
Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients

Abstract
1. Introduction
2. Physical Exercise
- (a)
- an increase in exercise tolerance (due to an increased cardiac and skeletal muscle strength, improved function and enhanced maximal oxygen consumption coupled with an increased capillary network);
- (b)
- an increased insulin sensitivity in adipose tissue, skeletal muscle and endothelium leading to a reduced risk of systemic insulin resistance in persons type 2 diabetes;
- (c)
- reductions in elevated body weight (due to an increased catabolism in muscles and adipose tissue) and blood pressure (due to an increased vascular density of arterioles and a reduction in systemic vascular resistance elicited by an increased release of vasodilatation promoting NO and prostacyclin from the vascular endothelium);
- (d)
- an increase in HDL and LDL cholesterol particles size and a decrease in VLDL particles size and
- (e)
- an improved response of the immune system with a delayed onset of immunesescence and reduced systemic inflammation (e.g., reduced numbers of exhausted/senescent T cells, an increased T-cell proliferative capacity, reduced blood levels of inflammatory cytokines, an increased neutrophil phagocytic activity, and an enhanced natural killer (NK) cell cytotoxic activity) [2,3,4].
3. Alzheimer’s Disease
3.1. Aethiology, Risk Factors and Diagnosis
3.2. AD Antimicrobial Aβ Peptides and Tau Protection Hypothesis of AD
- (a)
- some elderly persons with widespread Aβ products deposition and NFT, and with no signs of dementia at the time of death, do not have brain gliosis and neuroinflammation [45];
- (b)
- attenuation of pro-inflammatory immune pathways reduces Aβ products deposition [46];
- (c)
- (d)
- (e)
- (f)
- (g)
- Aβ is an anionic antimicrobial peptide (A-AMP); Aβ42 and Aβ42 products (e.g., AβO) have neurotoxic and antimicrobial effects that elicit disruption of cell and mitochondrial (MITO) membranes; in contrast to cationic AMP, A-AMPs are more likely to bind to eukaryotic, bran cell membranes, however, they are less susceptible to proteases secreted by bacteria when entrapped with AMPs and thus more effective against microbes then cationic AMP [54,55,56,57];
- (h)
- Aβ42 and their AβOs simultaneously bind Zn2+ or Cu2+ ions and these bindings enhance their specificity and affinity for microbial cell membranes; presumably, some subtypes of the post-translationally modified Aβ42 products have the lowest affinity for Zn2+ or Cu2+ ions and thus the highest affinity for brain cell membranes, are the most neurotoxic [52];
- (i)
- brains of patients with AD, have a higher level of brain microbial/vial pathogens burden (e.g., in the hippocampus), compared to normal control brain tissue, and inheritance of the APO ε4 allele increases the risk for both late-onset AD and central nervous system (CNS) infection [58];
- (j)
- (k)
- increased levels of interferon-induced transmembrane protein proteins, in human with late-onset Alzheimer’s disease or in animal models of AD, due to an increased release of pro-inflammatory cytokines from neurons and astrocytes in response to viral infections and/or ageing, have a dual effect: they attenuate viral cell entry by reducing cell membrane fluidity of viral fusion sites, thus preventing viral fusion pore formation, however, they also enhance production of Aβ40, Aβ42 and β-amyloid by binding and increasing the activity of γ-secretase [34,62];
- (l)
- infection of primary adult rodent hippocampal neuronal cultures with Herpes simplex virus 1 elicited a dual response: a transient increase in tau protein content and a long term, persistent increase in Aβ42 products deposition [63].
3.3. Brain Aβ Peptide Clearance Is Dependent on Aβ Peptide Catabolism in Peripheral Organs
4. Ageing
4.1. Ageing Is Associated with Systemic Low-Grade Chronic Inflammation
- (a)
- chronic infection with human immunodeficiency virus with accumulation of senescent CD8+ T cells responsible for increased levels of pro-inflammatory molecules [67];
- (b)
- a low PA, associated with a reduced release of cytokines and myokines from skeletal muscle cells during contraction, reduces the positive effect of these muscle molecules on attenuating systemic inflammation [67] and promotes the development of type 2 diabetes, sarcopenia, depression, and different types of dementia including Alzheimer’s disease [67];
- (c)
- an excessive increase in visceral adipose tissue (VAT) mass (due to adipocyte hypertrophia and/or hyperplasia), associated with a low PA and an inappropriate diet, promotes local hypoxia and increased activation of hypoxia-inducible factor1α, production of reactive oxygen species, and release of DAMPS followed by an increased secretion of pro-inflammatory molecules and chemokines by VAT adipocytes, endothelial cells and resident macrophages [74] which elicits an infiltration of VAT with additional immune cells, (i.e., monocytes, neutrophils, dendritic cells, B cells, T cells and NK lymphocytes, and a concomitant reduction in T regulatory cells); the overall effect is an enhanced VAT initiated inflammation and transition of this local inflammation to a SCI [67];
- (d)
- microbiome dysbiosis (e.g., changes in gut microbiota composition and gene pool, increased intestinal paracellular permeability and endotoxemia), is associated with multiple causative factors including overuse of drugs, lack of microbial exposure during or after birth, diabetes type 2, and obesity may lead to or sustain SCI [67];
- (e)
- a diet rich with processed food (with a high fat, sugar, salt and additives content and low on fresh fruits, vegetables, fibber content, micronutrients and long chain omega 3 fatty acids) is associated with SCI and microbiome dysbiosis [67]; examples are high-glycaemic-load foods common in processed food that promote activation of oxidative stress pathways that activate pro-inflammatory genes [67] and deficient intake of long chain omega 3 fatty acids, due to a diet of mainly processed foods, that reduces the human body’s ability to form inflammation attenuating molecules (e.g., resolvins, maresins and protectins) [67];
- (f)
- industrial toxicants (e.g., phthalates, bisphenols, polycyclic aromatic hydrocarbons and flame retardants1) promote inflammation, for example via oxidative stress, and increase the risk for neurodegenerative diseases, type 2 diabetes and metabolic syndrome among others [67].
4.2. Molecular Mechanisms of Inflammaging
4.3. Inflammaging and Neuroinflammation
4.4. Ageing Reduces the Efficienty of Innate and Adaptive Immunity Responses
- (a)
- an increased lifespan of macrophages due to sustained stimulation with PAMPS that bind to TLR (e.g., LPS), attenuated chemotaxis, superoxide production and expression of TLR in macrophages;
- (b)
- increased numbers of NK and NKT cells with reduced per-cell cytotoxicity and cytokine production;
- (c)
- increased levels of pro-inflammatory cytokines IL1, IL6 and TNFα in the extracellular space; and
- (d)
- reduced numbers, distribution, migration and MHC expression and signalling in dendritic cells. Immunosenescence-associated changes to the adaptive immunity were observed in B cells (reduced number and capacity for antibody production to new antigens) and T cells (increased number of memory cells, regulatory T cells, CD28 cells, release of Th1 cytokines; and reduced numbers of naive T cells, reduced CD4:CD8 ratio, reduced proliferation, release of Th1 cytokines, cytotoxicity, and T cell receptor variety) [90,91].
