
B Cells in Primary Membranous Nephropathy: Escape from Immune Tolerance and Implications for Patient Management



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Reconceptualizing Membranous Nephropathy as a B Cell Disorder
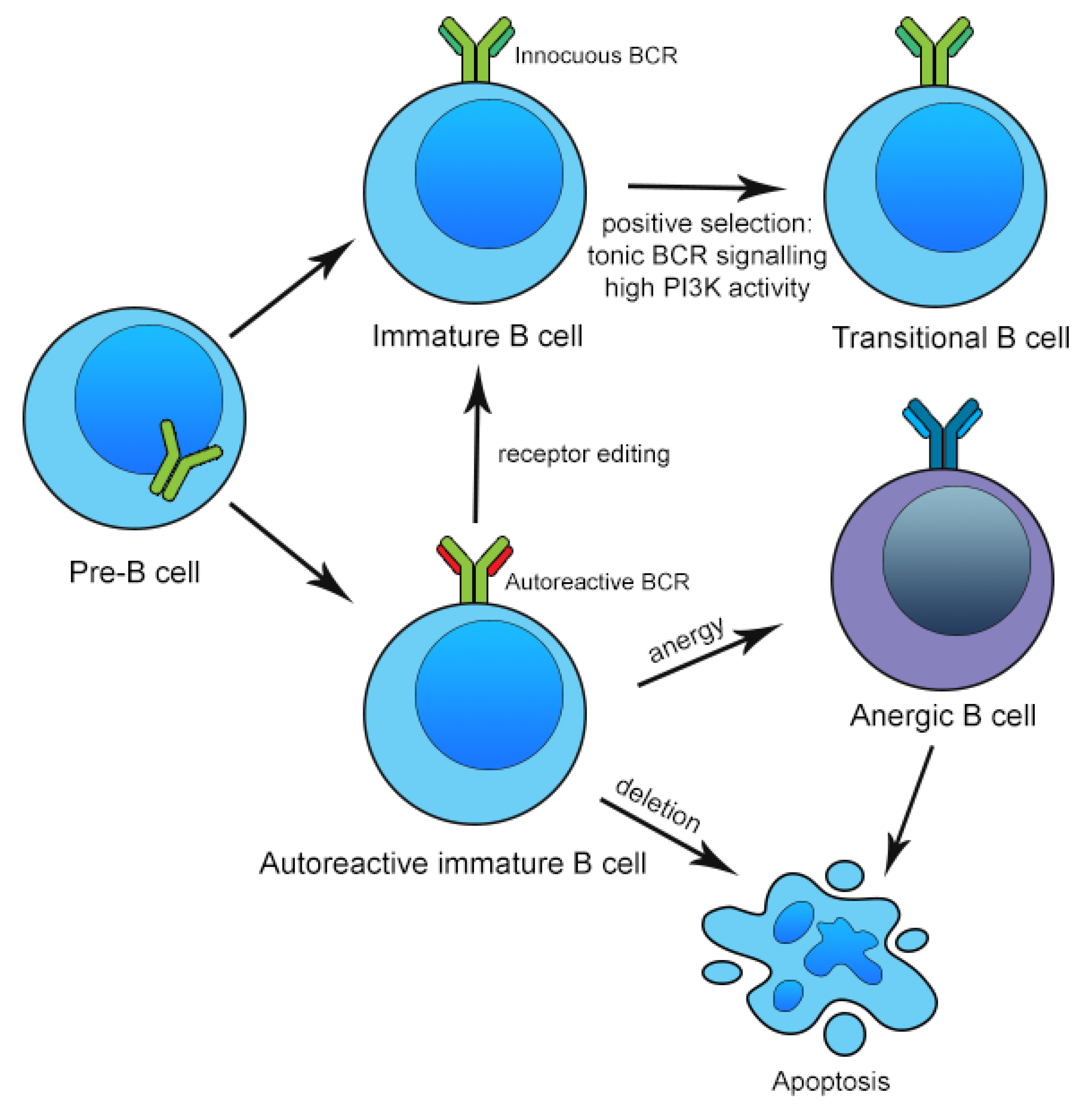
3. Mechanisms of Central and Peripheral B Cell Tolerance
4. Perturbations in Circulating B Cell Repertoire, Tolerance and Regulation in Membranous Nephropathy
5. Infiltrating B Cell Subsets and Intrarenal Tertiary Lymphoid Structures in Membranous Nephropathy
6. B Cells and the Treatment of Membranous Nephropathy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- McGrogan, A.; Franssen, C.F.; de Vries, C.S. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef]
- Tang, L.; Yao, J.; Kong, X.; Sun, Q.; Wang, Z.; Zhang, Y.; Wang, P.; Liu, Y.; Li, W.; Cui, M.; et al. Increasing prevalence of membranous nephropathy in patients with primary glomerular diseases: A cross-sectional study in China. Nephrology 2017, 22, 168–173. [Google Scholar] [CrossRef]
- Hu, R.; Quan, S.; Wang, Y.; Zhou, Y.; Zhang, Y.; Liu, L.; Zhou, X.J.; Xing, G. Spectrum of biopsy proven renal diseases in Central China: A 10-year retrospective study based on 34,630 cases. BMC Nephrol. 2020, 10, 10994. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Wood, C.; Wagner, I.; Agar, J.; Dowling, J.; Thomson, N.; Atkins, R. Idiopathic membranous nephropathy in an Australian population: The incidence of thromboembolism and its impact on the natural history. Nephron 1993, 63, 240–241. [Google Scholar] [CrossRef]
- Braun, N.; Schweisfurth, A.; Lohöfener, C.; Lange, C.; Gründemann, C.; Kundt, G.; Gröne, H.J. Epidemiology of glomerulonephritis in Northern Germany. Int. Urol. Nephrol. 2011, 43, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Luodelete, M.; Dong, C.; Li, B.; Zhang, W.; Nie, P.; Liu, J.; Chen, X.; Luo, P. Pathological spectrum of glomerular disease in patients with renal insufficiency: A single-center study in Northeastern China. Ren. Fail. 2019, 41, 473–480. [Google Scholar] [CrossRef]
- Chiu, H.F.; Chen, H.C.; Lu, K.C.; Shu, K.H. Distribution of glomerular diseases in Taiwan: Preliminary report of National Renal Biopsy Registry-publication on behalf of Taiwan Society of Nephrology. BMC Nephrol. 2018, 19, 6. [Google Scholar] [CrossRef]
- Gupta, S.; Connolly, J.; Pepper, R.J.; Walsh, S.B.; Yaqoob, M.M.; Kleta, R.; Ashman, N. Membranous nephropathy: A retrospective observational study of membranous nephropathy in north east and central London. BMC Nephrol. 2017, 18, 201. [Google Scholar] [CrossRef]
- Li, J.; Cui, Z.; Long, J.; Huang, W.; Wang, J.; Zhang, H.; Wang, H.; Zhang, L.; Ronco, P.; Zhao, M.H. Primary glomerular nephropathy among hospitalized patients in a national database in China. Nephrol. Dial. Transplant. 2018, 33, 2173–2181. [Google Scholar] [CrossRef]
- McQuarrie, E.P.; Mackinnon, B.; Stewart, G.A.; Geddes, C.C. Membranous nephropathy remains the commonest primary cause of nephrotic syndrome in a northern European Caucasian population. Nephrol. Dial. Transplant. 2010, 25, 1009–1010. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Turkmen, A.; Sumnu, A.; Cebeci, E.; Yazici, H.; Eren, N.; Seyahi, N.; Dilek, K.; Dede, F.; Derici, U.; Unsal, A.; et al. Epidemiological features of primary glomerular disease in Turkey: A multicenter study by the Turkish Society of Nephrology Glomerular Diseases Working Group. BMC Nephrol. 2020, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.T.; Chan, C.M.; Lim, C.; Choo, J.; Chin, Y.M.; Teng, W.L.; Loh, A.H.L.; Choong, H.L.; Tan, H.K.; Wong, K.S.; et al. Changes in primary glomerulonephritis in Singapore over four decades. Clin. Nephrol. 2019, 91, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, N. Primary glomerulonephritis in mainland China: An overview. Contrib. Nephrol. 2013, 181, 1–11. [Google Scholar] [CrossRef]
- Chang, J.H.; Kim, D.K.; Kim, H.W.; Park, S.Y.; Yoo, T.H.; Kim, B.S.; Kang, S.W.; Choi, K.H.; Han, D.S.; Jeong, H.J.; et al. Changing prevalence of glomerular diseases in Korean adults: A review of 20 years of experience. Nephrol. Dial. Transplant. 2009, 24, 2406–2410. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Taguchi, T.; Sugiyama, H.; Sato, H. Membranous nephropathy in Japan: Analysis of the Japan Renal Biopsy Registry (J-RBR). Clin. Exp. Nephrol. 2012, 16, 557–563. [Google Scholar] [CrossRef]
- Zeng, C.H.; Chen, H.M.; Wang, R.S.; Chen, Y.; Zhang, S.H.; Liu, L.; Li, L.S.; Liu, Z.H. Etiology and clinical characteristics of membranous nephropathy in Chinese patients. Am. J. Kidney Dis. 2008, 52, 691–698. [Google Scholar] [CrossRef]
- Zhu, P.; Zhou, F.D.; Wang, S.X.; Zhao, M.H.; Wang, H.Y. Increasing frequency of idiopathic membranous nephropathy in primary glomerular disease: A 10-year renal biopsy study from a single Chinese nephrology centre. Nephrology 2015, 20, 560–566. [Google Scholar] [CrossRef]
- Zuo, K.; Wu, Y.; Li, S.J.; Xu, F.; Zeng, C.H.; Liu, Z.H. Long-term outcome and prognostic factors of idiopathic membranous nephropathy in the Chinese population. Clin. Nephrol. 2013, 79, 445–453. [Google Scholar] [CrossRef]
