
Comprehensive Transcriptome Analysis Uncovers Distinct Expression Patterns Associated with Early Salinity Stress in Annual Ryegrass (Lolium Multiflorum L.)

 ,
,  ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
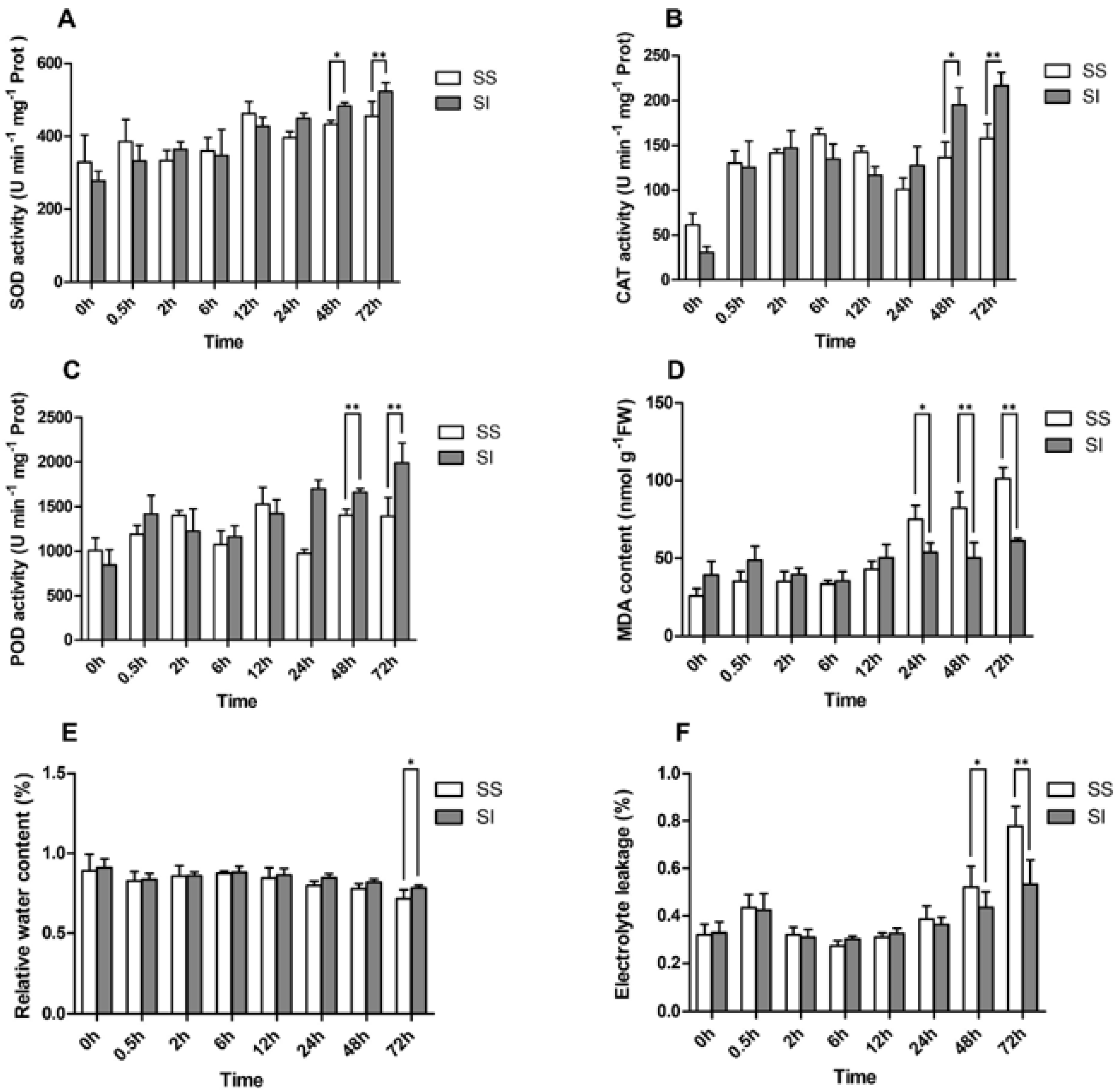
2.1. Phenotypic and Physiological Responses of L. Multiflorumi to Salt Stress
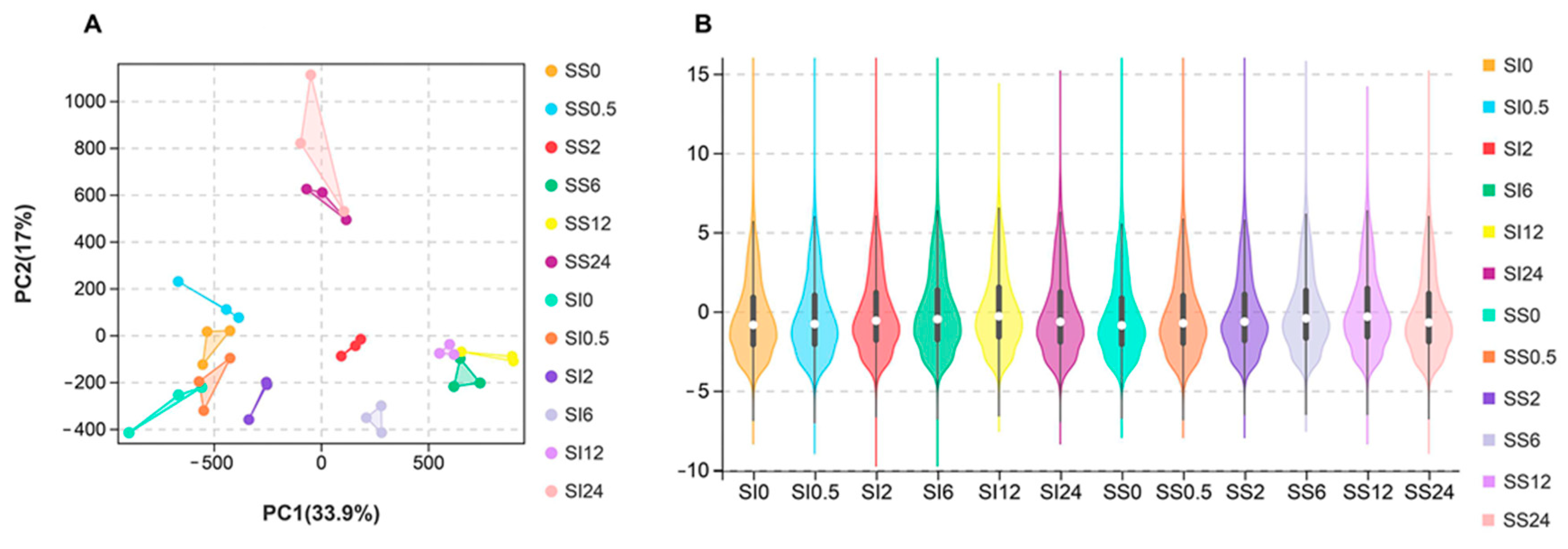
2.2. Processing and Mapping of the RNA-Seq Data
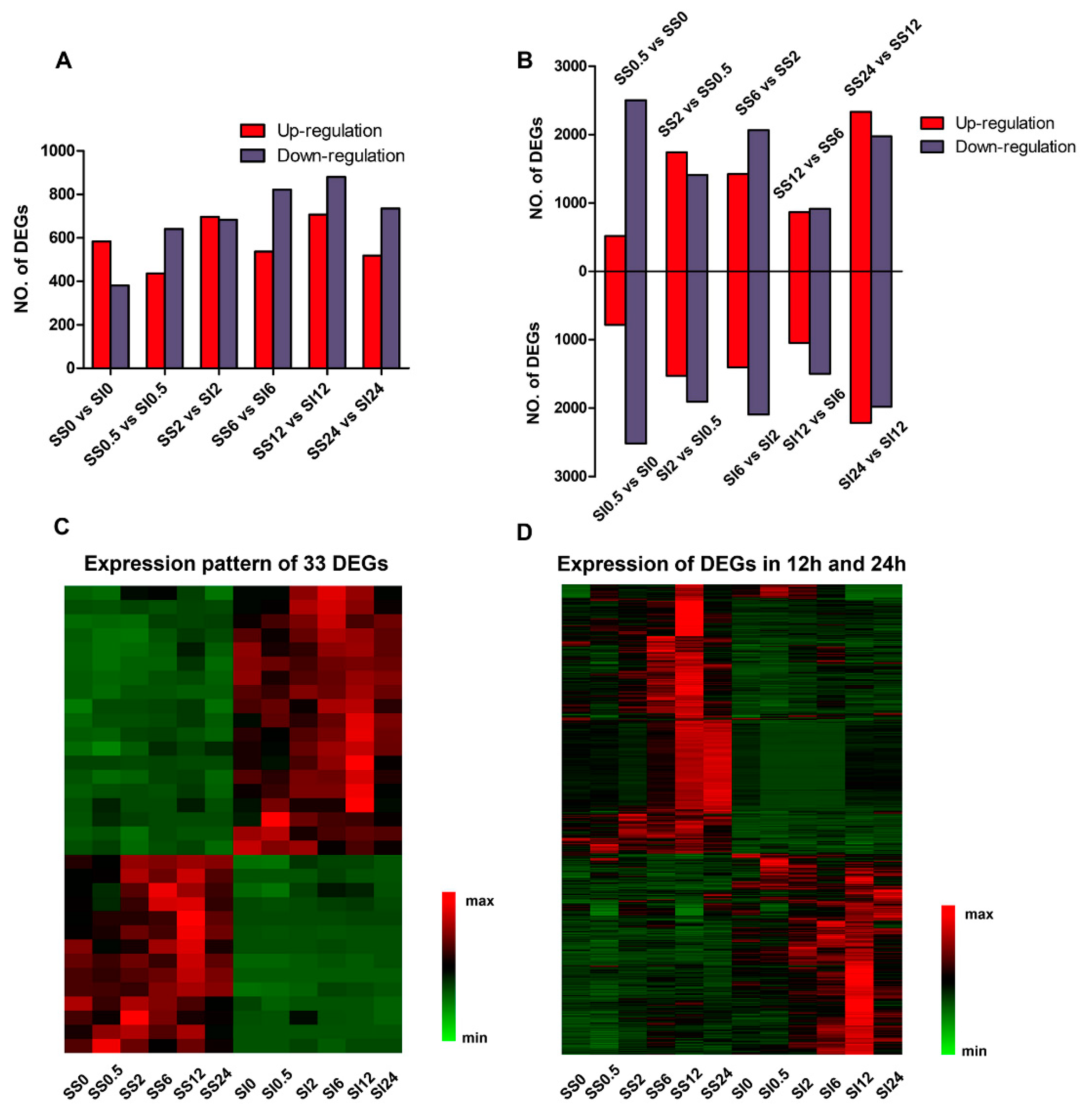
2.3. Identification of the Differentially Expressed Genes in the SS and SI Genotypes of L. Multiflorum under Salt Stress
2.4. Functional Characterization of the DEGs
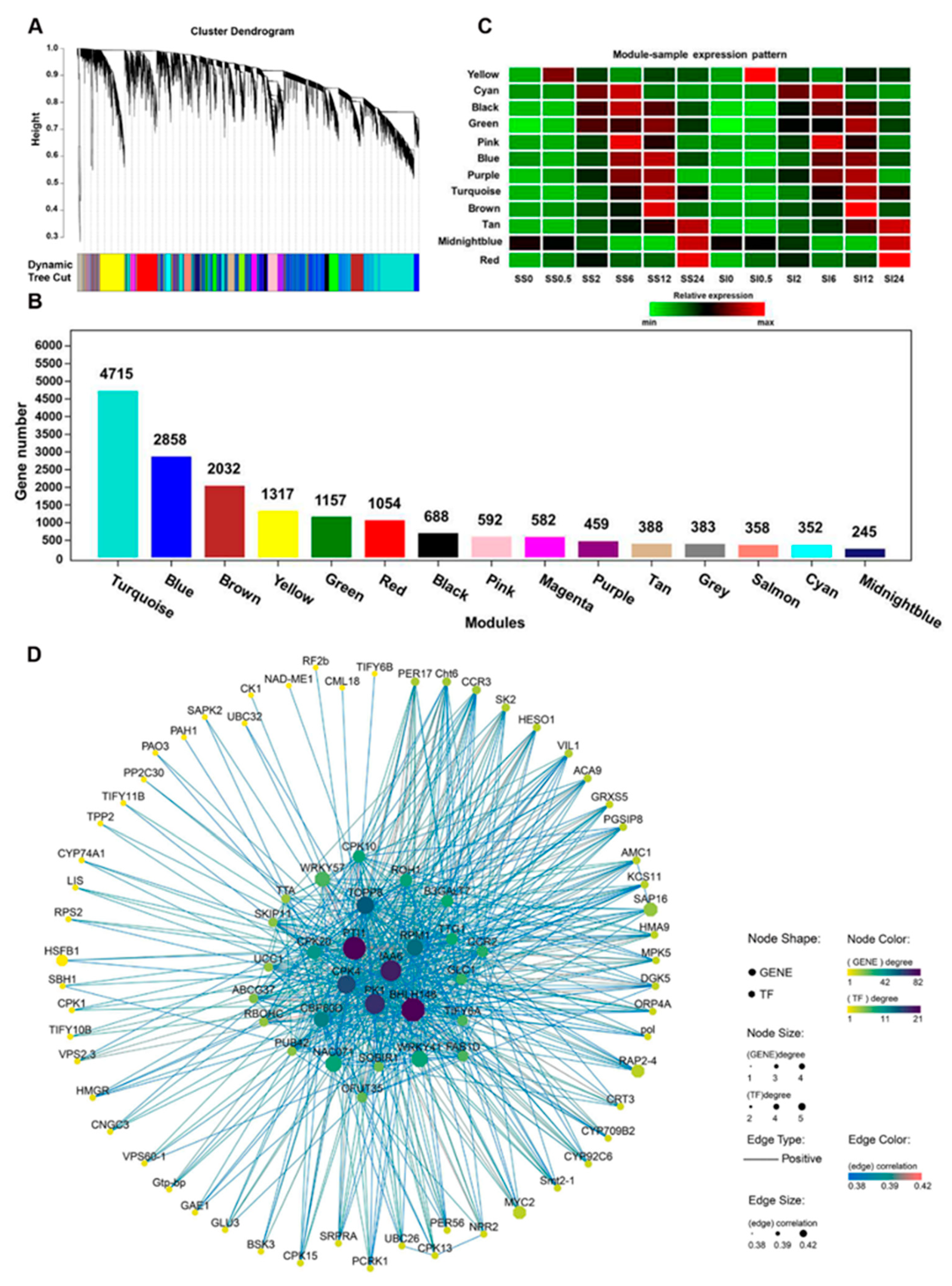
2.5. Weighted Gene Co-Expression Network Analysis of the DEGs
2.6. Identification of the Hub Genes in Different Stages
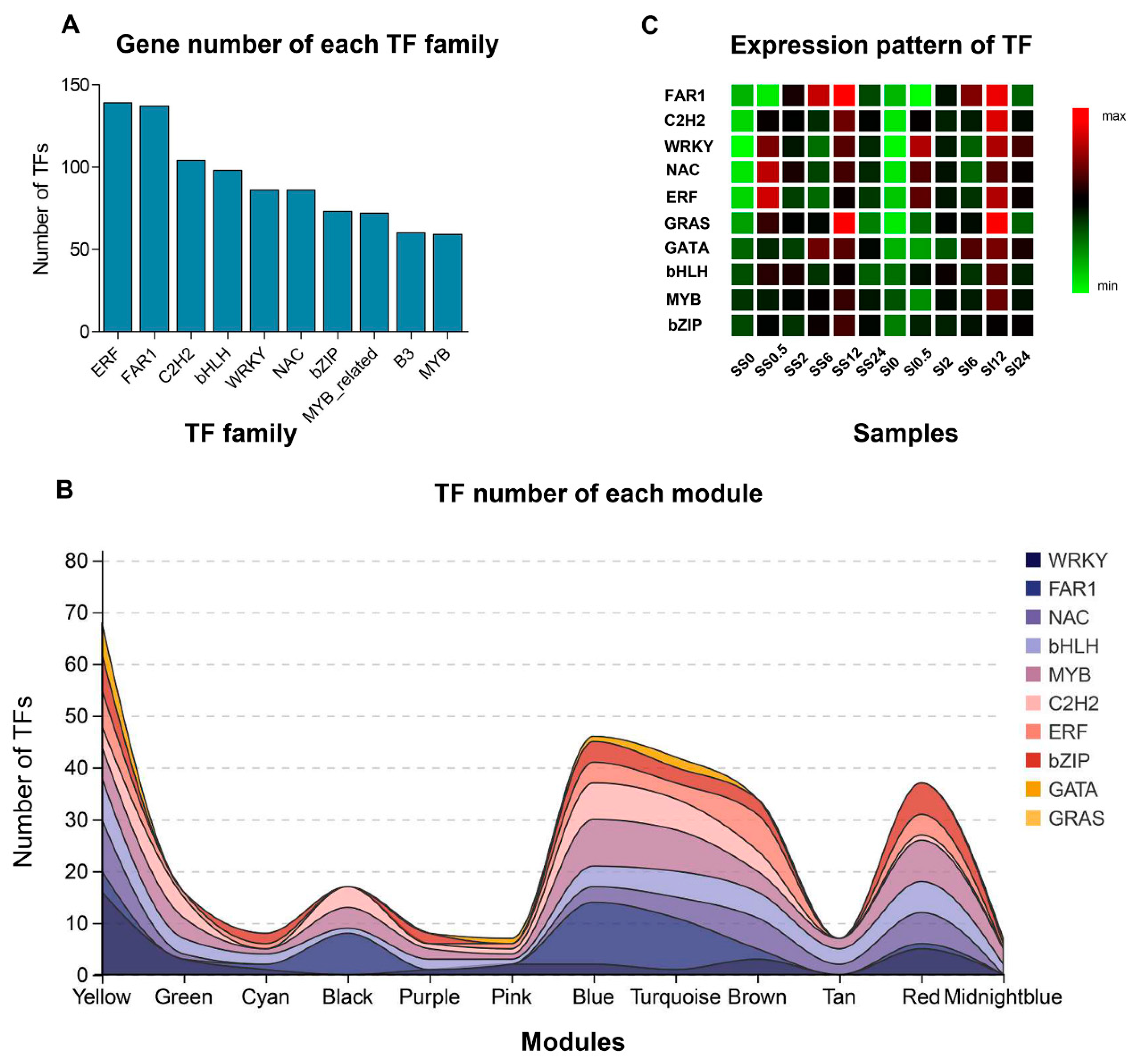
2.7. Identification of the Transcription Factors
2.8. Quantitative PCR (qPCR) Verification
3. Discussion
3.1. Regulation of Ionic Balance
3.2. Salinity Stress Sensing
3.3. Transcriptional Regulation
3.4. Metabolism Regulation
4. Materials and Methods
4.1. Determination of Relative Water Content
4.2. Determination of Electrolyte Leakage
4.3. Determination of Malondialdehyde Content and the Protective Antioxidant Enzyme Activity
4.4. Determination of the Na+ and K+ Concentrations
4.5. RNA Extraction, Library Construction, and Sequencing
4.6. Identification, de Novo Assembly, and Determination of the Expression Levels of the DEGs
4.7. Principal Component and Transcription Factors Analyses
4.8. Gene Ontology and KEGG Enrichment Analyses of the DEGs
4.9. Real-Time Quantitative PCR (qRT-PCR) Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bian, L.; Wang, J.; Liu, J.; Han, B. Spatiotemporal Changes of Soil Salinization in the Yellow River Delta of China from 2015 to 2019. Sustainability 2021, 13, 822. [Google Scholar] [CrossRef]
- El Moukhtari, A.; Cabassa-Hourton, C.; Farissi, M.; Savouré, A. How Does Proline Treatment Promote Salt Stress Tolerance During Crop Plant Development? Front. Plant Sci. 2020, 11, 1127. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, B. Protection of Halophytes and Their Uses for Cultivation of Saline-Alkali Soil in China. Biology 2021, 10, 353. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mao, B.; Yuan, D.; Chu, C.; Duan, M. Salt tolerance in rice: Physiological responses and molecular mechanisms. Crop J. 2021, 10, 13–25. [Google Scholar] [CrossRef]
- Ejaz, M.; Ali, M.; Hasan, H.; Khan, S.; Lal, A.; Qazi, N.; Shafique, A.; Dar, H.; Gul, A. Salt-regulating genes in wheat. In Climate Change and Food Security with Emphasis on Wheat; Academic Press: Cambridge, MA, USA, 2020; pp. 77–91. [Google Scholar]
- Zhang, M.; Liang, X.; Wang, L.; Cao, Y.; Song, W.; Shi, J.; Lai, J.; Jiang, C. A HAK family Na+ transporter confers natural variation of salt tolerance in maize. Nat. Plants 2019, 5, 1297–1308. [Google Scholar] [CrossRef]
- Phang, T.-H.; Shao, G.; Lam, H.-M. Salt Tolerance in Soybean. J. Integr. Plant Biol. 2008, 50, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, G. Exploration and Utilization of Salt-Tolerant Barley Germplasm. In Exploration, Identification and Utilization of Barley Germplasm; Zhang, G., Li, C., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 75–113. [Google Scholar]
- Zhang, F.; Zhu, G.; Du, L.; Shang, X.; Cheng, C.; Yang, B.; Hu, Y.; Cai, C.; Guo, W. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 2016, 6, 20582. [Google Scholar] [CrossRef] [Green Version]