4.5. Ageing and Obesity Modulate the Innate and Adaptive Immune Responses
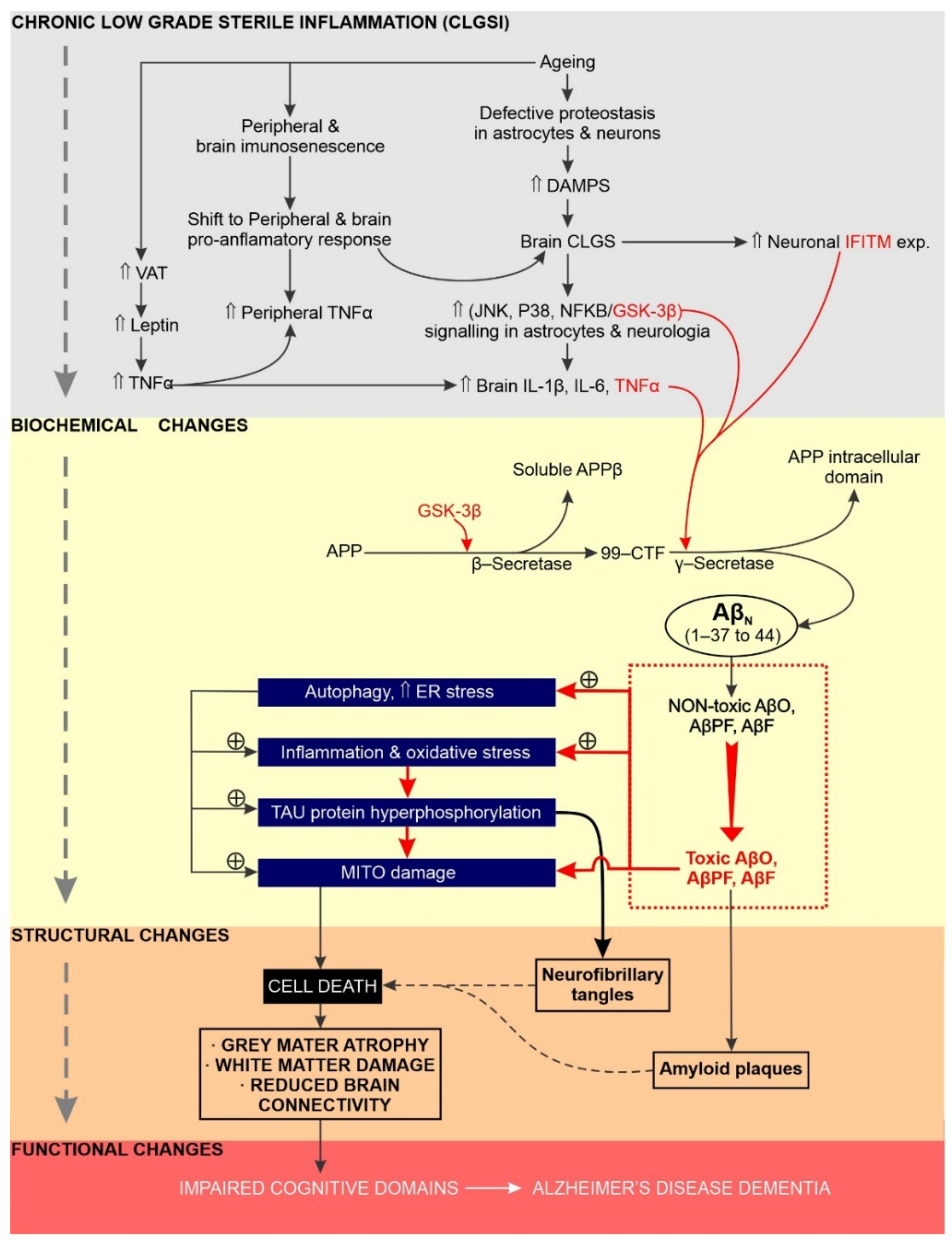
5. Chronic Neuroinflammation Has a Significant Impact on the Initiation, Sustainability and Progression of AD
5.1. Overview
5.2. Increased GSK Activity Promotes Chronic Neuroinflammation and AD Etiology
5.3. Does Excessive Endoplasmic Reticulum Stress Promote Chronic Neuroinflammation and AD?
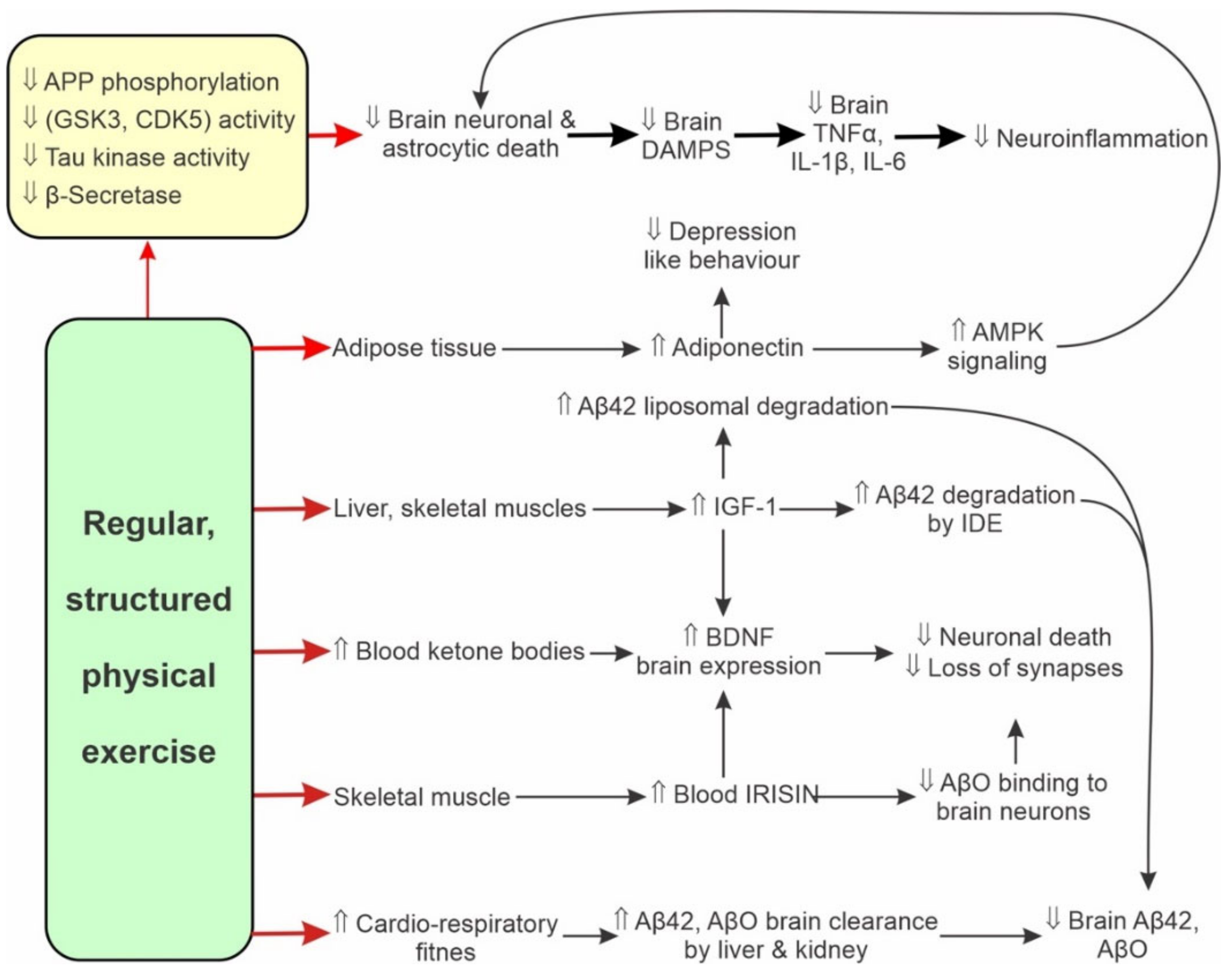
6. Physical Activity Delays Ageing-Related Changes
6.1. Human Studies
6.2. Effects of Physical Activity and Ageing on Proteostasis
7. Physical Activity Attenuates Expression of Pro-Inflammatory Markers
7.1. Overview
7.2. Animal Studies
7.3. Human Studies
8. Physical Activity Modulates Adaptive Immunity
9. Physical Activity Attenuates AD Neuroinflammation
9.1. Animal Studies
9.2. Human Studies
10. Physical Activity Attenuates AD Progression
10.1. Muscle Activity Modulates Cognition via Muscle-Brain Interactions
10.2. Human Studies on Old Age Health Subjects
10.3. Human Studies on Persons with AD
- (a)
- data stratification by sex on the effects of physical activity-related improvements in cognition;
- (b)
- measures to improve participants’ compliance with supervised training;
- (c)
- an increased number of participants;
- (d)
- extending the trail’s duration to 12 months or more;
- (e)
- robust inclusion and exclusion criteria for study participant selection; and
- (f)
- use of a standardised PA or PE regime and a comprehensive evaluation protocol for measuring physical activity-related improvements in cognition.