- Deegens, J.K.; Wetzels, J.F. Membranous nephropathy in the older adult: Epidemiology, diagnosis and management. Drugs Aging 2007, 24, 717–732. [Google Scholar] [CrossRef]
- Choi, J.Y.; Chin, H.J.; Lee, H.; Bae, E.H.; Chang, T.I.; Lim, J.H.; Jung, H.Y.; Cho, J.H.; Kim, C.D.; Kim, Y.L.; et al. Idiopathic membranous nephropathy in older patients: Clinical features and outcomes. PLoS ONE 2020, 15, e0240566. [Google Scholar] [CrossRef]
- Donadio, J.V., Jr. Treatment of glomerulonephritis in the elderly. Am. J. Kidney Dis. 1990, 16, 307–311. [Google Scholar] [CrossRef]
- Fogo, A.B.; Lusco, M.A.; Najafian, B.; Alpers, C.E. AJKD Atlas of Renal Pathology: Membranous Nephropathy. Am. J. Kidney Dis. 2015, 66, e15–e17. [Google Scholar] [CrossRef] [PubMed]
- Hladunewich, M.A.; Troyanov, S.; Calafati, J.; Cattran, D.C. The natural history of the non-nephrotic membranous nephropathy patient. Clin. J. Am. Soc. Nephrol. 2009, 4, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Glassock, R.J. Glomerular diseases: Membranous nephropathy–A modern view. Clin. J. Am. Soc. Nephrol. 2014, 9, 609–616. [Google Scholar] [CrossRef]
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997. [Google Scholar] [CrossRef]
- Moroni, G.; Ponticelli, C. Secondary Membranous Nephropathy. A Narrative Review. Front. Med. 2020, 7, 611317. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Glassock, R.J.; Nath, K.A.; Sethi, S.; Fervenza, F.C. A Proposal for a Serology-Based Approach to Membranous Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 421–430. [Google Scholar] [CrossRef]
- Ronco, P.; Debiec, H. Membranous nephropathy: Current understanding of various causes in light of new target antigens. Curr. Opin. Nephrol. Hypertens. 2021, 30, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.H., Jr.; Bonegio, R.G.; Lambeau, G.; Beck, D.M.; Powell, D.W.; Cummins, T.D.; Klein, J.B.; Salant, D.J. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N. Engl. J. Med. 2009, 361, 11–21. [Google Scholar] [CrossRef]
- Debiec, H.; Guigonis, V.; Mougenot, B.; Decobert, F.; Haymann, J.P.; Bensman, A.; Deschênes, G.; Ronco, P.M. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N. Engl. J. Med. 2002, 346, 2053–2060. [Google Scholar] [CrossRef]
- Kerjaschki, D. Pathomechanisms and molecular basis of membranous glomerulopathy. Lancet 2004, 364, 1194–1196. [Google Scholar] [CrossRef]
- Farquhar, M.G.; Saito, A.; Kerjaschki, D.; Orlando, R.A. The Heymann nephritis antigenic complex: Megalin (gp330) and RAP. J. Am. Soc. Nephrol. 1995, 6, 35–47. [Google Scholar] [CrossRef]
- Wang, Y.M.; Lee, V.W.S.; Wu, H.; Harris, D.C.H.; Alexander, S.I. Heymann nephritis in Lewis rats. J. Immunol. Res. 2015, 109, 15–29. [Google Scholar] [CrossRef]
- Li, X.; Wei, D.; Zhou, Z.; Wang, B.; Xu, Y.; Pan, J.; Yang, C.; Lu, J.; Qiu, Y. Anti-PLA2R Antibodies in Chinese Patients with Membranous Nephropathy. Med. Sci. Monit. 2016, 22, 1630–1636. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Joshi, M.; Chaturvedi, A.; Little, D.J.; Thurlow, J.S.; Waldman, M.; Olson, S.W. Detection of PLA2R Autoantibodies before the Diagnosis of Membranous Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.A.; McRae, J.L.; Dwyer, K.M. PLA2R and membranous nephropathy: A 3 year prospective Australian study. Nephrology 2016, 21, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Kumar, V.; Kumar, A.; Yadav, A.K.; Nada, R.; Kumar, H.; Kumar, V.; Rathi, M.; Kohli, H.S.; Gupta, K.L.; et al. PLA2R antibodies, glomerular PLA2R deposits and variations in PLA2R1 and HLA-DQA1 genes in primary membranous nephropathy in South Asians. Nephrol. Dial. Transplant. 2016, 31, 1486–1493. [Google Scholar] [CrossRef]
- Pang, L.; Zhang, A.M.; Li, H.X.; Du, J.L.; Jiao, L.L.; Duan, N.; Liu, Y.; Yu, D. Serum anti-PLA2R antibody and glomerular PLA2R deposition in Chinese patients with membranous nephropathy: A cross-sectional study. Medicine 2017, 96, e7218. [Google Scholar] [CrossRef]
- Wang, J.; Cui, Z.; Lu, J.; Probst, C.; Zhang, Y.M.; Wang, X.; Qu, Z.; Wang, F.; Meng, L.Q.; Cheng, X.Y.; et al. Circulating Antibodies against Thrombospondin Type-I Domain-Containing 7A in Chinese Patients with Idiopathic Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Tomas, N.M.; Beck, L.H., Jr.; Meyer-Schwesinger, C.; Seitz-Polski, B.; Ma, H.; Zahner, G.; Dolla, G.; Hoxha, E.; Helmchen, U.; Dabert-Gay, A.S.; et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N. Engl. J. Med. 2014, 371, 2277–2287. [Google Scholar] [CrossRef]
- Sethi, S. New ‘Antigens’ in Membranous Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 268–278. [Google Scholar] [CrossRef]
- Sethi, S.; Debiec, H.; Madden, B.; Vivarelli, M.; Charlesworth, M.C.; Ravindran, A.; Gross, L.; Ulinski, T.; Buob, D.; Tran, C.L.; et al. Semaphorin 3B-associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int. 2020, 98, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Madden, B.; Debiec, H.; Morelle, J.; Charlesworth, M.C.; Gross, L.; Negron, V.; Buob, D.; Chaudhry, S.; Jadoul, M.; et al. Protocadherin 7-Associated Membranous Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1249–1261. [Google Scholar] [CrossRef]