- Galvan-Ampudia, C.S.; Testerink, C. Salt stress signals shape the plant root. Curr. Opin. Plant Biol. 2011, 14, 296–302. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Raihan, M.R.H.; Masud, A.A.C.; Rahman, K.; Nowroz, F.; Rahman, M.; Nahar, K.; Fujita, M. Regulation of Reactive Oxygen Species and Antioxidant Defense in Plants under Salinity. Int. J. Mol. Sci. 2021, 22, 9326. [Google Scholar] [CrossRef]
- Noctor, F.G. Redox Homeostasis and Antioxidant Signaling: A Metabolic Interface between Stress Perception and Physiological Responses. Plant Cell 2005, 17, 1866–1875. [Google Scholar]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar]
- Tavanti, T.R.; de Melo, A.A.R.; Moreira, L.D.K.; Sanchez, D.E.J.; dos Santos Silva, R.; da Silva, R.M.; Dos Reis, A.R. Micronutrient fertilization enhances ROS scavenging system for alleviation of abiotic stresses in plants. Plant Physiol. Biochem. 2021, 160, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Zhu, J. Regulation of Abscisic Acid Biosynthesis. Plant Physiol. 2003, 133, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Yao, J.; Xu, R.; You, C.; Wang, X.; Hao, Y. An apple NAC transcription factor enhances salt stress tolerance by modulating the ethylene response. Physiol. Plant. 2018, 164, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Duarte-Delgado, D.; Said, D.; Schoof, H.; Oyiga, B.C.; Ballvora, A. Transcriptome profiling at osmotic and ionic phases of salt stress response in bread wheat uncovers trait-specific candidate genes. BMC Plant Biol. 2020, 20, 428. [Google Scholar] [CrossRef]
- Wang, D.; Lu, X.; Chen, X.; Wang, S.; Ye, W. Temporal salt stress-induced transcriptome alterations and regulatory mechanisms revealed by PacBio long-reads RNA sequencing in Gossypium hirsutum. BMC Genom. 2020, 21, 838. [Google Scholar] [CrossRef]
- Ismail, A.; El-Sharkawy, I.; Sherif, S. Salt Stress Signals on Demand: Cellular Events in the Right Context. Int. J. Mol. Sci. 2020, 21, 3918. [Google Scholar] [CrossRef]
- Zhu, J.K. Salt and Drought Stress Signal Transduction in Plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, P.M. Sodium (Na+) homeostasis and salt tolerance of plants. Environ. Exp. Bot. 2013, 92, 19–31. [Google Scholar] [CrossRef]
- Wang, X.; Ma, X.; Li, Z.; Nie, G.; Zhang, X. Analysis of Antioxidant Enzyme Activity and Antioxidant Genes Expression During Germination of Two Different Genotypes of Lolium multiflorum Under Salt Tolerance. Protein Pept. Lett. 2016, 23, 433–441. [Google Scholar] [CrossRef]
- Wu, T.Y.; Goh, H.Z.; Azodi, C.B.; Krishnamoorthi, S.; Urano, D. Evolutionarily conserved hierarchical gene regulatory networks for plant salt stress response. Nat. Plants 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hoang, X.L.T.; Nhi, D.N.H.; Thu, N.B.A.; Thao, N.P.; Tran, L.-S.P. Transcription Factors and Their Roles in Signal Transduction in Plants under Abiotic Stresses. Curr. Genom. 2017, 18, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, W.; Ma, T.; Niu, D.; Jin, J.B.; Wang, H.; Lin, R. A pair of light signaling factors FHY3 and FAR1 regulates plant immunity by modulating chlorophyll biosynthesis. J. Integr. Plant Biol. 2016, 58, 91–103. [Google Scholar] [CrossRef]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) Function as Transcriptional Activators in Abscisic Acid Signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Deng, X. Arabidopsis FHY3 defines a key phytochrome A signaling component directly interacting with its homologous partner FAR1. EMBO J. 2014, 21, 1339–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, R.; Dai, Y.; Yuan, L.; Sun, Q.; Zhang, S.; Wang, X. A B-box zinc finger protein, MdBBX10, enhanced salt and drought stresses tolerance in Arabidopsis. Plant Mol. Biol. 2019, 99, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, Z.; Yang, Y.; Chen, X.; Chen, G. Function Annotation of an SBP-box Gene in Arabidopsis Based on Analysis of Co-expression Networks and Promoters. Int. J. Mol. Sci. 2009, 10, 116–132. [Google Scholar] [CrossRef] [Green Version]