10.4. Animal Studies
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1RM | one repetition maximum |
| 99-CTF | 99-amino acid membrane bound C-terminal fragment |
| A-AMP | anionic antimicrobial peptide |
| Aβ40,42 | amyloid β peptide with 40 or 42 amino acid residues |
| AβF | amyloid β fibrils |
| AβO | soluble amyloid β oligomers |
| AβPF | amyloid β protofibrils |
| AD | Alzheimer’s disease |
| AKT | protein kinase B |
| AMPK | 5′ AMP-activated protein kinase |
| APH-AD | Antimicrobial Protection Hypothesis of AD |
| APO | apolipoprotein |
| APP | amyloid precursor protein |
| ATF6 | activating transcription factor 6 |
| ATP | adenosine triphosphate |
| BACE1 | β-secretase |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| CAA | cerebral amiloid pathology |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DAMPS | damage associated molecular patterns |
| DM | diabetes mellitus |
| eIF2α | eukaryotic translation initiation factor 2 subunit 1 |
| eIF2B | guanine nucleotide exchange factor (GEF) for its GTP-binding protein partner eIF2 |
| EOAD | early onset Alzheimer’s disease |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic-reticulum-associated protein degradation |
| FAD | familial Alzheimer’s disease |
| FNDC5 | fibronectin type III domain-containing protein 5 |
| GSH | glutathione |
| GSK3β | glycogen synthase kinase 3β |
| IDE | insulin-degrading enzyme |
| IFITM | interferon-induced transmembrane protein |
| IGF1 | insulin-like growth factor |
| IL | interleukin |
| iNOS | inducible nitric oxide synthases |
| JAK | cytokine-activated Janus kinase |
| JNK | c-Jun N-terminal Kinase |
| LBD | Lewy body dementia |
| LOAD | late onset Alzheimer’s disease |
| LRP1 | low density lipoprotein receptor-related protein 1 |
| LTD | long-term depression |
| LTP | long term potentiation |
| MCI | mild cognitive impairment |
| MITO | mitochondrial |
| NAD+ | nicotinamide adenine dinucleotide |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFT NLRP3 | neurofibrillary tangles NLR family pyrin domain containing 3 protein |
| NK | natural killer |
| NMDAR | N-methyl-D-aspartate receptor and ion channel |
| NRF2 | Nuclear factor-erythroid factor 2-related factor 2 |
| P38 | mitogen-activated protein kinase 38 |
| PA | physical activity |
| PAMPS | pathogen associated molecular patterns |
| PE | physical exercise |
| PCC | posterior cingulate cortex |
| PERK | protein kinase R-like endoplasmic reticulum kinase |
| PGC1α | peroxisome proliferator-activated receptor-γ coactivator |
| PI3K | phosphatidylinositol 3-kinase |
| PKA | protein kinase A |
| PKB | protein kinase B |
| PKC | protein kinase C |
| PRRS | pattern recognition receptors |
| PS1 | presenilin-1 |
| PS2 | presenilin-2 |
| RAGE | receptor for advanced glycation end products |
| ROS | reactive oxidative species |
| SASP | senescence-associated secretory phenotype |
| SAT | subcutaneous adipose tissue |
| SCI | systemic low-grade chronic inflammation |
| STAT3/5 | signal transducers and activators of transcription 3 and 5 |
| TLR | toll-like receptor |
| TNFα | tumour necrosis factor α |
| UPR | unfolded protein response |
| VAD | vascular dementia |
| VAT | visceral adipose tissue |
| VEGF | vascular endothelial growth factor |
References
- Caspersen, C.J.; Powell, K.E.; Christenson, G.M. Physical activity, exercise, and physical fitness: Definitions and distinctions for health-related research. Public Health Rep. 1985, 100, 126–131. [Google Scholar]
- Myers, J. Exercise and Cardiovascular Health. Circulation 2003, 107, e2–e5. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C.; Wentz, L.M. The compelling link between physical activity and the body’s defense system. J. Sport Health Sci. 2019, 8, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Nystoriak, M.A.; Bhatnagar, A. Cardiovascular Effects and Benefits of Exercise. Front. Cardiovasc. Med. 2018, 5, 135. [Google Scholar] [CrossRef]
- Timmons, J.F.; Minnock, D.; Hone, M.; Cogan, K.E.; Murphy, J.C.; Egan, B. Comparison of time-matched aerobic, resistance, or concurrent exercise training in older adults. Scand. J. Med. Sci. Sports 2018, 28, 2272–2283. [Google Scholar] [CrossRef] [PubMed]
- Leal, L.G.; Lopes, M.A.; Batista, M.L., Jr. Physical Exercise-Induced Myokines and Muscle-Adipose Tissue Crosstalk: A Review of Current Knowledge and the Implications for Health and Metabolic Diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Nagy, E.E.; Moreau, K.L. Aerobic exercise training and vascular function with ageing in healthy men and women. J. Physiol. 2019, 597, 4901–4914. [Google Scholar] [CrossRef]
- Garcia-Morales, V.; Gonzalez-Acedo, A.; Melguizo-Rodriguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodriguez, J.J. Current understanding of the physiopathology, diagnosis and therapeutic approach to alzheimer’s disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and management. Med Clin. North Am. 2019, 103, 263–293. [Google Scholar] [CrossRef]
- Meng, Q.; Lin, M.-S.; Tzeng, I.-S. Relationship Between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef]
- Barnes, J.N. Exercise, cognitive function, and aging. Adv. Physiol. Educ. 2015, 39, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Chapman, S.B.; Aslan, S.; Spence, J.S.; DeFina, L.F.; Keebler, M.W.; Didehbani, N.; Lu, H. Shorter term aerobic exercise improves brain, cognition, and cardiovascular fitness in aging. Front. Aging Neurosci. 2013, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Duzel, E.; van Praag, H.; Sendtner, M. Can physical exercise in old age improve memory and hippocampal function? Brain 2016, 139, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Prakash, R.S.; Voss, M.W.; Chaddock, L.; Hu, L.; Morris, K.S.; White, S.M.; Wójcicki, T.R.; McAuley, E.; Kramer, A.F. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus 2009, 19, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, I. Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure 2020, 82, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Capelli, V.; Husain, M. Cognition and dementia in older patients with epilepsy. Brain 2018, 141, 1592–1608. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Z.-R.; Zhang, H.-W.; Tseng, C.-H.; Peng, H.-C.; Kok, V.C.; Li, G.P.; Hsiung, C.A.; Hsu, C.-Y. Late-onset epilepsy and subsequent increased risk of dementia. Aging 2021, 13, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Won, E.; Kim, Y.-K. Neuroinflammation-Associated Alterations of the Brain as Potential Neural Biomarkers in Anxiety Disorders. Int. J. Mol. Sci. 2020, 21, 6546. [Google Scholar] [CrossRef]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Khan, T.K. An Algorithm for Preclinical Diagnosis of Alzheimer’s Disease. Front. Neurosci. 2018, 12, 275. [Google Scholar] [CrossRef]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Reiman, E.M. Characterizing the Preclinical Stages of Alzheimer’s Disease and the Prospect of Presymptomatic Intervention. J. Alzheimer’s Dis. 2013, 33, S405–S416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tang, W.; Chao, F.-L.; Zhou, C.-N.; Jiang, L.; Zhang, Y.; Liang, X.; Tang, J.; Qi, Y.-Q.; Yang, H.; et al. Four-month treadmill exercise prevents the decline in spatial learning and memory abilities and the loss of spinophilin-immunoreactive puncta in the hippocampus of APP/PS1 transgenic mice. Neurobiol. Dis. 2019, 136, 104723. [Google Scholar] [CrossRef] [PubMed]
- Ströhle, A.; Schmidt, D.K.; Schultz, F.; Fricke, N.; Staden, T.; Hellweg, R.; Priller, J.; Rapp, M.A.; Rieckmann, N. Drug and Exercise Treatment of Alzheimer Disease and Mild Cognitive Impairment: A Systematic Review and Meta-Analysis of Effects on Cognition in Randomized Controlled Trials. Am. J. Geriatr. Psychiatry 2015, 23, 1234–1249. [Google Scholar] [CrossRef]