- Sethi, S.; Madden, B.J.; Debiec, H.; Charlesworth, M.C.; Gross, L.; Ravindran, A.; Hummel, A.M.; Specks, U.; Fervenza, F.C.; Ronco, R. Exostosin 1/Exostosin 2-Associated Membranous Nephropathy. J. Am. Soc. Nephrol. 2019, 30, 1123–1136. [Google Scholar] [CrossRef]
- Sethi, S.; Debiec, H.; Madden, B.; Charlesworth, M.C.; Morelle, J.; Gross, L.; Ravindran, A.; Buob, D.; Jadoul, M.; Fervenza, F.C.; et al. Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int. 2020, 97, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sun, L.; Dong, H.; Wang, Y.; Xu, X.; Zhao, Z.; Cheng, W.; Liu, X.; Zhao, X.; Geng, Y.; et al. Neural Epidermal Growth Factor-Like 1 Protein-Positive Membranous Nephropathy in Chinese Patients. Clin. J. Am. Soc. Nephrol. 2021, 16, 727–735. [Google Scholar] [CrossRef]
- Dahan, K.; Debiec, H.; Plaisier, E.; Cachanado, M.; Rousseau, A.; Wakselman, L.; Michel, P.A.; Mihout, F.; Dussol, B.; Matignon, M.; et al. Rituximab for Severe Membranous Nephropathy: A 6-Month Trial with Extended Follow-Up. J. Am. Soc. Nephrol. 2017, 28, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cui, Z.; Zhang, Y.M.; Qu, Z.; Wang, F.; Meng, L.Q.; Cheng, X.Y.; Liu, G.; Zhou, F.D.; Zhao, M.H. Rituximab for non-responsive idiopathic membranous nephropathy in a Chinese cohort. Nephrol. Dial. Transplant. 2018, 33, 1558–1563. [Google Scholar] [CrossRef]
- Lu, W.; Gong, S.; Li, J.; Luo, H.; Wang, Y. Efficacy and safety of rituximab in the treatment of membranous nephropathy: A systematic review and meta-analysis. Medicine 2020, 99, e19804. [Google Scholar] [CrossRef]
- Zhang, J.; Bian, L.; Ma, F.Z.; Jia, Y.; Lin, P. Efficacy and safety of rituximab therapy for membranous nephropathy: A meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8021–8029. [Google Scholar] [CrossRef]
- Huang, L.; Dong, Q.R.; Zhao, Y.J.; Hu, G.C. Rituximab for the management of idiopathic membranous nephropathy: A meta-analysis. Int. Urol. Nephrol. 2021, 53, 111–119. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Cravedi, P.; Chianca, A.; Perna, A.; Ruggiero, B.; Gaspari, F.; Rambaldi, A.; Marasà, M.; Remuzzi, G. Rituximab in idiopathic membranous nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Gauckler, P.; Shin, J.I.; Alberici, F.; Audard, V.; Bruchfeld, A.; Busch, M.; Cheung, C.K.; Crnogorac, M.; Delbarba, E.; Eller, K.; et al. Rituximab in Membranous Nephropathy. Kidney Int. Rep. 2021, 6, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Fervenza, F.C.; Appel, G.B.; Barbour, S.J.; Rovin, B.H.; Lafayette, R.A.; Aslam, N.; Jefferson, J.A.; Gipson, P.E.; Rizk, D.V.; Sedor, J.R.; et al. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy. N. Engl. J. Med. 2019, 381, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Van den Brand, J.; Ruggenenti, P.; Chianca, A.; Hofstra, J.M.; Perna, A.; Ruggiero, B.; Wetzels, J.F.M.; Remuzzi, G. Safety of Rituximab Compared with Steroids and Cyclophosphamide for Idiopathic Membranous Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 2729–2737. [Google Scholar] [CrossRef] [PubMed]
- Fervenza, F.C.; Abraham, R.S.; Erickson, S.B.; Irazabal, M.V.; Eirin, A.; Specks, U.; Nachman, P.H.; Bergstralh, E.J.; Leung, N.; Cosio, F.G.; et al. Rituximab therapy in idiopathic membranous nephropathy: A 2-year study. Clin. J. Am. Soc. Nephrol. 2010, 5, 2188–2198. [Google Scholar] [CrossRef]
- Fervenza, F.C.; Cosio, F.G.; Erickson, S.B.; Specks, U.; Herzenberg, A.M.; Dillon, J.J.; Leung, N.; Cohen, I.M.; Wochos, D.N.; Bergstralh, E.; et al. Rituximab treatment of idiopathic membranous nephropathy. Kidney Int. 2008, 73, 117–125. [Google Scholar] [CrossRef]
- Sethi, S.; Kumar, S.; Lim, K.; Jordan, S.C. Obinutuzumab is Effective for the Treatment of Refractory Membranous Nephropathy. Kidney Int. Rep. 2020, 5, 1515–1518. [Google Scholar] [CrossRef]
- Klomjit, N.; Fervenza, F.C.; Zand, L. Successful Treatment of Patients With Refractory PLA(2)R-Associated Membranous Nephropathy With Obinutuzumab: A Report of 3 Cases. Am. J. Kidney Dis. 2020, 76, 883–888. [Google Scholar] [CrossRef]
- Barrett, C.; Willcocks, L.C.; Jones, R.B.; Tarzi, R.M.; Henderson, R.B.; Cai, G.; Gisbert, S.I.; Belson, A.S.; Savage, C.O. Effect of belimumab on proteinuria and anti-phospholipase A2 receptor autoantibody in primary membranous nephropathy. Nephrol. Dial. Transplant. 2020, 35, 599–606. [Google Scholar] [CrossRef]
- Podestà, M.A.; Ruggiero, B.; Remuzzi, G.; Ruggenenti, P. Ofatumumab for multirelapsing membranous nephropathy complicated by rituximab-induced serum-sickness. BMJ Case Rep. 2020, 13, e232896. [Google Scholar] [CrossRef]
- Schatz, D.G.; Swanson, P.C. V(D)J recombination: Mechanisms of initiation. Annu. Rev. Genet. 2011, 45, 167–202. [Google Scholar] [CrossRef]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Nemazee, D.A.; Bürki, K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature 1989, 337, 562–566. [Google Scholar] [CrossRef]