- Bogamuwa, S.P.; Jang, J.-C. Tandem CCCH Zinc Finger Proteins in Plant Growth, Development and Stress Response. Plant Cell Physiol. 2014, 55, 1367–1375. [Google Scholar] [CrossRef]
- Kiełbowicz-Matuk, A. Involvement of plant C2H2-type zinc finger transcription factors in stress responses. Plant Sci. Int. J. Exp. Plant Biol. 2012, 185–186, 78–85. [Google Scholar] [CrossRef]
- Jin, M.; Gan, S.; Jiao, J.; He, Y.; Liu, H.; Yin, X.; Zhu, Q.; Rao, J. Genome-Wide Analysis of the bZIP Gene Family and the Role of AchnABF1 from Postharvest Kiwifruit (Actinidia chinensis cv. Hongyang) in Osmotic and Freezing Stress Adaptations. Plant Sci. 2021, 308, 110927. [Google Scholar] [CrossRef]
- Chatterjee, P.; Samaddar, S.; Niinemets, Ü.; Sa, T. Brevibacterium linens RS16 confers salt tolerance to Oryza sativa genotypes by regulating antioxidant defense and H + ATPase activity. Microbiol. Res. 2018, 215, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.C.D.; Locato, V.; Gara, L.D. Redox regulation in plant programmed cell death. Plant Cell Environ. 2012, 35, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanratana, M.; Joe, M.M.; Choudhury, A.R.; Anandham, R.; Krishnamoorthy, R.; Kim, K.; Jeon, S.; Choi, J.; Choi, J.; Sa, T. Physiological response of tomato plant to chitosan-immobilized aggregated Methylobacterium oryzae CBMB20 inoculation under salinity stress. 3 Biotech 2019, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Agathokleous, E.; Kitao, M.; Edward, J.C. Hormesis: A Compelling Platform for Sophisticated Plant Science. Trends Plant Sci. 2019, 24, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Agathokleous, E. Accumulator plants and hormesis. Environ. Pollut. 2021, 274, 116526. [Google Scholar] [CrossRef]
- Benito, B.; Haro, R.; Amtmann, A.; Cuin, T.A.; Dreyer, I. The twins K+ and Na+ in plants. J. Plant Physiol. 2014, 171, 723–731. [Google Scholar] [CrossRef]
- Lima, L.; Frosi, G.; Lopes, R.; Santos, M.G. Remobilization of leaf Na+ content and use of nonstructural carbohydrates vary depending on the time when salt stress begins in woody species. Plant Physiol. Biochem. 2020, 158, 385–395. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt-tress responses. New Phytol. 2018, 217, 523–539. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chen, X.; Lu, X.; Zhao, B.; Yang, Y.; Liu, J. Integrative analysis of transcriptome and metabolome reveal mechanism of tolerance to salt stress in oat (Avena sativa L.). Plant Physiol. Biochem. 2021, 160, 315–328. [Google Scholar] [CrossRef]
- Garriga, M.; Raddatz, N.; Véry, A.-A.; Sentenac, H.; Rubio-Meléndez, M.E.; González, W.; Dreyer, I. Cloning and functional characterization of HKT1 and AKT1 genes of Fragaria spp.-relationship to plant response to salt stress. J. Plant Physiol. 2017, 210, 9–17. [Google Scholar] [CrossRef]
- Zhang, M.; Cao, Y.; Wang, Z.; Wang, Z.Q.; Shi, J.; Liang, X.; Song, W.; Chen, Q.; Lai, J.; Jiang, C. A retrotransposon in an HKT1 family sodium transporter causes variation of leaf Na+ exclusion and salt tolerance in maize. New Phytol. 2018, 217, 1161–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kader, A.; Seidel, T.; Golldack, D.; Lindberg, S. Expressions of OsHKT1, OsHKT2, and OsVHA are differentially regulated under NaCl stress in salt-sensitive and salt-tolerant rice (Oryza sativa L.) cultivars. J. Exp. Bot. 2006, 57, 4257–4268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santa-María, G.; Rubio, F.; Dubcovsky, J.; Rodriguez-Navarro, A. The HAK1 gene of barley is a member of a large gene family and encodes a high-affinity potassium transporter. Plant Cell 1998, 9, 2281–2289. [Google Scholar]