- Tabei, K.-I.; Satoh, M.; Ogawa, J.-I.; Tokita, T.; Nakaguchi, N.; Nakao, K.; Kida, H.; Tomimoto, H. Cognitive Function and Brain Atrophy Predict Non-pharmacological Efficacy in Dementia: The Mihama-Kiho Scan Project2. Front. Aging Neurosci. 2018, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. cAMP, cGMP and Amyloid β: Three Ideal Partners for Memory Formation. Trends Neurosci. 2018, 41, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Hoogmartens, J.; Cacace, R.; Van Broeckhoven, C. Insight into the genetic etiology of Alzheimer’s disease: A comprehensive review of the role of rare variants. Alzheimer’s Dementia Diagn. Assess. Dis. Monit. 2021, 13, e12155. [Google Scholar] [CrossRef]
- Vitek, M.P.; Araujo, J.A.; Fossel, M.; Greenberg, B.D.; Howell, G.R.; Rizzo, S.J.S.; Seyfried, N.T.; Tenner, A.J.; Territo, P.R.; Windisch, M.; et al. Translational animal models for Alzheimer’s disease: An Alzheimer’s Association Business Consortium Think Tank. Alzheimer’s Dementia: Transl. Res. Clin. Interv. 2020, 6, e12114. [Google Scholar] [CrossRef]
- Rabinovici, G.D. Late-onset Alzheimer Disease. Contin. Lifelong Learn. Neurol. 2019, 25, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Early-onset Alzheimer Disease and Its Variants. Contin. Lifelong Learn. Neurol. 2019, 25, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Growdon, J.H. Is Alzheimer’s Disease Risk Modifiable? J. Alzheimer’s Dis. 2019, 67, 795–819. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.Y.; Yan, R. Activity of Alzheimer’s γ-secretase is linked to changes of interferon-induced transmembrane proteins (IFITM) in innate immunity. Mol. Neurodegener. 2020, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Eimer, W.A.; Kumar, D.K.V.; Shanmugam, N.K.N.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56.e53–63.e53. [Google Scholar] [CrossRef]
- Luna, S.; Cameron, D.J.; Ethell, D. Amyloid-β and APP Deficiencies Cause Severe Cerebrovascular Defects: Important Work for an Old Villain. PLoS ONE 2013, 8, e75052. [Google Scholar] [CrossRef]
- Dominguez, D.; Tournoy, J.; Hartmann, D.; Huth, T.; Cryns, K.; Deforce, S.; Serneels, L.; Camacho, I.E.; Marjaux, E.; Craessaerts, K.; et al. Phenotypic and Biochemical Analyses of BACE1- and BACE2-deficient Mice. J. Biol. Chem. 2005, 280, 30797–30806. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients With Mild Alzheimer DiseaseA Randomized Controlled Trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef]
- Chu, H.; Pazgier, M.; Jung, G.; Nuccio, S.-P.; Castillo, P.A.; de Jong, M.F.; Winter, M.G.; Winter, S.E.; Wehkamp, J.; Shen, B.; et al. Human α-Defensin 6 Promotes Mucosal Innate Immunity Through Self-Assembled Peptide Nanonets. Science 2012, 337, 477–481. [Google Scholar] [CrossRef]
- Tsai, P.-W.; Yang, C.-Y.; Chang, H.-T.; Lan, C.-Y. Characterizing the Role of Cell-Wall β-1,3-Exoglucanase Xog1p in Candida albicans Adhesion by the Human Antimicrobial Peptide LL-37. PLoS ONE 2011, 6, e21394. [Google Scholar] [CrossRef]
- Wallin, C.; Jarvet, J.; Biverstål, H.; Wärmländer, S.; Danielsson, J.; Gräslund, A.; Abelein, A. Metal ion coordination delays amyloid-β peptide self-assembly by forming an aggregation–inert complex. J. Biol. Chem. 2020, 295, 7224–7234. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Pittman, J.; Zerweck, J.; Venkata, B.S.; Moore, P.C.; Sachleben, J.R.; Meredith, S.C. β-Amyloid aggregation and heterogeneous nucleation. Protein Sci. 2019, 28, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, J.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Stein, T.; Tai, H.-C.; Icardo, O.D.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Reed, Q.W.; Cramer, P.E.; Landreth, G.E. Deletion of CD14 Attenuates Alzheimer’s Disease Pathology by Influencing the Brain’s Inflammatory Milieu. J. Neurosci. 2010, 30, 15369–15373. [Google Scholar] [CrossRef]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer’s Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef]
- Chen, V.C.-H.; Wu, S.-I.; Huang, K.-Y.; Yang, Y.-H.; Kuo-You, H.; Liang, H.-Y.; Huang, K.-L.; Gossop, M. Herpes Zoster and Dementia: A nationwide population-based cohort study. J. Clin. Psychiatry 2018, 79, 16m11312. [Google Scholar] [CrossRef]
- Tzeng, N.-S.; Chung, C.-H.; Lin, F.-H.; Chiang, C.-P.; Yeh, C.-B.; Huang, S.-Y.; Lu, R.-B.; Chang, H.-A.; Kao, Y.-C.; Yeh, H.-W.; et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections—a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Brown, C.M.; Colton, C.A. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 2009, 30, 1350–1360. [Google Scholar] [CrossRef]
- Trumble, B.C.; Stieglitz, J.; Blackwell, A.D.; Allayee, H.; Beheim, B.; Finch, C.E.; Gurven, M.; Kaplan, H. Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. 2016, 31, 1508–1515. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimer’s Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic Protein-Membrane Interactions: Mechanistic Insight from Model Systems. Angew. Chem. Int. Ed. Engl. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.; Phoenix, D.A. Anionic Antimicrobial Peptides from Eukaryotic Organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of Copper Interactions with Alzheimer Amyloid β Peptides: Identification of an attomolar-affinity copper binding site on amyloid beta1-42. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial Dysfunction: Different Routes to Alzheimer’s Disease Therapy. Oxidative Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef] [PubMed]
- Urosevic, N.; Martins, R.N. Infection and Alzheimer’s Disease: The APOE ε4 Connection and Lipid Metabolism. J. Alzheimer’s Dis. 2008, 13, 421–435. [Google Scholar] [CrossRef]
- Felsky, D.; Patrick, E.; Schneider, J.A.; Mostafavi, S.; Gaiteri, C.; Patsopoulos, N.; Bennett, D.A.; De Jager, P.L. Polygenic analysis of inflammatory disease variants and effects on microglia in the aging brain. Mol. Neurodegener. 2018, 13, 38. [Google Scholar] [CrossRef]
- Pimenova, A.A.; Raj, T.; Goate, A.M. Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 300–310. [Google Scholar] [CrossRef]
- Afridi, R.; Lee, W.-H.; Suk, K. Microglia Gone Awry: Linking Immunometabolism to Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.-Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Powell-Doherty, R.D.; Abbott, A.R.N.; Nelson, L.A.; Bertke, A.S. Amyloid-β and p-Tau Anti-Threat Response to Herpes Simplex Virus 1 Infection in Primary Adult Murine Hippocampal Neurons. J. Virol. 2020, 94, e01874-19. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Park, T.J.; Huang, X.; Kim, M.O. Abnormal amyloid beta metabolism in systemic abnormalities and Alzheimer’s pathology: Insights and therapeutic approaches from periphery. Ageing Res. Rev. 2021, 71, 101451. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 2021, 91, 740–755. [Google Scholar] [CrossRef]
- Hughes, C.; Choi, M.L.; Yi, J.-H.; Kim, S.-C.; Drews, A.; George-Hyslop, P.S.; Bryant, C.; Gandhi, S.; Cho, K.; Klenerman, D. Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun. Biol. 2020, 3, 79. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.-I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Sendama, W. The effect of ageing on the resolution of inflammation. Ageing Res. Rev. 2020, 57, 101000. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B.; Paganelli, R. Aging, Obesity, and Inflammatory Age-Related Diseases. Front. Immunol. 2017, 8, 1745. [Google Scholar] [CrossRef] [PubMed]