- Hartley, S.B.; Cooke, M.P.; Fulcher, D.A.; Harris, A.W.; Cory, S.; Basten, A.; Goodnow, C.C. Elimination of self-reactive B lymphocytes proceeds in two stages: Arrested development and cell death. Cell 1993, 72, 325–335. [Google Scholar] [CrossRef]
- Luning Prak, E.T.; Monestier, M.; Eisenberg, R.A. B cell receptor editing in tolerance and autoimmunity. Ann. N. Y. Acad. Sci. 2011, 1217, 96–121. [Google Scholar] [CrossRef]
- Tze, L.E.; Schram, B.R.; Lam, K.P.; Hogquist, K.A.; Hippen, K.L.; Liu, J.; Shinton, S.A.; Otipoby, K.L.; Rodine, P.R.; Vegoe, A.L.; et al. Basal immunoglobulin signaling actively maintains developmental stage in immature B cells. PLoS Biol. 2005, 3, e82. [Google Scholar] [CrossRef]
- Tiegs, S.L.; Russell, D.M.; Nemazee, D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 1993, 177, 1009–1020. [Google Scholar] [CrossRef]
- Halverson, R.; Torres, R.M.; Pelanda, R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat. Immunol. 2004, 5, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Gay, D.; Saunders, T.; Camper, S.; Weigert, M. Receptor editing: An approach by autoreactive B cells to escape tolerance. J. Exp. Med. 1993, 177, 999–1008. [Google Scholar] [CrossRef]
- Goodnow, C.C.; Crosbie, J.; Adelstein, S.; Lavoie, T.B.; Smith-Gill, S.J.; Brink, R.A.; Pritchard-Briscoe, H.; Wotherspoon, J.S.; Loblay, R.H.; Raphael, K.; et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988, 334, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Cambier, J.C.; Gauld, S.B.; Merrell, K.T.; Vilen, B.J. B-cell anergy: From transgenic models to naturally occurring anergic B cells? Nat. Rev. Immunol. 2007, 7, 633–643. [Google Scholar] [CrossRef]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant autoantibody production by early human B cell precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef]
- Pillai, S.; Cariappa, A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 2009, 9, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.D.; Srivastava, B.; Allman, D. Regulation of peripheral B cell maturation. Cell. Immunol. 2006, 239, 92–102. [Google Scholar] [CrossRef]
- Rowland, S.L.; Leahy, K.F.; Halverson, R.; Torres, R.M.; Pelanda, R. BAFF receptor signaling aids the differentiation of immature B cells into transitional B cells following tonic BCR signaling. J. Immunol. 2010, 185, 4570–4581. [Google Scholar] [CrossRef]
- Cyster, J.G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 2005, 23, 127–159. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.S.; Wardemann, H.; Chelnis, J.; Cunningham-Rundles, C.; Meffre, E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J. Exp. Med. 2004, 200, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Kerfoot, S.M.; Yaari, G.; Patel, J.R.; Johnson, K.L.; Gonzalez, D.G.; Kleinstein, S.H.; Haberman, A.M. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 2011, 34, 947–960. [Google Scholar] [CrossRef]
- Shlomchik, M.J.; Weisel, F. Germinal center selection and the development of memory B and plasma cells. Immunol. Rev. 2012, 247, 52–63. [Google Scholar] [CrossRef]
- Papavasiliou, F.N.; Schatz, D.G. Somatic hypermutation of immunoglobulin genes: Merging mechanisms for genetic diversity. Cell 2002, 109, S35–S44. [Google Scholar] [CrossRef]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Alt, F.W. Class-switch recombination: Interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004, 4, 541–552. [Google Scholar] [CrossRef]
- Papa, I.; Vinuesa, C.G. Synaptic Interactions in Germinal Centers. Front. Immunol. 2018, 9, 1858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Garcia-Ibanez, L.; Toellner, K.M. Regulation of germinal center B-cell differentiation. Immunol. Rev. 2016, 270, 8–19. [Google Scholar] [CrossRef]
- Cerutti, A.; Cols, M.; Puga, I. Activation of B cells by non-canonical helper signals. EMBO. Rep. 2012, 13, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef]
- Vincent, F.B.; Saulep-Easton, D.; Figgett, W.A.; Fairfax, K.A.; Mackay, F. The BAFF/APRIL system: Emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013, 24, 203–215. [Google Scholar] [CrossRef]
- Smulski, C.R.; Eibel, H. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front. Immunol. 2018, 9, 2285. [Google Scholar] [CrossRef]
- Avery, D.T.; Kalled, S.L.; Ellyard, J.I.; Ambrose, C.; Bixler, S.A.; Thien, M.; Brink, R.; Mackay, F.; Hodgkin, P.D.; Tangye, S.G. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Investig. 2003, 112, 286–297. [Google Scholar] [CrossRef]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The BAFF/APRIL system in SLE pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [Google Scholar] [CrossRef]
- Cyster, J.G.; Hartley, S.B.; Goodnow, C.C. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature 1994, 371, 389–395. [Google Scholar] [CrossRef]