- Li, H.; Chang, J.; Chen, H.; Wang, Z.; Gu, X.; Wei, C.; Zhang, Y.; Ma, J.; Yang, J.; Zhang, X. Exogenous Melatonin Confers Salt Stress Tolerance to Watermelon by Improving Photosynthesis and Redox Homeostasis. Front. Plant Sci. 2017, 8, 295. [Google Scholar] [CrossRef] [Green Version]
- Silveira, J.A.G.; Carvalho, F.E.L. Proteomics, photosynthesis and salt resistance in crops: An integrative view. J. Proteom. 2016, 143, 24–35. [Google Scholar] [CrossRef]
- Shabala, S.; Wu, H.; Bosea, J. Salt stress sensing and early signalling events in plant roots: Current knowledge and hypothesis. Plant. Sci. 2015, 241, 109–119. [Google Scholar] [CrossRef]
- Valmonte, G.R.; Arthur, K.; Higgins, C.M.; MacDiarmid, R.M. Calcium-Dependent Protein Kinases in Plants: Evolution, Expression and Function. Plant Cell Physiol. 2014, 55, 551–569. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ji, W.; Zhu, Y.; Gao, P.; Li, Y.; Cai, H.; Bai, X.; Guo, D. GsCBRLK, a calcium/calmodulin-binding receptor-like kinase, is a positive regulator of plant tolerance to salt and ABA stress. J. Exp. Bot. 2010, 61, 2519–2533. [Google Scholar] [CrossRef] [Green Version]
- Kader, M.A.; Lindberg, S.; Seidel, T.; Golldack, D.; Yemelyanov, V. Sodium sensing induces different changes in free cytosolic calcium concentration and pH in salt-tolerant and -sensitive rice (Oryza sativa) cultivars. Physiol. Plant 2007, 130, 99–111. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y. Unraveling salt stress signaling in plants. J. Integr. Plant Biol. 2018, 60, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Van Zelm, E.; Zhang, Y.; Testerink, C. Salt Tolerance Mechanisms of Plants. Annu. Rev. Plant Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Barrena, M.J.; Martínez-Ripoll, M.; Albert, A. Structural Biology of a Major Signaling Network that Regulates Plant Abiotic Stress: The CBL-CIPK Mediated Pathway. Int. J. Mol. Sci. 2013, 14, 5734–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javid, M.G.; Sorooshzadeh, A.; Moradi, F.; Sanavy, S.A.M.M.; Allahdadi, I. The role of phytohormones in alleviating salt stress in crop plants. Aust. J. Crop Sci. 2011, 6, 726–734. [Google Scholar]
- Peng, J.; Li, Z.; Wen, X.; Li, W.; Shi, H.; Yang, L.; Zhu, H.; Guo, H.; Yu, H. Salt-Induced Stabilization of EIN3/EIL1 Confers Salinity Tolerance by Deterring ROS Accumulation in Arabidopsis. PLoS Genet. 2014, 10, e1004664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Niu, L.; Yu, J.; Liao, W.; Xie, J.; Lv, J.; Feng, Z.; Hu, L.; Dawuda, M. The Involvement of Ethylene in Calcium-Induced Adventitious Root Formation in Cucumber under Salt Stress. Int. J. Mol. Sci. 2019, 20, 1047. [Google Scholar] [CrossRef] [Green Version]
- Jalil, S.U.; Ansari, M.I. Role of Phytohormones in Recuperating Salt Stress. In Salt Stress, Microbes, and Plant Interactions: Mechanisms and Molecular Approaches; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Gibbs, D.J.; Lee, S.C.; Isa, N.M.; Gramuglia, S.; Fukao, T.; Bassel, G.W.; Correia, C.S.; Corbineau, F.; Theodoulou, F.L.; Bailey-Serres, J. Homeostatic response to hypoxia is regulated by the N-end rule pathway in plants. Nature 2011, 479, 415–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licausi, F.; Kosmacz, M.; Weits, D.A.; Giuntoli, B.; Giorgi, F.M.; Voesenek, L.; Perata, P.; Dongen, J. Oxygen sensing in plants is mediated by an N-end rule pathway for protein destabilization. Nature 2011, 479, 419–422. [Google Scholar] [CrossRef]