- Orellana, A.M.; Vasconcelos, A.R.; Leite, J.A.; Lima, L.D.S.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.; Scavone, C. Age-related neuroinflammation and changes in AKT-GSK-3β and WNT/ β-CATENIN signaling in rat hippocampus. Aging 2015, 7, 1094–1111. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Lazarus, D.D.; Moldawer, L.L.; Lowry, S.F. Insulin-like growth factor-1 activity is inhibited by interleukin-1 alpha, tumor necrosis factor-alpha, and interleukin-6. Lymphokine Cytokine Res. 1993, 12, 219–223. [Google Scholar]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.-Y.; Baldwin, A.S., Jr. NF-kappa B-Induced Loss of MyoD Messenger RNA: Possible Role in Muscle Decay and Cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- Hahn, W.S.; Kuzmicic, J.; Burrill, J.S.; Donoghue, M.A.; Foncea, R.; Jensen, M.D.; Lavandero, S.; Arriaga, E.A.; Bernlohr, D.A. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1033–E1045. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; Van Der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Espinal, M.P.; Muñoz, J.P.; Richaud-Patin, Y.; Carasa, I.F.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Piacentini, R.; Puma, D.D.L.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 2017, 65, 1302–1316. [Google Scholar] [CrossRef]
- Paouri, E.; Tzara, O.; Zenelak, S.; Georgopoulos, S. Genetic Deletion of Tumor Necrosis Factor-α Attenuates Amyloid-β Production and Decreases Amyloid Plaque Formation and Glial Response in the 5XFAD Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 165–181. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Sun, L.-N.; Qi, J.-S.; Gao, R. Physical exercise reserved amyloid-beta induced brain dysfunctions by regulating hippocampal neurogenesis and inflammatory response via MAPK signaling. Brain Res. 2018, 1697, 1–9. [Google Scholar] [CrossRef]
- Ray, D.; Yung, R. Immune senescence, epigenetics and autoimmunity. Clin. Immunol. 2018, 196, 59–63. [Google Scholar] [CrossRef]
- Bauer, M.E.; De la Fuente, M. The role of oxidative and inflammatory stress and persistent viral infections in immunosenescence. Mech. Ageing Dev. 2016, 158, 27–37. [Google Scholar] [CrossRef]
- Akha, A.A.S. Aging and the immune system: An overview. J. Immunol. Methods 2018, 463, 21–26. [Google Scholar] [CrossRef]
- Łuc, M.; Misiak, B.; Pawłowski, M.; Stanczykiewicz, B.; Zabłocka, A.; Szcześniak, D.; Pałęga, A.; Rymaszewska, J. Gut microbiota in dementia. Critical review of novel findings and their potential application. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 104, 110039. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Ageing and obesity similarly impair antibody responses. Clin. Exp. Immunol. 2017, 187, 64–70. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef]
- Yarlagadda, A.; Alfson, E.; Clayton, A.H. The Blood Brain Barrier and the Role of Cytokines in Neuropsychiatry. Psychiatry 2009, 6, 18–22. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. eBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.M.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- He, N.; Jin, W.-L.; Lok, K.-H.; Wang, Y.; Yin, M.; Wang, Z.-J. Amyloid-β1–42 oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013, 4, e924. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-Y.; Ding, Y.-X.; Bian, G.-L.; Chen, L.-W.; Yao, X.-Y.; Lin, Y.-B.; Wang, Z.; Chen, B.-Y. Reactive Astrocytes Display Pro-inflammatory Adaptability with Modulation of Notch-PI3K-AKT Signaling Pathway Under Inflammatory Stimulation. Neuroscience 2020, 440, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- García-González, P.; Cabral-Miranda, F.; Hetz, C.; Osorio, F. Interplay Between the Unfolded Protein Response and Immune Function in the Development of Neurodegenerative Diseases. Front. Immunol. 2018, 9, 2541. [Google Scholar] [CrossRef]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef]
- Füger, P.; Hefendehl, J.; Veeraraghavalu, K.; Wendeln, A.-C.; Schlosser, C.; Obermüller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2020, 155, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, J.; Malm, T.; Goldsteins, G. Glycogen Synthase Kinase-3β: A Mediator of Inflammation in Alzheimer’s Disease? Int. J. Alzheimer’s Dis. 2011, 2011, 129753. [Google Scholar] [CrossRef]
- Martin, S.A.; Souder, D.C.; Miller, K.N.; Clark, J.P.; Sagar, A.K.; Eliceiri, K.; Puglielli, L.; Beasley, T.M.; Anderson, R.M. GSK3β Regulates Brain Energy Metabolism. Cell Rep. 2018, 23, 1922.e4–1931.e4. [Google Scholar] [CrossRef] [PubMed]
- Souder, D.C.; Anderson, R.M. An expanding GSK3 network: Implications for aging research. GeroScience 2019, 41, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. et Biophys. Acta 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. Gsk-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Tomioka, S.; Sorimachi, H.; Saido, T.C.; Maruyama, K.; Okuyama, A.; Fujisawa-Sehara, A.; Ohno, S.; Suzuki, K.; Ishiura, S. Membrane-anchored metalloprotease mdc9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem. J. 1999, 343 Pt 2, 371–375. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, Y.; Zhao, B. Roles of Glycogen Synthase Kinase 3 in Alzheimer’s Disease. Curr. Alzheimer Res. 2012, 9, 864–879. [Google Scholar] [CrossRef]
- Uemura, K.; Kuzuya, A.; Shimozono, Y.; Aoyagi, N.; Ando, K.; Shimohama, S.; Kinoshita, A. GSK3β Activity Modifies the Localization and Function of Presenilin 1. J. Biol. Chem. 2007, 282, 15823–15832. [Google Scholar] [CrossRef]
- Sirerol-Piquer, M.; Gomez-Ramos, P.; Hernandez, F.; Perez, M.; Morán, M.A.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J.; García-Verdugo, J.M. GSK3β overexpression induces neuronal death and a depletion of the neurogenic niches in the dentate gyrus. Hippocampus 2011, 21, 910–922. [Google Scholar] [CrossRef]
- Giese, K.P. GSK-3: A key player in neurodegeneration and memory. IUBMB Life 2009, 61, 516–521. [Google Scholar] [CrossRef]
- Hooper, C.; Markevich, V.; Plattner, F.; Killick, R.; Schofield, E.; Engel, T.; Hernandez, F.; Anderton, B.; Rosenblum, K.; Bliss, T.; et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 2007, 25, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.; Zhang, J.; Pu, M.; Zhou, X.; Dong, L.; Mao, X.; Shi, G.; Zou, J.; Wu, J.; Jiang, D.; et al. Modulation of GSK-3β/β-Catenin Signaling Contributes to Learning and Memory Impairment in a Rat Model of Depression. Int. J. Neuropsychopharmacol. 2018, 21, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J. Regulation of cell–cell adhesion by the cadherin–catenin complex. Biochem. Soc. Trans. 2008, 36, 149–155. [Google Scholar] [CrossRef]
- Vallée, A.; LeCarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2011, 15, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sato, S.; Murayama, O.; Murayama, M.; Park, J.M.; Yamaguchi, H.; Takashima, A. Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci. Lett. 2002, 321, 61–64. [Google Scholar] [CrossRef]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. Differential Regulation of STAT Family Members by Glycogen Synthase Kinase-3. J. Biol. Chem. 2008, 283, 21934–21944. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gannon, M.; Chen, Y.; Yan, S.; Zhang, S.; Feng, W.; Tao, J.; Sha, B.; Liu, Z.; Saito, T.; et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Transl. Med. 2020, 12, eaay6931. [Google Scholar] [CrossRef]
- Jolivalt, C.; Lee, C.; Beiswenger, K.; Smith, J.; Orlov, M.; Torrance, M.; Masliah, E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: Parallels with Alzheimer’s disease and correction by insulin. J. Neurosci. Res. 2008, 86, 3265–3274. [Google Scholar] [CrossRef] [PubMed]
- Kurochkin, I.V.; Goto, S. Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef]
- Qiu, W.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading Enzyme Regulates Extracellular Levels of Amyloid β-Protein by Degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid -protein, and the -amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Magdesian, M.H.; Carvalho, M.M.V.F.; Mendes, F.A.; Saraiva, L.M.; Juliano, M.A.; Juliano, L.; Garcia-Abreu, J.; Ferreira, S.T. Amyloid-β Binds to the Extracellular Cysteine-rich Domain of Frizzled and Inhibits Wnt/β-Catenin Signaling. J. Biol. Chem. 2008, 283, 9359–9368. [Google Scholar] [CrossRef]