- Salzer, U.; Jennings, S.; Grimbacher, B. To switch or not to switch--the opposing roles of TACI in terminal B cell differentiation. Eur. J. Immunol. 2007, 37, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.N.; Wright, J.A.; Kleiman, E.; Boucher, J.C.; Castro, I.; Clark, E.S. B-lymphocyte tolerance and effector function in immunity and autoimmunity. Immunol. Res. 2013, 57, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Lund, F.E.; Randall, T.D. Effector and regulatory B cells: Modulators of CD4+ T cell immunity. Nat. Rev. Immunol. 2010, 10, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Dasgupta, S.; Bandyopadhyay, M. Regulatory B cells in infection, inflammation, and autoimmunity. Cell. Immunol. 2020, 352, 104076. [Google Scholar] [CrossRef]
- Wang, B.; Zuo, K.; Wu, Y.; Huang, Q.; Qin, W.S.; Zeng, C.H.; Li, L.S.; Liu, Z.H. Correlation between B lymphocyte abnormality and disease activity in patients with idiopathic membranous nephropathy. J. Int. Med. Res. 2011, 39, 86–95. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Languille, E.; Debiec, H.; Hygino, J.; Dahan, K.; Simon, T.; Klatzmann, D.; Ronco, P. B- and T-cell subpopulations in patients with severe idiopathic membranous nephropathy may predict an early response to rituximab. Kidney Int. 2017, 92, 227–237. [Google Scholar] [CrossRef]
- Cantarelli, C.; Jarque, M.; Angeletti, A.; Manrique, J.; Hartzell, S.; O’Donnell, T.; Merritt, E.; Laserson, U.; Perin, L.; Donadei, C.; et al. A Comprehensive Phenotypic and Functional Immune Analysis Unravels Circulating Anti-Phospholipase A2 Receptor Antibody Secreting Cells in Membranous Nephropathy Patients. Kidney Int. Rep. 2020, 5, 1764–1776. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, Y.; Yang, K.; Crew, R.; Wang, H.; Jiang, Y. Higher frequencies of circulating ICOS(+), IL-21(+) T follicular helper cells and plasma cells in patients with new-onset membranous nephropathy. Autoimmunity 2017, 50, 458–467. [Google Scholar] [CrossRef]
- Odendahl, M.; Jacobi, A.; Hansen, A.; Feist, E.; Hiepe, F.; Burmester, G.R.; Lipsky, P.E.; Radbruch, A.; Dörner, T. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J. Immunol. 2000, 165, 5970–5979. [Google Scholar] [CrossRef]
- Su, Z.; Jin, Y.; Zhang, Y.; Guan, Z.; Li, H.; Chen, X.; Xie, C.; Zhang, C.; Liu, X.; Li, P.; et al. The Diagnostic and Prognostic Potential of the B-Cell Repertoire in Membranous Nephropathy. Front. Immunol. 2021, 12, 635326. [Google Scholar] [CrossRef]
- Tipton, C.M.; Fucile, C.F.; Darce, J.; Chida, A.; Ichikawa, T.; Gregoretti, I.; Schieferl, S.; Hom, J.; Jenks, S.; Feldman, R.J.; et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 2015, 16, 755–765. [Google Scholar] [CrossRef]
- Demaison, C.; David, D.; Fautrel, B.; Theze, J. V(H) gene-family representation in peripheral activated B cells from systemic lupus erythematosus (SLE) patients. Clin. Exp. Immunol. 1996, 104, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Stadinski, B.D.; Shekhar, K.; Gómez-Touriño, I.; Jung, J.; Sasaki, K.; Sewell, A.K.; Peakman, M.; Chakraborty, A.K.; Huseby, E.S. Hydrophobic CDR3 residues promote the development of self-reactive T cells. Nat. Immunol. 2016, 17, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Meffre, E.; Milili, M.; Blanco-Betancourt, C.; Antunes, H.; Nussenzweig, M.C.; Schiff, C. Immunoglobulin heavy chain expression shapes the B cell receptor repertoire in human B cell development. J. Clin. Investig. 2001, 108, 879–886. [Google Scholar] [CrossRef]
- Khass, M.; Schelonka, R.L.; Liu, C.R.; Elgavish, A.; Morel, L.; Burrows, P.D.; Schroeder, H.W., Jr. Alterations in B cell development, CDR-H3 repertoire and dsDNA-binding antibody production among C57BL/6 ΔD-iD mice congenic for the lupus susceptibility loci sle1, sle2 or sle3. Autoimmunity 2017, 50, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.M.; Wang, Y.; Zheng, G.; Zhang, G.Y.; Zhou, J.J.; Tan, T.K.; Cao, Q.; Hu, M.; Watson, D.; et al. DNA vaccine encoding CD40 targeted to dendritic cells in situ prevents the development of Heymann nephritis in rats. Kidney Int. 2013, 83, 223–232. [Google Scholar] [CrossRef]
- Biancone, L.; Andres, G.; Ahn, H.; DeMartino, C.; Stamenkovic, I. Inhibition of the CD40-CD40ligand pathway prevents murine membranous glomerulonephritis. Kidney Int. 1995, 48, 458–468. [Google Scholar] [CrossRef][Green Version]
- Oleinika, K.; Mauri, C.; Salama, A.D. Effector and regulatory B cells in immune-mediated kidney disease. Nat. Rev. Nephrol. 2019, 15, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Liu, Z.; Dai, H.; Liu, W.; Feng, Z.; Zhao, Q.; Gao, Y.; Liu, F.; Zhang, N.; Dong, X.; et al. The Potential Role of Regulatory B Cells in Idiopathic Membranous Nephropathy. J. Immunol. Res. 2020, 2020, 7638365. [Google Scholar] [CrossRef]
- Ramachandran, R.; Kaundal, U.; Girimaji, N.; Rakha, A.; Rathi, M.; Gupta, K.L.; Kohli, H.S.; Jha, V. Regulatory B Cells Are Reduced and Correlate With Disease Activity in Primary Membranous Nephropathy. Kidney Int. Rep. 2020, 5, 872–878. [Google Scholar] [CrossRef]
- Blair, P.A.; Noreña, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Yang, S.H.; Jo, H.A.; Oh, Y.J.; Park, M.; Kim, J.Y.; Lee, H.; Lee, J.P.; Lee, S.H.; Joo, K.W.; et al. BAFF and APRIL expression as an autoimmune signature of membranous nephropathy. Oncotarget 2018, 9, 3292–3302. [Google Scholar] [CrossRef]