- Jisha, V.; Lavanya, D.; Jyothilakshmi, V.; Axel, M.; Saivishnupriya, K.; Rajeshwari, R.; Pandey, G.K. Overexpression of an AP2/ERF Type Transcription Factor OsEREBP1 Confers Biotic and Abiotic Stress Tolerance in Rice. PLoS ONE 2015, 10, e0127831. [Google Scholar] [CrossRef]
- Corrêa, L.; Riaño-Pachón, D.; Schrago, C.G.; Santos, R.; Mueller-Roeber, B.; Vincentz, M. The Role of bZIP Transcription Factors in Green Plant Evolution: Adaptive Features Emerging from Four Founder Genes. PLoS ONE 2008, 3, e2944. [Google Scholar] [CrossRef]
- Jones, S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004, 5, 226. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, T.; Zhang, W.; Li, X. SOS3 mediates lateral root development under low salt stress through regulation of auxin redistribution and maxima in Arabidopsis. New Phytol. 2011, 189, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Yan, J.; Li, C.; Li, G.; Wu, Y.; Zheng, S. Transcription factor WRKY46 modulates the development of Arabidopsis lateral roots in osmotic/salt stress conditions via regulation of ABA signaling and auxin homeostasis. Plant J. 2015, 84, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, Y.-S.; Kim, S.-G.; Jung, J.-H.; Woo, J.-C.; Park, C.-M. Integration of auxin and salt signals by the NAC transcription factor NTM2 during seed germination in Arabidopsis. Plant Physiol. 2011, 156, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, Y.; Tanaka, M.; Ogawa, D.; Kurata, K.; Kurotani, K.-i.; Habu, Y.; Ando, T.; Sugimoto, K.; Mitsuda, N.; Katoh, E.; et al. RICE SALT SENSITIVE3 forms a ternary complex with JAZ and class-C bHLH factors and regulates jasmonate-induced gene expression and root cell elongation. Plant Cell 2013, 25, 1709–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filgueiras, C.C.; Martins, A.D.; Pereira, R.V.; Willett, D.S. The Ecology of Salicylic Acid Signaling: Primary, Secondary and Tertiary Effects with Applications in Agriculture. Int. J. Mol. Sci. 2019, 20, 5851. [Google Scholar] [CrossRef] [Green Version]
- Ahanger, M.A.; Aziz, U.; Alsahli, A.A.; Alyemeni, M.N.; Ahmad, P. Influence of Exogenous Salicylic Acid and Nitric Oxide on Growth, Photosynthesis, and Ascorbate-Glutathione Cycle in Salt Stressed Vigna angularis. Biomolecules 2020, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.L.; Li, N.; Xu, K. Crosstalk of phenylpropanoid biosynthesis with hormone signaling in Chinese cabbage is key to counteracting salt stress. Environ. Exp. Bot. 2020, 179, 104209. [Google Scholar] [CrossRef]
- Chu, X.; Wang, C.; Chen, X.; Lu, W.; Li, H.; Wang, X.; Hao, L.; Guo, X. The Cotton WRKY Gene GhWRKY41 Positively Regulates Salt and Drought Stress Tolerance in Transgenic Nicotiana benthamiana. PLoS ONE 2015, 11, e0157026. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, N.; Gong, S.; Lu, R.; Li, Y.; Li, X. Overexpression of a cotton (Gossypium hirsutum) WRKY gene, GhWRKY34, in Arabidopsis enhances salt-tolerance of the transgenic plants. Plant Physiol. Biochem. 2015, 96, 311–320. [Google Scholar] [CrossRef]
- Liang, Q.; Wu, Y.; Wang, K.; Bai, Z.; Liu, Q.; Pan, Y.; Zhang, L.; Jiang, B. Chrysanthemum WRKY gene DgWRKY5 enhances tolerance to salt stress in transgenic chrysanthemum. Sci. Rep. 2017, 7, 4799. [Google Scholar] [CrossRef] [Green Version]
- Imène, H.; Yordan, M.; Eva, i.k.; Dobrev, P.I.; Emna, G.; Franco-Zorrilla, J.M.; Irene, L.V.; Roberto, S.; André, C.; Abdelmounaim, E. The Solanum lycopersicum WRKY3 Transcription Factor SlWRKY3 Is Involved in Salt Stress Tolerance in Tomato. Front. Plant Sci. 2017, 8, 1343. [Google Scholar]