- Hernández, F.; de Barreda, E.G.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Hernández, F.; Gómez-Ramos, A.; Smith, M.; Perry, G.; Avila, J. Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. Eur. J. Biochem. 2002, 269, 1484–1489. [Google Scholar] [CrossRef]
- Zhao, Q.-F.; Yu, J.-T.; Tan, L. S-Nitrosylation in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 268–280. [Google Scholar] [CrossRef]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef]
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol. Dis. 2006, 23, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef]
- Ferreiro, E.; Oliveira, C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-? peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Ferreiro, E.; Pereira, C.M.F.; Oliveira, C. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: Involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 2008, 155, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.O.; Ferreiro, E.; Martins, I.; Santana, I.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M. Amyloid β-induced ER stress is enhanced under mitochondrial dysfunction conditions. Neurobiol. Aging 2012, 33, 824.e5–824.e16. [Google Scholar] [CrossRef]
- Ohno, M. PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer’s disease. Brain Res. Bull. 2018, 141, 72–78. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the Translation Initiation Factor eIF2α Increases BACE1 Levels and Promotes Amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef]
- Moreno-Agostino, D.; Daskalopoulou, C.; Wu, Y.-T.; Koukounari, A.; Haro, J.M.; Tyrovolas, S.; Panagiotakos, D.B.; Prince, M.; Prina, A.M. The impact of physical activity on healthy ageing trajectories: Evidence from eight cohort studies. Int. J. Behav. Nutr. Phys. Act. 2020, 17, 92. [Google Scholar] [CrossRef] [PubMed]
- Boraxbekk, C.-J.; Salami, A.; Wåhlin, A.; Nyberg, L. Physical activity over a decade modifies age-related decline in perfusion, gray matter volume, and functional connectivity of the posterior default-mode network—A multimodal approach. NeuroImage 2016, 131, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.M.; Petrov, M.E.; Lee, R.E. Aerobic physical activity to improve memory and executive function in sedentary adults without cognitive impairment: A systematic review and meta-analysis. Prev. Med. Rep. 2021, 23, 101496. [Google Scholar] [CrossRef]
- Angevaren, M.; Aufdemkampe, G.; Verhaar, H.J.J.; Aleman, A.; Vanhees, L. Physical activity and enhanced fitness to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst. Rev. 2008, 16, CD005381. [Google Scholar] [CrossRef]
- Lee, P.G.; Jackson, E.A.; Richardson, C.R. Exercise Prescriptions in Older Adults. Am. Fam. Physician 2017, 95, 425–432. [Google Scholar] [PubMed]
- Norton, S.; Matthews, F.; E Barnes, D.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Andreotti, D.Z.; Silva, J.D.N.; Matumoto, A.M.; Orellana, A.M.; De Mello, P.S.; Kawamoto, E.M. Effects of Physical Exercise on Autophagy and Apoptosis in Aged Brain: Human and Animal Studies. Front. Nutr. 2020, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Macian, F. Autophagy and the immune function in aging. Curr. Opin. Immunol. 2014, 29, 97–104. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef]
- De Mello, N.P.; Orellana, A.M.; Mazucanti, C.H.; de Morais Lima, G.; Scavone, C.; Kawamoto, E.M. Insulin and Autophagy in Neurodegeneration. Front. Neurosci. 2019, 13, 491. [Google Scholar] [CrossRef] [PubMed]
- Batatinha, H.A.P.; Diniz, T.A.; de Souza Teixeira, A.A.; Krüger, K.; Rosa-Neto, J.C. Regulation of autophagy as a therapy for immunosenescence-driven cancer and neurodegenerative diseases: The role of exercise. J. Cell. Physiol. 2019, 234, 14883–14895. [Google Scholar] [CrossRef] [PubMed]
- Mooren, F.C.; Krüger, K. Exercise, Autophagy, and Apoptosis. Prog. Mol. Biol. Transl. Sci. 2015, 135, 407–422. [Google Scholar] [CrossRef]
- Møller, A.B.; Vendelbo, M.H.; Christensen, B.; Clasen, B.F.; Bak, A.M.; Jørgensen, J.O.L.; Moller, N.; Jessen, N. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J. Appl. Physiol. 2015, 118, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, C.; Jamart, C.; Benoit, N.; Naslain, D.; Prémont, C.; Prévet, J.; Van Thienen, R.; Deldicque, L.; Francaux, M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 2015, 29, 3515–3526. [Google Scholar] [CrossRef]
- Tachtsis, B.; Smiles, W.; Lane, S.C.; Hawley, J.; Camera, D.M. Acute Endurance Exercise Induces Nuclear p53 Abundance in Human Skeletal Muscle. Front. Physiol. 2016, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Estébanez, B.; de Paz, J.A.; Cuevas, M.J.; González-Gallego, J. Endoplasmic Reticulum Unfolded Protein Response, Aging and Exercise: An Update. Front. Physiol. 2018, 9, 1744. [Google Scholar] [CrossRef] [PubMed]
- Passos, E.; Ascensão, A.; Martins, M.J.; Magalhães, J. Endoplasmic Reticulum Stress Response in Non-alcoholic Steatohepatitis: The Possible Role of Physical Exercise. Metabolism 2015, 64, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Smiles, W.; Hawley, J.A.; Camera, D. Effects of skeletal muscle energy availability on protein turnover responses to exercise. J. Exp. Biol. 2016, 219, 214–225. [Google Scholar] [CrossRef][Green Version]
- Metsios, G.S.; Moe, R.H.; Kitas, G.D. Exercise and inflammation. Best Pr. Res. Clin. Rheumatol. 2020, 34, 101504. [Google Scholar] [CrossRef]
- Dethlefsen, C.; Pedersen, K.S.; Hojman, P. Every exercise bout matters: Linking systemic exercise responses to breast cancer control. Breast Cancer Res. Treat. 2017, 162, 399–408. [Google Scholar] [CrossRef]
- Verheggen, R.J.H.M.; Maessen, M.F.; Green, D.J.; Hermus, A.R.M.M.; Hopman, M.T.E.; Thijssen, D.H.T. A systematic review and meta-analysis on the effects of exercise training versus hypocaloric diet: Distinct effects on body weight and visceral adipose tissue. Obes. Rev. 2016, 17, 664–690. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Irrcher, I.; Ljubicic, V.; Joseph, A.-M. Coordination of metabolic plasticity in skeletal muscle. J. Exp. Biol. 2006, 209, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef]
- Paolucci, E.M.; Loukov, D.; Bowdish, D.M.E.; Heisz, J.J. Exercise reduces depression and inflammation but intensity matters. Biol. Psychol. 2018, 133, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, P.; Dicarlo, M.; Zerlotin, R.; Zecca, C.; Dell’Abate, M.; Buccoliero, C.; Logroscino, G.; Colucci, S.; Grano, M. FNDC5/Irisin System in Neuroinflammation and Neurodegenerative Diseases: Update and Novel Perspective. Int. J. Mol. Sci. 2021, 22, 1605. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Tsai, C.-L.; Sun, H.-S.; Kuo, Y.-M.; Pai, M.-C. The Role of Physical Fitness in Cognitive-Related Biomarkers in Persons at Genetic Risk of Familial Alzheimer’s Disease. J. Clin. Med. 2019, 8, 1639. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Kuzuya, A.; Tanigawa, K.; Araki, M.; Kawai, R.; Ma, B.; Sasakura, Y.; Maesako, M.; Tashiro, Y.; Miyamoto, M.; et al. Fibronectin type III domain-containing protein 5 interacts with APP and decreases amyloid β production in Alzheimer’s disease. Mol. Brain 2018, 11, 61. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise Induces Hippocampal BDNF through a PGC-1α/FNDC5 Pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal Muscle PGC-1α1 Modulates Kynurenine Metabolism and Mediates Resilience to Stress-Induced Depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef]