- Netti, G.S.; Infante, B.; Spadaccino, F.; Godeas, G.; Corallo, M.G.; Prisciandaro, C.; Croce, L.; Rotondi, M.; Gesualdo, L.; Stallone, G.; et al. Serum Levels of BAFF and APRIL Predict Clinical Response in Anti-PLA2R-Positive Primary Membranous Nephropathy. J. Immunol. Res. 2019, 2019, 8483650. [Google Scholar] [CrossRef]
- Alexopoulos, E.; Seron, D.; Hartley, R.B.; Nolasco, F.; Cameron, J.S. Immune mechanisms in idiopathic membranous nephropathy: The role of the interstitial infiltrates. Am. J. Kidney Dis. 1989, 13, 404–412. [Google Scholar] [CrossRef]
- Cohen, C.D.; Calvaresi, N.; Armelloni, S.; Schmid, H.; Henger, A.; Ott, U.; Rastaldi, M.P.; Kretzler, M. CD20-positive infiltrates in human membranous glomerulonephritis. J. Nephrol. 2005, 18, 328–333. [Google Scholar]
- Fleig, S.V.; Konen, F.F.; Schröder, C.; Schmitz, J.; Gingele, S.; Bräsen, J.H.; Lovric, S.; Schmidt, B.M.W.; Haller, H.; Skripuletz, T.; et al. Long-term B cell depletion associates with regeneration of kidney function. Immunity Inflamm. Dis. 2021, 9, 1479–1488. [Google Scholar] [CrossRef]
- Segerer, S.; Schlöndorff, D. B cells and tertiary lymphoid organs in renal inflammation. Kidney Int. 2008, 73, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Pipi, E.; Nayar, S.; Gardner, D.H.; Colafrancesco, S.; Smith, C.; Barone, F. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front. Immunol. 2018, 9, 1952. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Cheng, Y.; Chang, D.; Liu, T.; Liu, L.; Pei, G.; Zhang, N.; Wang, Z.; Guo, K.; Chen, W.; et al. Tertiary lymphoid organs are associated with the progression of kidney damage and regulated by interleukin-17A. Theranostics 2021, 11, 117–131. [Google Scholar] [CrossRef]
- Bombardieri, M.; Lewis, M.; Pitzalis, C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Kolovou, K.; Laskari, K.; Roumelioti, M.; Tektonidou, M.G.; Panayiotidis, P.; Boletis, J.N.; Marinaki, S.; Sfikakis, P.P. B-cell oligoclonal expansions in renal tissue of patients with immune-mediated glomerular disease. Clin. Immunol. 2020, 217, 108488. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, A.; Iyoda, M.; Shibata, T.; Sugisaki, T. Th2 cytokines increase and stimulate B cells to produce IgG4 in idiopathic membranous nephropathy. Kidney Int. 2005, 68, 302–310. [Google Scholar] [CrossRef]
- Clatworthy, M.R.; Watson, C.J.; Plotnek, G.; Bardsley, V.; Chaudhry, A.N.; Bradley, J.A.; Smith, K.G. B-cell-depleting induction therapy and acute cellular rejection. N. Engl. J. Med. 2009, 360, 2683–2685. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, J.E.; Carriazo, S.; Ortiz, A. Treatment of idiopathic membranous nephropathy in adults: KDIGO 2012, cyclophosphamide and cyclosporine A are out, rituximab is the new normal. Clin. Kidney J. 2019, 12, 629–638. [Google Scholar] [CrossRef]
- Floege, J.; Barbour, S.J.; Cattran, D.C.; Hogan, J.J.; Nachman, P.H.; Tang, S.C.W.; Wetzels, J.F.M.; Cheung, M.; Wheeler, D.C.; Winkelmayer, W.C.; et al. Management and treatment of glomerular diseases (part 1): Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019, 95, 268–280. [Google Scholar] [CrossRef]
- Ponticelli, C.; Escoli, R.; Moroni, G. Does cyclophosphamide still play a role in glomerular diseases? Autoimmun. Rev. 2018, 17, 1022–1027. [Google Scholar] [CrossRef]
- Farouk, S.S.; Rein, J.L. The Many Faces of Calcineurin Inhibitor Toxicity-What the FK? Adv. Chronic Kidney Dis. 2020, 27, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Hurd, E.R.; Giuliano, V.J. The effect of cyclophosphamide on B and T lymphocytes in patients with connective tissue diseases. Arthritis Rheum. 1975, 18, 67–75. [Google Scholar] [CrossRef]
- Górski, A.; Wasik, M. Evidence that cyclophosphamide is a strong inhibitor of human B cell proliferation. Transplantation 1987, 43, 441–442. [Google Scholar] [CrossRef]
- Zhu, L.P.; Cupps, T.R.; Whalen, G.; Fauci, A.S. Selective effects of cyclophosphamide therapy on activation, proliferation, and differentiation of human B cells. J. Clin. Investig. 1987, 79, 1082–1090. [Google Scholar] [CrossRef]
- Cupps, T.R.; Edgar, L.C.; Fauci, A.S. Suppression of human B lymphocyte function by cyclophosphamide. J. Immunol. 1982, 128, 2453–2457. [Google Scholar]
- Morikawa, K.; Oseko, F.; Morikawa, S. The distinct effects of FK506 on the activation, proliferation, and differentiation of human B lymphocytes. Transplantation 1992, 54, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- De Bruyne, R.; Bogaert, D.; De Ruyck, N.; Lambrecht, B.N.; Van Winckel, M.; Gevaert, P.; Dullaers, M. Calcineurin inhibitors dampen humoral immunity by acting directly on naive B cells. Clin. Exp. Immunol. 2015, 180, 542–550. [Google Scholar] [CrossRef]
- Suzuki, N.; Sakane, T.; Tsunematsu, T. Effects of a novel immunosuppressive agent, FK506, on human B cell activation. Clin. Exp. Immunol. 1990, 79, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Wicker, L.S.; Boltz, R.C., Jr.; Matt, V.; Nichols, E.A.; Peterson, L.B.; Sigal, N.H. Suppression of B cell activation by cyclosporin A, FK506 and rapamycin. Eur. J. Immunol. 1990, 20, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Kumar, V.; Bharati, J.; Rovin, B.; Nada, R.; Kumar, V.; Rathi, M.; Jha, V.; Gupta, K.L.; Kohli, H.S. Long-Term Follow-Up of Cyclical Cyclophosphamide and Steroids Versus Tacrolimus and Steroids in Primary Membranous Nephropathy. Kidney. Int. Rep. 2021, 6, 2653–2660. [Google Scholar] [CrossRef]