- Xu, X.; Pan, Y.; Wang, C.; Ying, Q.; Song, H.; Wang, H. Overexpression of DnWRKY11 enhanced salt and drought stress tolerance of transgenic tobacco. Biologia 2014, 69, 994–1000. [Google Scholar] [CrossRef]
- Fang, X.; Li, W.; Yuan, H.; Chen, H.; Bo, C.; Ma, Q.; Cai, R. Mutation of ZmWRKY86 confers enhanced salt stress tolerance in maize. Plant Physiol. Biochem. 2021, 167, 840–850. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, Y.; Zhen, W.; Mei, C.; Kai, L.; Yu, Y.; Shan, L.; Zhang, X.; Wang, X.; Zhang, D. Cooperation of three WRKY-domain transcription factors WRKY18, WRKY40, and WRKY60 in repressing two ABA-responsive genes ABI4 and ABI5 in Arabidopsis. J. Exp. Bot. 2012, 63, 6371–6392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, A.; Sun, H.; Hakim; Yang, X.; Zhang, X. A novel cotton WRKY gene, GhWRKY6-like, improves salt tolerance by activating the ABA signaling pathway and scavenging of reactive oxygen species. Physiol. Plant. 2017, 162, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, R.; Jiang, Q.; Sun, X.; Zhang, H.; Hu, Z. GmNAC06, a NAC domain transcription factor enhances salt stress tolerance in soybean. Plant Mol. Biol. 2021, 105, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Chen, Y.; Chen, J.; Dai, X.; Zhang, W. Physiological mechanisms underlying OsNAC5-dependent tolerance of rice plants to abiotic stress. Planta 2011, 234, 331–345. [Google Scholar] [CrossRef]
- Yao, W.; Zhao, K.; Cheng, Z.; Li, X.; Zhou, B.; Jiang, T. Transcriptome Analysis of Poplar Under Salt Stress and Over-Expression of Transcription Factor NAC57 Gene Confers Salt Tolerance in Transgenic Arabidopsis. Front. Plant Sci. 2018, 9, 1121. [Google Scholar] [CrossRef]
- Singh, P.; Mohanta, T.K.; Sinha, A.K. Unraveling the intricate nexus of molecular mechanisms governing rice root development: OsMPK3/6 and auxin-cytokinin interplay. PLoS ONE 2015, 10, e0123620. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Yang, W.; Liao, P.; Guo, Y.; Kumar, A.; Gao, W. Transcriptome analysis reveals differentially expressed ERF transcription factors associated with salt response in cotton. Plant Sci. 2019, 281, 72–81. [Google Scholar] [CrossRef]
- Zhang, M.; Smith, J.A.C.; Harberd, N.P.; Jiang, C. The regulatory roles of ethylene and reactive oxygen species (ROS) in plant salt stress responses. Plant Mol. Biol. 2016, 91, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chory, J. Phytochrome signaling mechanisms and the control of plant development. Trends in Cell Biology 2011, 21, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Tian, T.; Lin, R.; Deng, X.; Wang, H.; Li, G. Arabidopsis FHY3 and FAR1 Regulate Light-Induced myo-Inositol Biosynthesis and Oxidative Stress Responses by Transcriptional Activation of MIPS1. Mol. Plant 2016, 9, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Z.; Liu, X.; Zhu, T.; Xie, K.; Hou, Q.; Yan, T.; Niu, C.; Zhang, S.; Yang, M.; et al. ZmFAR1 and ZmABCG26 Regulated by microRNA Are Essential for Lipid Metabolism in Maize Anther. Int. J. Mol. Sci. 2021, 22, 7916. [Google Scholar] [CrossRef]
- Ma, L.; Xue, N.; Fu, X.; Zhang, H.; Li, G. Arabidopsis thaliana Far-Red Elongated Hypocotyls3 (FHY3) and Far-Red-Impaired Response1 (FAR1) modulate starch synthesis in response to light and sugar. New Phytol. 2017, 213, 1682–1696. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Wang, Y.; Wang, S. The non-DNA binding bHLH transcription factor Paclobutrazol Resistances are involved in the regulation of ABA and salt responses in Arabidopsis. Plant Physiol. Biochem. 2019, 139, 239–245. [Google Scholar] [CrossRef]