- Schlittler, M.; Goiny, M.; Agudelo, L.Z.; Venckunas, T.; Brazaitis, M.; Skurvydas, A.; Kamandulis, S.; Ruas, J.; Erhardt, S.; Westerblad, H.; et al. Endurance exercise increases skeletal muscle kynurenine aminotransferases and plasma kynurenic acid in humans. Am. J. Physiol. Cell Physiol. 2016, 310, C836–C840. [Google Scholar] [CrossRef] [PubMed]
- Karstoft, K.; Pedersen, B.K. Exercise and type 2 diabetes: Focus on metabolism and inflammation. Immunol. Cell Biol. 2016, 94, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.E.; Pasiakos, S.M.; Blesso, C.N.; Fussell, M.A.; Rodriguez, N.R. Divergent Roles of Inflammation in Skeletal Muscle Recovery From Injury. Front. Physiol. 2020, 11, 87. [Google Scholar] [CrossRef]
- Kang, E.-B.; Kwon, I.-S.; Koo, J.-H.; Kim, E.-J.; Kim, C.-H.; Lee, J.; Yang, C.-H.; Lee, Y.-I.; Cho, I.-H.; Cho, J.-Y. Treadmill exercise represses neuronal cell death and inflammation during Aβ-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis 2013, 18, 1332–1347. [Google Scholar] [CrossRef]
- Cerqueira, É.; Marinho, D.A.; Neiva, H.P.; Lourenço, O. Inflammatory Effects of High and Moderate Intensity Exercise—A Systematic Review. Front. Physiol. 2020, 10, 1550. [Google Scholar] [CrossRef]
- Ihalainen, J.K.; Schumann, M.; Eklund, D.; Hämäläinen, M.; Moilanen, E.; Paulsen, G.; Häkkinen, K.; Mero, A.A. Combined aerobic and resistance training decreases inflammation markers in healthy men. Scand. J. Med. Sci. Sports 2018, 28, 40–47. [Google Scholar] [CrossRef]
- Cabanas-Sánchez, V.; Guallar-Castillón, P.; Higueras-Fresnillo, S.; García-Esquinas, E.; Rodríguez-Artalejo, F.; Martinez-Gomez, D. Physical Activity, Sitting Time, and Mortality From Inflammatory Diseases in Older Adults. Front. Physiol. 2018, 9, 898. [Google Scholar] [CrossRef]
- Beavers, K.M.; Ambrosius, W.T.; Nicklas, B.J.; Rejeski, W.J. Independent and Combined Effects of Physical Activity and Weight Loss on Inflammatory Biomarkers in Overweight and Obese Older Adults. J. Am. Geriatr. Soc. 2013, 61, 1089–1094. [Google Scholar] [CrossRef]
- Beavers, K.M.; Hsu, F.-C.; Isom, S.; Kritchevsky, S.B.; Church, T.; Goodpaster, B.; Pahor, M.; Nicklas, B.J. Long-Term Physical Activity and Inflammatory Biomarkers in Older Adults. Med. Sci. Sports Exerc. 2010, 42, 2189–2196. [Google Scholar] [CrossRef]
- Nicklas, B.J.; Hsu, F.-C.; Brinkley, T.J.; Church, T.S.; Goodpaster, B.H.; Kritchevsky, S.; Pahor, M. Exercise Training and Plasma C-Reactive Protein and Interleukin-6 in Elderly People. J. Am. Geriatr. Soc. 2008, 56, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Qiu, P.; Xia, R.; Lin, H.; Ye, B.; Tao, J.; Chen, L. Effect of Aerobic Exercise on Inflammatory Markers in Healthy Middle-Aged and Older Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Aging Neurosci. 2019, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Sardeli, A.V.; Tomeleri, C.M.; Cyrino, E.S.; Fernhall, B.; Cavaglieri, C.R.; Chacon-Mikahil, M.P.T. Effect of resistance training on inflammatory markers of older adults: A meta-analysis. Exp. Gerontol. 2018, 111, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Cronin, O.; Keohane, D.M.; Molloy, M.G.; Shanahan, F. The effect of exercise interventions on inflammatory biomarkers in healthy, physically inactive subjects: A systematic review. QJM 2017, 110, 629–637. [Google Scholar] [CrossRef]
- Campbell, J.P.; Turner, J.E. Debunking the Myth of Exercise-Induced Immune Suppression: Redefining the Impact of Exercise on Immunological Health Across the Lifespan. Front. Immunol. 2018, 9, 648. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.C.; Jesse, C.R.; Del Fabbro, L.; de Gomes, M.G.; Goes, A.T.R.; Filho, C.B.; Luchese, C.; Pereira, A.A.M.; Boeira, S.P. Swimming exercise prevents behavioural disturbances induced by an intracerebroventricular injection of amyloid-β 1-42 peptide through modulation of cytokine/NF-kappaB pathway and indoleamine-2,3-dioxygenase in mouse brain. Behav. Brain Res. 2017, 331, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-L.; Zhao, G.; Zhang, H.; Shi, L.-D. Long-term treadmill exercise inhibits the progression of Alzheimer’s disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav. Brain Res. 2013, 256, 261–272. [Google Scholar] [CrossRef]
- Yu, F.; Xu, B.; Song, C.; Ji, L.; Zhang, X. Treadmill exercise slows cognitive deficits in aging rats by antioxidation and inhibition of amyloid production. NeuroReport 2013, 24, 342–347. [Google Scholar] [CrossRef]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.-S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.-H. Retinoic Acid Attenuates -Amyloid Deposition and Rescues Memory Deficits in an Alzheimer’s Disease Transgenic Mouse Model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef]
- Phiel, C.; Wilson, C.A.; Lee, V.M.-Y.; Klein, P.S. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Lim, G.P.; Harris-White, M.E.; Yang, F.; Ambegaokar, S.S.; Ubeda, O.J.; Glabe, C.G.; Teter, B.; Frautschy, S.A.; Cole, G.M. Antibodies against β-amyloid reduce aβ oligomers, glycogen synthase kinase-3β activation and τ phosphorylation in vivo and in vitro. J. Neurosci. Res. 2005, 83, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.A.; Sun, Q.; Gamblin, T.C. Tau phosphorylation by GSK-3β promotes tangle-like filament morphology. Mol. Neurodegener. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Salkovic-Petrisic, M.; Tribl, F.; Schmidt, M.; Hoyer, S.; Riederer, P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J. Neurochem. 2006, 96, 1005–1015. [Google Scholar] [CrossRef]
- Su, Y.; Ryder, J.; Li, B.; Wu, X.; Fox, N.; Solenberg, P.; Brune, K.; Paul, S.; Zhou, Y.; Liu, F.; et al. Lithium, a Common Drug for Bipolar Disorder Treatment, Regulates Amyloid-β Precursor Protein Processing. Biochemistry 2004, 43, 6899–6908. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Russo-Neustadt, A.A. Exercise activates the phosphatidylinositol 3-kinase pathway. Brain Res. Mol. 2005, 135, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Leem, Y.-H.; Lim, H.-J.; Shim, S.-B.; Cho, J.-Y.; Kim, B.-S.; Han, P.-L. Repression of tau hyperphosphorylation by chronic endurance exercise in aged transgenic mouse model of tauopathies. J. Neurosci. Res. 2009, 87, 2561–2570. [Google Scholar] [CrossRef]
- Um, H.-S.; Kang, E.-B.; Koo, J.-H.; Kim, H.-T.; Lee, J.; Kim, E.-J.; Yang, C.-H.; An, G.-Y.; Cho, I.-H.; Cho, J.-Y. Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s disease. Neurosci. Res. 2011, 69, 161–173. [Google Scholar] [CrossRef]
- Luan, K.; Rosales, J.L.; Lee, K.-Y. Viewpoint: Crosstalks between neurofibrillary tangles and amyloid plaque formation. Ageing Res. Rev. 2013, 12, 174–181. [Google Scholar] [CrossRef]
- Souza, L.C.; Filho, C.B.; Goes, A.T.R.; Del Fabbro, L.; de Gomes, M.G.; Savegnago, L.; Oliveira, M.S.; Jesse, C.R. Neuroprotective Effect of Physical Exercise in a Mouse Model of Alzheimer’s Disease Induced by β-Amyloid1–40 Peptide. Neurotox. Res. 2013, 24, 148–163. [Google Scholar] [CrossRef]
- El-Kader, S.M.A.; Al-Jiffri, O.H. Aerobic exercise improves quality of life, psychological well-being and systemic inflammation in subjects with Alzheimer’s disease. Afr. Health Sci. 2016, 16, 1045–1055. [Google Scholar] [CrossRef]
- Jensen, C.S.; Bahl, J.M.; Østergaard, L.B.; Høgh, P.; Wermuth, L.; Heslegrave, A.; Zetterberg, H.; Heegaard, N.H.; Hasselbalch, S.G.; Simonsen, A.H. Exercise as a potential modulator of inflammation in patients with Alzheimer’s disease measured in cerebrospinal fluid and plasma. Exp. Gerontol. 2019, 121, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; LeRoith, D. Insulin and insulin-like growth factor receptors in the brain: Physiological and pathological aspects. Eur. Neuropsychopharmacol. 2014, 24, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Frozza, R.L.; De Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Ribeiro, F.C.; Sudo, F.K.; Drummond, C.; Assunção, N.; VanderBorght, B.; Tovar-Moll, F.; Mattos, P.; De Felice, F.G.; Ferreira, S.T. Cerebrospinal fluid irisin correlates with amyloid-β, BDNF, and cognition in Alzheimer’s disease. Alzheimer’s Dementia Diagn. Assess. Dis. Monit. 2020, 12, e12034. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Song, J. The Role of Irisin in Alzheimer’s Disease. J. Clin. Med. 2018, 7, 407. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Cao, L.; Wang, L.-M.; Guo, C.; Ye, J.-L.; Chai, Y.-F.; Yan, Z.-Y. Development of Neurotrophic Molecules for Treatment of Neurode-generation. Curr. Protein Pept. Sci. 2001, 2, 261–276. [Google Scholar] [CrossRef]