- Sentís, A.; Diekmann, F.; Llobell, A.; de Moner, N.; Espinosa, G.; Yagüe, J.; Campistol, J.M.; Mirapeix, E.; Juan, M. Kinetic analysis of changes in T- and B-lymphocytes after anti-CD20 treatment in renal pathology. Immunobiology 2017, 222, 620–630. [Google Scholar] [CrossRef]
- Beck, L.H., Jr.; Fervenza, F.C.; Beck, D.M.; Bonegio, R.G.; Malik, F.A.; Erickson, S.B.; Cosio, F.G.; Cattran, D.C.; Salant, D.J. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1543–1550. [Google Scholar] [CrossRef]
- Cravedi, P.; Ruggenenti, P.; Sghirlanzoni, M.C.; Remuzzi, G. Titrating rituximab to circulating B cells to optimize lymphocytolytic therapy in idiopathic membranous nephropathy. Clin. J. Am. Soc. Nephrol. 2007, 2, 932–937. [Google Scholar] [CrossRef]
- Fenoglio, R.; Baldovino, S.; Sciascia, S.; De Simone, E.; Del Vecchio, G.; Ferro, M.; Quattrocchio, G.; Naretto, C.; Roccatello, D. Efficacy of low or standard rituximab-based protocols and comparison to Ponticelli’s regimen in membranous nephropathy. J. Nephrol. 2021, 34, 565–571. [Google Scholar] [CrossRef]
- Moroni, G.; Depetri, F.; Del Vecchio, L.; Gallelli, B.; Raffiotta, F.; Giglio, E.; Brunini, F.; D’Amico, M.; Longhi, S.; Radice, A.; et al. Low-dose rituximab is poorly effective in patients with primary membranous nephropathy. Nephrol. Dial. Transplant. 2017, 32, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Teisseyre, M.; Boyer-Suavet, S.; Crémoni, M.; Brglez, V.; Esnault, V.; Seitz-Polski, B. Analysis and Management of Rituximab Resistance in PLA2R1-Associated Membranous Nephropathy. Kidney Int. Rep. 2021, 6, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Boyer-Suavet, S.; Andreani, M.; Cremoni, M.; Brglez, V.; Benzaken, S.; Bernard, G.; Nachman, P.; Esnault, V.; Seitz-Polski, B. Rituximab bioavailability in primary membranous nephropathy. Nephrol. Dial. Transplant. 2019, 34, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Fogueri, U.; Cheungapasitporn, W.; Bourne, D.; Fervenza, F.C.; Joy, M.S. Rituximab Exhibits Altered Pharmacokinetics in Patients With Membranous Nephropathy. Ann. Pharmacother. 2019, 53, 357–363. [Google Scholar] [CrossRef]
- Thiel, J.; Rizzi, M.; Engesser, M.; Dufner, A.K.; Troilo, A.; Lorenzetti, R.; Voll, R.E.; Venhoff, N. B cell repopulation kinetics after rituximab treatment in ANCA-associated vasculitides compared to rheumatoid arthritis, and connective tissue diseases: A longitudinal observational study on 120 patients. Arthritis Res. Ther. 2017, 19, 101. [Google Scholar] [CrossRef]
- Pozdzik, A.; Beukinga, I.; Gu-Trantien, C.; Willard-Gallo, K.; Nortier, J.; Pradier, O. Circulating (CD3(-)CD19(+)CD20(-)IgD(-)CD27(high)CD38(high)) Plasmablasts: A Promising Cellular Biomarker for Immune Activity for Anti-PLA2R1 Related Membranous Nephropathy? Mediat. Inflamm. 2016, 2016, 7651024. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.; Carsetti, R.; Serafinelli, J.; Rocca, S.; Massella, L.; Gargiulo, A.; Lo Russo, A.; Capponi, C.; Cotugno, N.; Porzio, O.; et al. Prolonged Impairment of Immunological Memory After Anti-CD20 Treatment in Pediatric Idiopathic Nephrotic Syndrome. Front. Immunol. 2019, 10, 1653. [Google Scholar] [CrossRef] [PubMed]
- Roll, P.; Palanichamy, A.; Kneitz, C.; Dorner, T.; Tony, H.P. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2377–2386. [Google Scholar] [CrossRef]
- Gudbrandsdottir, S.; Brimnes, M.; Køllgaard, T.; Hasselbalch, H.C.; Nielsen, C.H. Effects of rituximab and dexamethasone on regulatory and proinflammatory B-cell subsets in patients with primary immune thrombocytopenia. Eur. J. Haematol. 2018, 100, 45–52. [Google Scholar] [CrossRef]
- Quan, C.; ZhangBao, J.; Lu, J.; Zhao, C.; Cai, T.; Wang, B.; Yu, H.; Qiao, J.; Lu, C. The immune balance between memory and regulatory B cells in NMO and the changes of the balance after methylprednisolone or rituximab therapy. J. Neuroimmunol. 2015, 282, 45–53. [Google Scholar] [CrossRef]
- Ehrenstein, M.R.; Wing, C. The BAFFling effects of rituximab in lupus: Danger ahead? Nat. Rev. Rheumatol. 2016, 12, 367–372. [Google Scholar] [CrossRef]
- Vallerskog, T.; Heimbürger, M.; Gunnarsson, I.; Zhou, W.; Wahren-Herlenius, M.; Trollmo, C.; Malmström, V. Differential effects on BAFF and APRIL levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R167. [Google Scholar] [CrossRef]
- Cambridge, G.; Perry, H.C.; Nogueira, L.; Serre, G.; Parsons, H.M.; De La Torre, I.; Dickson, M.C.; Leandro, M.J.; Edwards, J.C. The effect of B-cell depletion therapy on serological evidence of B-cell and plasmablast activation in patients with rheumatoid arthritis over multiple cycles of rituximab treatment. J. Autoimmun. 2014, 50, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Lavie, F.; Miceli-Richard, C.; Ittah, M.; Sellam, J.; Gottenberg, J.E.; Mariette, X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: Insights into a new regulating system of BAFF production. Ann. Rheum. Dis. 2007, 66, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Furie, R.; Teng, Y.K.O.; Contreras, G.; Malvar, A.; Yu, X.; Ji, B.; Green, Y.; Gonzalez-Rivera, T.; Bass, D.; et al. A secondary analysis of the Belimumab International Study in Lupus Nephritis trial examined effects of belimumab on kidney outcomes and preservation of kidney function in patients with lupus nephritis. Kidney Int. 2021, in press. [Google Scholar] [CrossRef]
- Yang, M.; Sun, L.; Wang, S.; Ko, K.H.; Xu, H.; Zheng, B.J.; Cao, X.; Lu, L. Novel function of B cell-activating factor in the induction of IL-10-producing regulatory B cells. J. Immunol. 2010, 184, 3321–3325. [Google Scholar] [CrossRef] [PubMed]
- Ginzler, E.M.; Wax, S.; Rajeswaran, A.; Copt, S.; Hillson, J.; Ramos, E.; Singer, N.G. Atacicept in combination with MMF and corticosteroids in lupus nephritis: Results of a prematurely terminated trial. Arthritis Res. Ther. 2012, 14, R33. [Google Scholar] [CrossRef]
- Boyer-Suavet, S.; Andreani, M.; Lateb, M.; Savenkoff, B.; Brglez, V.; Benzaken, S.; Bernard, G.; Nachman, P.H.; Esnault, V.; Seitz-Polski, B. Neutralizing Anti-Rituximab Antibodies and Relapse in Membranous Nephropathy Treated With Rituximab. Front. Immunol. 2019, 10, 3069. [Google Scholar] [CrossRef]
- Podestà, M.A.; Gennarini, A.; Portalupi, V.; Rota, S.; Alessio, M.G.; Remuzzi, G.; Ruggenenti, P. Accelerating the Depletion of Circulating Anti-Phospholipase A2 Receptor Antibodies in Patients with Severe Membranous Nephropathy: Preliminary Findings with Double Filtration Plasmapheresis and Ofatumumab. Nephron 2020, 144, 30–35. [Google Scholar] [CrossRef]
- Leandro, M.J. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res. Ther. 2013, 15 (Suppl. S1), S3. [Google Scholar] [CrossRef] [PubMed]
- Redfield, R.R.; Jordan, S.C.; Busque, S.; Vincenti, F.; Woodle, E.S.; Desai, N.; Reed, E.F.; Tremblay, S.; Zachary, A.A.; Vo, A.A.; et al. Safety, pharmacokinetics, and pharmacodynamic activity of obinutuzumab, a type 2 anti-CD20 monoclonal antibody for the desensitization of candidates for renal transplant. Am. J. Transplant. 2019, 19, 3035–3045. [Google Scholar] [CrossRef]
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, B.F.; Moser, K.; Hauser, A.E.; Peddinghaus, A.; Voigt, C.; Eilat, D.; Radbruch, A.; Hiepe, F.; Manz, R.A. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J. Exp. Med. 2004, 199, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Juárez, G.; Rojas-Rivera, J.; Logt, A.V.; Justino, J.; Sevillano, A.; Caravaca-Fontán, F.; Ávila, A.; Rabasco, C.; Cabello, V.; Varela, A.; et al. The STARMEN trial indicates that alternating treatment with corticosteroids and cyclophosphamide is superior to sequential treatment with tacrolimus and rituximab in primary membranous nephropathy. Kidney Int. 2021, 99, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Debiec, H.; Ruggiero, B.; Chianca, A.; Pellé, T.; Gaspari, F.; Suardi, F.; Gagliardini, E.; Orisio, S.; Benigni, A.; et al. Anti-Phospholipase A2 Receptor Antibody Titer Predicts Post-Rituximab Outcome of Membranous Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 2545–2558. [Google Scholar] [CrossRef] [PubMed]
- Zonozi, R.; Laliberte, K.; Huizenga, N.R.; Rosenthal, J.K.; Jeyabalan, A.; Collins, A.B.; Cortazar, F.B.; Niles, J.L. Combination of Rituximab, Low-Dose Cyclophosphamide, and Prednisone for Primary Membranous Nephropathy: A Case Series With Extended Follow Up. Am. J. Kidney Dis. 2021, 78, 793–803. [Google Scholar] [CrossRef]
- Hartono, C.; Chung, M.; Kuo, S.F.; Seshan, S.V.; Muthukumar, T. Bortezomib therapy for nephrotic syndrome due to idiopathic membranous nephropathy. J. Nephrol. 2014, 27, 103–106. [Google Scholar] [CrossRef]
- Salhi, S.; Ribes, D.; Colombat, M.; Fortenfant, F.; Faguer, S. Bortezomib plus dexamethasone for rituximab-resistant PLA2R(+) membranous nephropathy. Kidney Int. 2021, 100, 708–709. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
So, B.Y.F.; Yap, D.Y.H.; Chan, T.M. B Cells in Primary Membranous Nephropathy: Escape from Immune Tolerance and Implications for Patient Management. Int. J. Mol. Sci. 2021, 22, 13560. https://doi.org/10.3390/ijms222413560
So BYF, Yap DYH, Chan TM. B Cells in Primary Membranous Nephropathy: Escape from Immune Tolerance and Implications for Patient Management. International Journal of Molecular Sciences. 2021; 22(24):13560. https://doi.org/10.3390/ijms222413560
Chicago/Turabian StyleSo, Benjamin Y. F., Desmond Y. H. Yap, and Tak Mao Chan. 2021. "B Cells in Primary Membranous Nephropathy: Escape from Immune Tolerance and Implications for Patient Management" International Journal of Molecular Sciences 22, no. 24: 13560. https://doi.org/10.3390/ijms222413560
APA StyleSo, B. Y. F., Yap, D. Y. H., & Chan, T. M. (2021). B Cells in Primary Membranous Nephropathy: Escape from Immune Tolerance and Implications for Patient Management. International Journal of Molecular Sciences, 22(24), 13560. https://doi.org/10.3390/ijms222413560