- Paola, D.D.; Zuluaga, D.L.; Sonnante, G. The miRNAome of durum wheat: Isolation and characterisation of conserved and novel microRNAs and their target genes. BMC Genom. 2016, 17, 505. [Google Scholar] [CrossRef] [Green Version]
- Wen, F.; Yue, Y.; He, T.; Gao, X.; Zhou, Z.; Long, X. Identification of miR390-TAS3-ARF pathway in response to salt stress in Helianthus tuberosus L. Gene 2020, 738, 144460. [Google Scholar] [CrossRef]
- Wang, S.; Shi, M.; Zhang, Y.; Xie, X.; Sun, P.; Fang, C.; Zhao, J. FvMYB24, a strawberry R2R3-MYB transcription factor, improved salt stress tolerance in transgenic Arabidopsis. Biochem. Biophys. Res. Commun. 2021, 569, 93–99. [Google Scholar] [CrossRef]
- Li, B.; Fan, R.; Guo, S.; Wang, P.; Zhu, X.; Fan, Y.; Chen, Y.; He, K.; Kumar, A.; Shi, J.; et al. The Arabidopsis MYB transcription factor, MYB111 modulates salt responses by regulating flavonoid biosynthesis. Environ. Exp. Bot. 2019, 166, 103807. [Google Scholar] [CrossRef]
- Dong, W.; Gao, T.; Wang, Q.; Chen, J.; Lv, J.; Song, Y. Salinity stress induces epigenetic alterations to the promoter of MsMYB4 encoding a salt-induced MYB transcription factor. Plant Physiol. Biochem. 2020, 155, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Jiang, T.; Xub, L.; Liu, H.; Mao, H.; Wang, X.; Jiao, B.; Duan, Y.; Wang, Q.; Dong, Q.; et al. A salt-stress-regulator from the Poplar R2R3 MYB family integrates the regulation of lateral root emergence and ABA signaling to mediate salt stress tolerance in Arabidopsis. Plant Physiol. Biochem. 2017, 114, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Guo, J.; Feng, R.; Jia, J.; Han, W.; Gong, H. The regulatory role of silicon on carbohydrate metabolism in Cucumis sativus L. under salt stress. Plant Soil 2016, 406, 231–249. [Google Scholar] [CrossRef]
- Khelil, A.; Menuc, T.; Ricard, B. Adaptive response to salt involving carbohydrate metabolism in leaves of a salt-sensitive tomato cultivar. Plant Physiol. Biochem. 2007, 45, 551–559. [Google Scholar] [CrossRef]
- Kholghi, M.; Toorchi, M.; Bandehagh, A.; Ostendorp, A.; Ostendorp, S.; Hanhart, P.; Kehr, J. Comparative proteomic analysis of salt-responsive proteins in canola roots by 2-DE and MALDI-TOF MS. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 227–236. [Google Scholar] [CrossRef]
- Zhang, T.; Gong, H.; Wen, X.; Lu, C. Salt stress induces a decrease in excitation energy transfer from phycobilisomes to photosystem II but an increase to photosystem I in the cyanobacterium Spirulina platensis. J. Plant Physiol. 2010, 167, 951–958. [Google Scholar] [CrossRef]
- Narayanan, S.; Tamura, P.J.; Roth, M.R.; Prasad, P.V.V.; Welti, R. Wheat leaf lipids during heat stress: I. High day and night temperatures result in major lipid alterations. Plant Cell Environ. 2016, 39, 787–803. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Li, M.; Zhang, X.; Yang, Q.; Huang, B. Up-regulation of lipid metabolism and glycine betaine synthesis are associated with choline-induced salt tolerance in halophytic seashore paspalum. Plant Cell Environ. 2020, 43, 159–173. [Google Scholar] [CrossRef]
- Hu, Q.; Cui, H.; Ma, C.; Li, Y.; Yang, C.; Wang, K.; Sun, Y. Lipidomic metabolism associated with acetic acid priming-induced salt tolerance in Carex rigescens. Plant Physiol. Biochem. 2021, 167, 665–677. [Google Scholar] [CrossRef]
- Uchendu, E.E.; Leonard, S.W.; Traber, M.G.; Reed, B.M. Vitamins C and E improve regrowth and reduce lipid peroxidation of blackberry shoot tips following cryopreservation. Plant Cell Rep. 2010, 29, 25–35. [Google Scholar] [CrossRef]
- Sattler, S.E.; Gilliland, L.U.; Magallanes-Lundback, M.; Pollard, M.; DellaPenna, D. Vitamin E Is Essential for Seed Longevity and for Preventing Lipid Peroxidation during Germination. Plant Cell 2004, 16, 1419–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, J.A.G.; Melo, A.R.B.; Viégas, R.A.; Oliveira, J.T.A. Salinity-induced effects on nitrogen assimilation related to growth in cowpea plants. Environ. Exp. Bot. 2001, 46, 171–179. [Google Scholar] [CrossRef]
- Fan, J.; Xu, J.; Zhang, W.; Amee, M.; Liu, D.; Chen, L. Salt-Induced Damage is Alleviated by Short-Term Pre-Cold Treatment in Bermudagrass (Cynodon dactylon). Plants 2019, 8, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, F.; Su, Q.; Jiang, H.; Cui, J.; He, X.; Wu, Z.; Zhang, Z.; Liu, J.; Zhao, Y. Effects of strigolactone on photosynthetic and physiological characteristics in salt-stressed rice seedlings. Sci. Rep. 2020, 10, 6183. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, F.R.; Oliveira, J.T.A.; Martins-Miranda, A.S.; Viégas, R.A.; Silveira, J.A.G. Superoxide dismutase, catalase and peroxidase activities do not confer protection against oxidative damage in salt-stressed cowpea leaves. New Phytol. 2010, 163, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Sousa, R.A.; Silva, J.; Baccan, N.; Cadore, S. Determination of metals in bottled coconut water using an inductively coupled plasma optical emission spectrometer. J. Food Compos. Anal. 2005, 18, 399–408. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2009, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, C.T.; Franz, M.; Kazi, F.; Donaldson, S.L.; Morris, Q.; Bader, G.D. Cytoscape Web: An interactive web-based network browser. Bioinformatics 2010, 2347–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panahi, B.; Hejazi, M.A. Weighted gene co-expression network analysis of the salt-responsive transcriptomes reveals novel hub genes in green halophytic microalgae Dunaliella salina. Sci. Rep. 2021, 11, 1607. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Schefe, J.H.; Lehmann, K.E.; Buschmann, I.R.; Unger, T.; Funke-Kaiser, H. Quantitative real-time RT-PCR data analysis: Current concepts and the novel “gene expression’sCTdifference” formula. J. Mol. Med. 2006, 84, 901–910. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, G.; Xiao, P.; Wang, X.; Huang, L.; Nie, G.; Li, Z.; Peng, Y.; Li, D.; Zhang, X. Comprehensive Transcriptome Analysis Uncovers Distinct Expression Patterns Associated with Early Salinity Stress in Annual Ryegrass (Lolium Multiflorum L.). Int. J. Mol. Sci. 2022, 23, 3279. https://doi.org/10.3390/ijms23063279
Feng G, Xiao P, Wang X, Huang L, Nie G, Li Z, Peng Y, Li D, Zhang X. Comprehensive Transcriptome Analysis Uncovers Distinct Expression Patterns Associated with Early Salinity Stress in Annual Ryegrass (Lolium Multiflorum L.). International Journal of Molecular Sciences. 2022; 23(6):3279. https://doi.org/10.3390/ijms23063279
Chicago/Turabian StyleFeng, Guangyan, Pengqing Xiao, Xia Wang, Linkai Huang, Gang Nie, Zhou Li, Yan Peng, Dandan Li, and Xinquan Zhang. 2022. "Comprehensive Transcriptome Analysis Uncovers Distinct Expression Patterns Associated with Early Salinity Stress in Annual Ryegrass (Lolium Multiflorum L.)" International Journal of Molecular Sciences 23, no. 6: 3279. https://doi.org/10.3390/ijms23063279
APA StyleFeng, G., Xiao, P., Wang, X., Huang, L., Nie, G., Li, Z., Peng, Y., Li, D., & Zhang, X. (2022). Comprehensive Transcriptome Analysis Uncovers Distinct Expression Patterns Associated with Early Salinity Stress in Annual Ryegrass (Lolium Multiflorum L.). International Journal of Molecular Sciences, 23(6), 3279. https://doi.org/10.3390/ijms23063279