- Longo, F.M.; Yang, T.; Knowles, J.K.; Xie, Y.; Moore, L.A.; Massa, S.M. Small Molecule Neurotrophin Receptor Ligands: Novel Strategies for Targeting Alzheimers Disease Mechanisms. Curr. Alzheimer Res. 2007, 4, 503–506. [Google Scholar] [CrossRef]
- Nascimento, C.M.C.; Pereira, J.R.; de Andrade, L.P.; Garuffi, M.; Ayan, C.; Kerr, D.S.; Talib, L.L.; Cominetti, M.R.; Stella, F. Physical Exercise Improves Peripheral BDNF Levels and Cognitive Functions in Mild Cognitive Impairment Elderly with Different BDNF Val66Met Genotypes. J. Alzheimer’s Dis. 2015, 43, 81–91. [Google Scholar] [CrossRef]
- Latsko, M.S.; Gilman, T.L.; Matt, L.M.; Nylocks, K.M.; Coifman, K.G.; Jasnow, A.M. A Novel Interaction between Tryptophan Hydroxylase 2 (TPH2) Gene Polymorphism (rs4570625) and BDNF Val66Met Predicts a High-Risk Emotional Phenotype in Healthy Subjects. PLoS ONE 2016, 11, e0162585. [Google Scholar] [CrossRef]
- Cagni, F.C.; Campêlo, C.L.D.C.; Coimbra, D.G.; Barbosa, M.R.; Júnior, L.G.O.; Neto, A.B.S.; Ribeiro, A.M.; Júnior, C.D.O.G.; de Andrade, T.; Silva, R.H. Association of BDNF Val66MET Polymorphism With Parkinson’s Disease and Depression and Anxiety Symptoms. J. Neuropsychiatry Clin. Neurosci. 2017, 29, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Imai, A.S.S.-I.; Guarente, L. The brain, sirtuins, and ageing. Nat. Rev. Neurosci. 2017, 18, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.G.; Dennis, A.; Rawlings, N.B.; Stagg, C.; Matthews, L.; Morris, M.; Kolind, S.H.; Foxley, S.; Jenkinson, M.; Nichols, T.E.; et al. Multi-modal characterization of rapid anterior hippocampal volume increase associated with aerobic exercise. NeuroImage 2015, 131, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Maass, A.; Düzel, S.; Brigadski, T.; Goerke, M.; Becke, A.; Sobieray, U.; Neumann, K.; Lövdén, M.; Lindenberger, U.; Bäckman, L.; et al. Relationships of peripheral IGF-1, VEGF and BDNF levels to exercise-related changes in memory, hippocampal perfusion and volumes in older adults. NeuroImage 2016, 131, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Forti, L.N.; Van Roie, E.; Njemini, R.; Coudyzer, W.; Beyer, I.; Delecluse, C.; Bautmans, I. Dose-and gender-specific effects of resistance training on circulating levels of brain derived neurotrophic factor (BDNF) in community-dwelling older adults. Exp. Gerontol. 2015, 70, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Lavin, K.M.; Perkins, R.K.; Jemiolo, B.; Raue, U.; Trappe, S.W.; Trappe, T.A. Effects of aging and lifelong aerobic exercise on basal and exercise-induced inflammation in women. J. Appl. Physiol. 2020, 129, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef]
- Farina, N.; Rusted, J.; Tabet, N. The effect of exercise interventions on cognitive outcome in Alzheimer’s disease: A systematic review. Int. Psychogeriatrics 2014, 26, 9–18. [Google Scholar] [CrossRef]
- Forbes, D.; Forbes, S.C.; Blake, C.M.; Thiessen, E.J.; Forbes, S. Exercise programs for people with dementia. Cochrane Database Syst. Rev. 2015, 15, CD006489. [Google Scholar] [CrossRef]
- Hoffmann, K.; Sobol, N.A.; Frederiksen, K.S.; Beyer, N.; Vogel, A.; Vestergaard, K.; Brændgaard, H.; Gottrup, H.; Lolk, A.; Wermuth, L.; et al. Moderate-to-High Intensity Physical Exercise in Patients with Alzheimer’s Disease: A Randomized Controlled Trial. J. Alzheimer’s Dis. 2016, 50, 443–453. [Google Scholar] [CrossRef]
- Morris, J.K.; Vidoni, E.D.; Johnson, D.K.; Van Sciver, A.; Mahnken, J.D.; Honea, R.A.; Wilkins, H.M.; Brooks, W.M.; Billinger, S.A.; Swerdlow, R.H.; et al. Aerobic exercise for Alzheimer’s disease: A randomized controlled pilot trial. PLoS ONE 2017, 12, e0170547. [Google Scholar] [CrossRef]
- Yu, J.-T.; Xu, W.; Tan, C.-C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L.; et al. Evidence-based prevention of Alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.L.; Cyarto, E.V.; Ellis, K.A.; Ames, D.; Desmond, P.; Phal, P.; Sharman, M.J.; Szoeke, C.; Rowe, C.C.; Masters, C.L.; et al. A Randomized Controlled Trial of Adherence to a 24-Month Home-Based Physical Activity Program and the Health Benefits for Older Adults at Risk of Alzheimer’s Disease: The AIBL Active-Study. J. Alzheimer’s Dis. 2019, 70, S187–S205. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.-X.; Liang, J.-H.; Xu, Y.; Wang, Y.-Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: A meta-analysis. BMC Geriatr. 2019, 19, 181. [Google Scholar] [CrossRef]
- Hansson, O.; Svensson, M.; Gustavsson, A.-M.; Andersson, E.; Yang, Y.; Nägga, K.; Hållmarker, U.; James, S.; Deierborg, T. Midlife physical activity is associated with lower incidence of vascular dementia but not Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Ngandu, T.; Rusanen, M.; Antikainen, R.; Bäckman, L.; Havulinna, S.; Hänninen, T.; Laatikainen, T.; Lehtisalo, J.; Levälahti, E.; et al. Multidomain lifestyle intervention benefits a large elderly population at risk for cognitive decline and dementia regardless of baseline characteristics: The FINGER trial. Alzheimer’s Dement. 2018, 14, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Lamb, S.E.; Sheehan, B.; Atherton, N.; Nichols, V.; Collins, H.; Mistry, D.; Dosanjh, S.; Slowther, A.M.; Khan, I.; Petrou, S.; et al. Dementia And Physical Activity (DAPA) trial of moderate to high intensity exercise training for people with dementia: Randomised controlled trial. BMJ 2018, 361, k1675. [Google Scholar] [CrossRef]
- Karssemeijer, E.G.A.; Aaronson, J.A.; Bossers, W.J.; Smits, T.; Olde Rikkert, M.G.M.; Kessels, R.P.C. Positive effects of combined cognitive and physical exercise training on cognitive function in older adults with mild cognitive impairment or dementia: A meta-analysis. Ageing Res. Rev. 2017, 40, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tsitkanou, S.; Della Gatta, P.; Foletta, V.; Russell, A. The Role of Exercise as a Non-pharmacological Therapeutic Approach for Amyotrophic Lateral Sclerosis: Beneficial or Detrimental? Front. Neurol. 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Agata, V. Effects of Physical Activity on Amyotrophic Lateral Sclerosis. J. Funct. Morphol. Kinesiol. 2020, 5, 29. [Google Scholar] [CrossRef]
- Ferri, A.; Lanfranconi, F.; Corna, G.; Bonazzi, R.; Marchese, S.; Magnoni, A.; Tremolizzo, L. Tailored Exercise Training Counteracts Muscle Disuse and Attenuates Reductions in Physical Function in Individuals With Amyotrophic Lateral Sclerosis. Front. Physiol. 2019, 10, 1537. [Google Scholar] [CrossRef]
- Tsukita, K.; Sakamaki-Tsukita, H.; Takahashi, R. Long-term Effect of Regular Physical Activity and Exercise Habits in Patients With Early Parkinson Disease. Neurology 2022, 98, e859–e871. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-S.; Yang, S.; Tan, Z.-X.; Wang, M.-M.; Xing, Y.; Dong, F.; Zhang, F. The benefits and mechanisms of exercise training for Parkinson’s disease. Life Sci. 2020, 245, 117345. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, A.O.; Filho, A.S.; Murillo-Rodriguez, E.; Rocha, N.; Carta, M.G.; Machado, S. Physical Exercise For Parkinson’s Disease: Clinical And Experimental Evidence. Clin. Pract. Epidemiology Ment. Health 2018, 14, 89–98. [Google Scholar] [CrossRef]
- Julian, T.H.; Glascow, N.; