires from COVID-19 Patients Showed Signatures Associated with Severity of Disease. Immunity 2020, 53, 442–455.e4. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Oh, J.E.; Lee, H.K. Single Cell Transcriptomic Re-analysis of Immune Cells in Bronchoalveolar Lavage Fluids Reveals the Correlation of B Cell Characteristics and Disease Severity of Patients with SARS-CoV-2 Infection. Immune Netw. 2021, 21, e10. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; Joly, F.; Vazquez, T.; Benech, P.; Rosenzwajg, M.; Carpentier, W.; Saadoun, D. Expansion of functionally anergic CD21-/low marginal zone-like B cell clones in hepatitis C virus infec-tion-related autoimmunity. J. Immunol. 2011, 187, 6550–6563. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Bertoni, F.; Zucca, E. Marginal-Zone Lymphomas. N. Engl. J. Med. 2022, 386, 568–581. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Trifonov, V.; Fangazio, M.; Bruscaggin, A.; Rasi, S.; Spina, V.; Gaidano, G. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regu-lating marginal zone development. J. Exp. Med. 2012, 209, 1537–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Deaglio, S.; Dominguez-Sola, D.; Rasi, S.; Vaisitti, T.; Agostinelli, C.; Spina, V.; Bruscaggin, A.; Monti, S.; Cerri, M.; et al. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood 2011, 118, 4930–4934. [Google Scholar] [CrossRef]
- Sindel, A.; Al-Juhaishi, T.; Yazbeck, V. Marginal Zone Lymphoma: State-of-the-Art Treatment. Curr. Treat. Options Oncol. 2019, 20, 90. [Google Scholar] [CrossRef]
- Cerhan, J.R.; Habermann, T.M. Epidemiology of marginal zone lymphoma. Ann. Lymphoma 2021, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.M.; Morton, L.M.; Shiels, M.S.; Clarke, C.A.; Engels, E.A. Risk of non-Hodgkin lymphoma subtypes in HIV-infected people during the HAART era: A popula-tion-based study. Aids 2014, 28, 2313–2318. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.A.; Gadalla, S.; Morton, L.M.; Landgren, O.; Pfeiffer, R.; Warren, J.L.; Berndt, S.I.; Ricker, W.; Parsons, R.; Engels, E.A. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int. J. Cancer 2009, 125, 398–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, F.; Lortholary, O.; Hermine, O. Infection-associated lymphomas derived from marginal zone B cells: A model of antigen-driven lymphopro-liferation. Blood 2006, 107, 3034–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roggero, E.; Zucca, E.; Mainetti, C.; Bertoni, F.; Valsangiacomo, C.; Pedrinis, E.; Borisch, B.; Piffaretti, J.-C.; Cavalli, F.; Isaacson, P.G. Eradication of Borrelia burgdorferi infection in primary marginal zone B-cell lymphoma of the skin. Hum. Pathol. 2000, 31, 263–268. [Google Scholar] [CrossRef]
- Arcaini, L.; Zibellini, S.; Passamonti, F.; Rattotti, S.; Lucioni, M.; Invernizzi, R.; Merli, M.; Rizzi, S.; Boveri, E.; Rumi, E.; et al. Splenic marginal zone lymphoma: Clinical clustering of immunoglobulin heavy chain repertoires. Blood Cells Mol. Dis. 2009, 42, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.K.; Sahota, S.S.; Ottensmeier, C.H.; Zhu, D.; Forconi, F.; Hamblin, T.J. The occurrence and significance of V gene mutations in B cell—Derived human malignancy. Adv. Cancer Res. 2001, 83, 81–116. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Marusawa, H.; Endo, Y.; Chiba, T. Inflammation-mediated genomic instability: Roles of activation-induced cytidine deaminase in carcinogen-esis. Cancer Sci. 2012, 103, 1201–1206. [Google Scholar] [CrossRef]
- Deutsch, A.J.; Rinner, B.; Wenzl, K.; Pichler, M.; Troppan, K.; Steinbauer, E.; Neumeister, P. NR4A1-mediated apoptosis suppresses lymphomagenesis and is associated with a favorable can-cer-specific survival in patients with aggressive B-cell lymphomas. Blood 2014, 123, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Taddesse-Heath, L.; Pittaluga, S.; Sorbara, L.; Bussey, M.; Raffeld, M.; Jaffe, E.S. Marginal Zone B-Cell Lymphoma in Children and Young Adults. Am. J. Surg. Pathol. 2003, 27, 522–531. [Google Scholar] [CrossRef]
- Bertoni, F.; Rossi, D.; Zucca, E. Recent advances in understanding the biology of marginal zone lymphoma. F1000Research 2018, 7, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanujam, M.; Davidson, A. BAFF blockade for systemic lupus erythematosus: Will the promise be fulfilled? Immunol. Rev. 2008, 223, 156–174. [Google Scholar] [CrossRef]
- Boudreaux, S.P.; Duren, R.P.; Call, S.G.; Nguyen, L.; Freire, P.R.; Narayanan, P.; Redell, M.S.; Conneely, O.M. Drug targeting of NR4A nuclear receptors for treatment of acute myeloid leukemia. Leukemia 2018, 33, 52–63. [Google Scholar] [CrossRef] [PubMed]
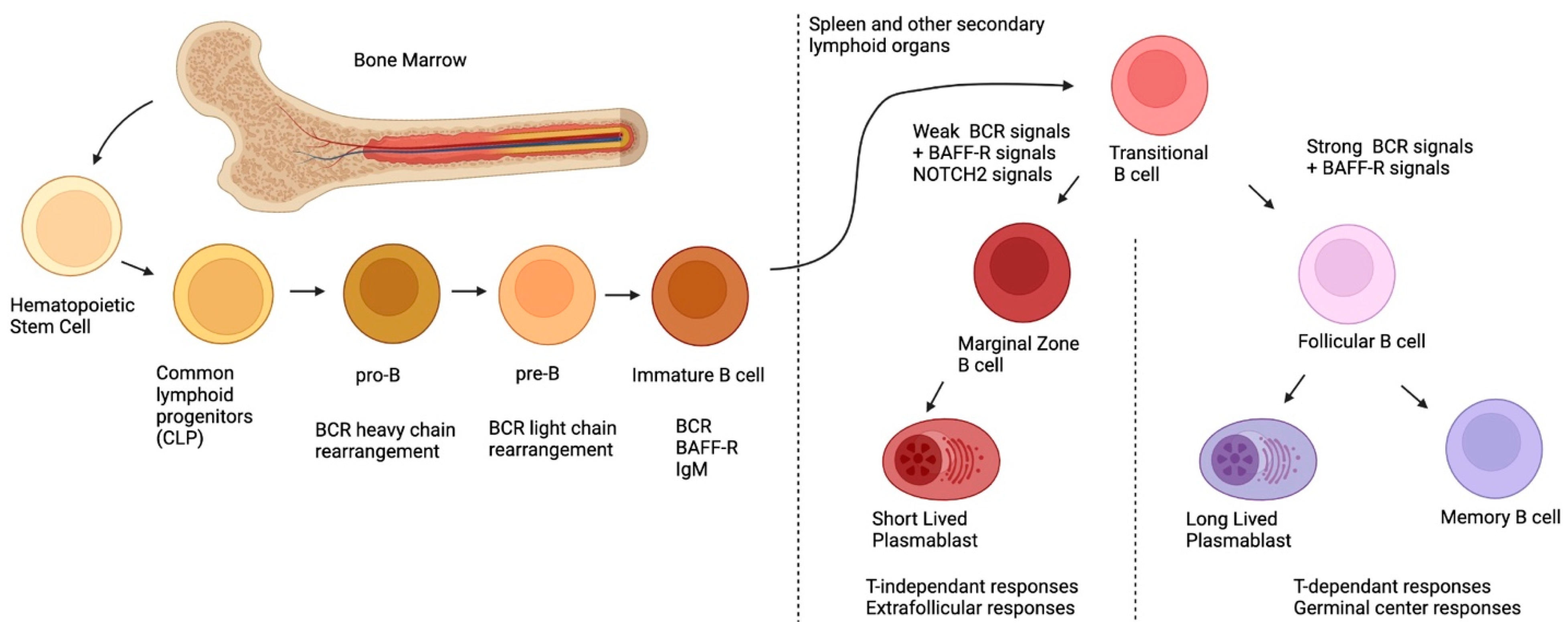

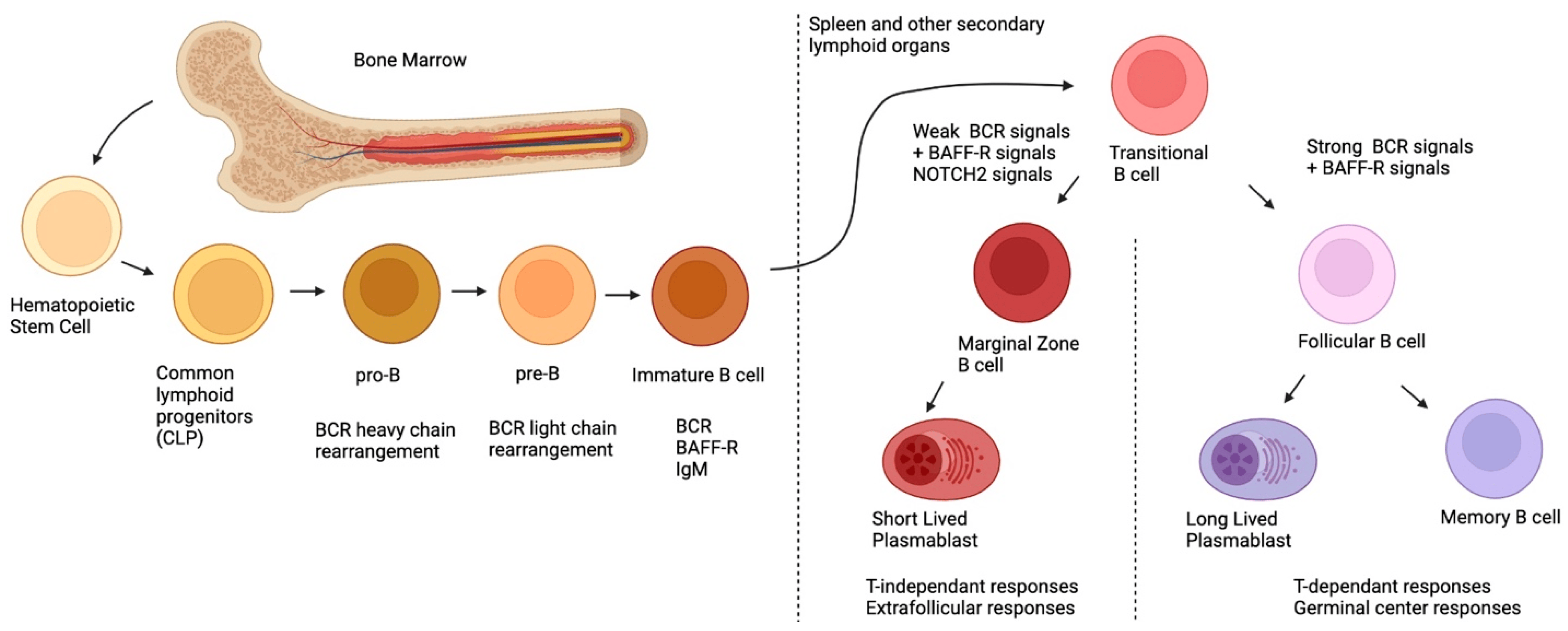{kind=link}
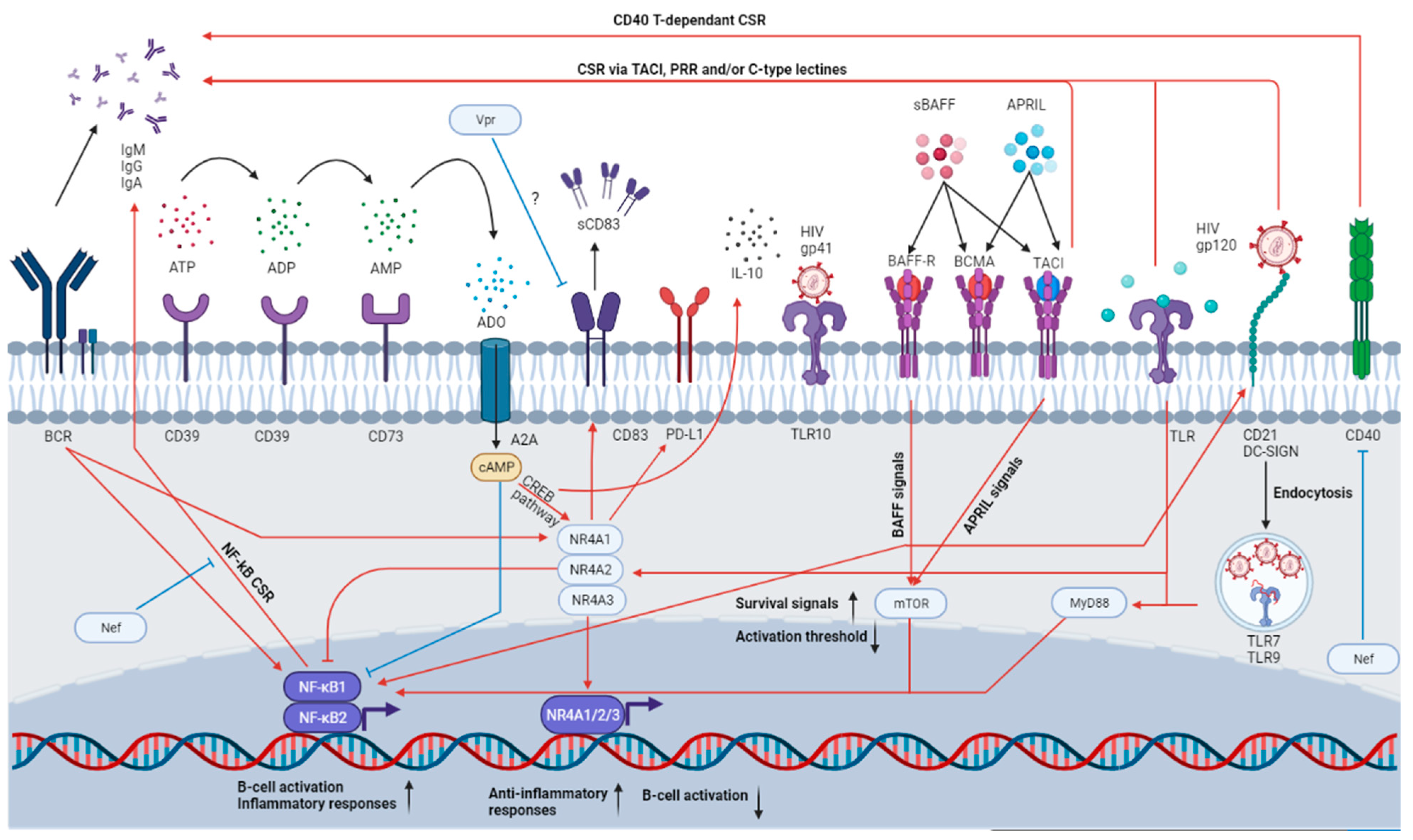{kind=link}
| Species | Population | Phenotype | Mechanism of Suppression | References |
|---|---|---|---|---|
| Mouse | B10 | CD19 + CD5 + CD1dhi | IL-10 | [52,53] |
| MZ B-cells | IgMhi IgDlo CD21hi CD23-CD1dhi | IL-10 | [54] | |
| T2-MZP | B220 + CD21hi CD1dhi IgMhi CD23+ | IL-10 | [55] | |
| B1a | CD90-CD5+ | IL-10 | [56] | |
| Plasma cells | CD19 + CD138 + IgM+ | IL10, IL-35 | [57] | |
| Plasmablasts | CD138 + CD44hi | IL-10 | [58] | |
| Tim-1 + B-cells | CD19 + Tim-1+ | IL-10 | [59] | |
| IL-35-Bregs | CD5 + CD1dhi FcγIibhi | IL-35 | [60] | |
| GITRL + B-cells | - | GITRL | [61] | |
| Killer B-cells | CD19 + CD5 + FasL+ | FasL, TGF-β | [62,63] | |
| PD-L1hi B-cells | CD19 + PD-L1hi | PD-L1 | [64] | |
| - | B220 + CD39 + CD73+ | ADO, CD39 + CD73 + Extracellular vesicules | [65,66] | |
| GIFT-15 B-cells | B220 + CD21 + CD22 + CD23 + CD24 + CD1d + CD138 + IgM + IgD+ | IL-10 | [67] | |
| Human | MZp | CD19 + CD1c + CD21lo IgMhi CD27 + CD10+ | CD83, PD-L1, IL-10 | [4,6] |
| Transitional B-cells | CD19 + CD24hi CD38hi | IL-10 | [68] | |
| Memory B-cells | CD19 + CD24hi CD27+ | IL-10 | [69] | |
| Br1 | CD25hi CD71hi CD73lo | IL-10 | [70] | |
| TIM1 + B-cells | CD19 + TIM1+ | IL-10 | [71] | |
| Plasmablast | CD19lo CD27hi CD38hi | IL-10 | [72,73] | |
| IgA + B-cells | CD19 + IgA+ | IL-10, PD-L1 | [74] | |
| Exhausted B-cells | CD19 + CD95+ | CD95 | [75] | |
| Killer B-cells | CD19 + CD38 + IgM + FasL+ | FasL | [76] | |
| PD-L1 B-cells | CD19 + PD-L1+ | PD-L1 | [63] | |
| CD39high | CD19 + CD39highCD73+ | ADO | [77] | |
| iBreg | - | TGF-β, IDO | [78] |
| mRNA Expression | Confirmed Protein Expression |
|---|---|
| NR4A1, NR4A2, NR4A3, CD83 CD39, CD73, TGF-β, IL-10, PD-L1, IL-10R, IL-27β, IL-12 p35, HLA-G | NR4A1, NR4A3, CD83, CD39, CD73, PD-L1, IL-10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doyon-Laliberté, K.; Aranguren, M.; Poudrier, J.; Roger, M. Marginal Zone B-Cell Populations and Their Regulatory Potential in the Context of HIV and Other Chronic Inflammatory Conditions. Int. J. Mol. Sci. 2022, 23, 3372. https://doi.org/10.3390/ijms23063372
Doyon-Laliberté K, Aranguren M, Poudrier J, Roger M. Marginal Zone B-Cell Populations and Their Regulatory Potential in the Context of HIV and Other Chronic Inflammatory Conditions. International Journal of Molecular Sciences. 2022; 23(6):3372. https://doi.org/10.3390/ijms23063372
Chicago/Turabian StyleDoyon-Laliberté, Kim, Matheus Aranguren, Johanne Poudrier, and Michel Roger. 2022. "Marginal Zone B-Cell Populations and Their Regulatory Potential in the Context of HIV and Other Chronic Inflammatory Conditions" International Journal of Molecular Sciences 23, no. 6: 3372. https://doi.org/10.3390/ijms23063372
APA StyleDoyon-Laliberté, K., Aranguren, M., Poudrier, J., & Roger, M. (2022). Marginal Zone B-Cell Populations and Their Regulatory Potential in the Context of HIV and Other Chronic Inflammatory Conditions. International Journal of Molecular Sciences, 23(6), 3372. https://doi.org/10.3390/ijms23063372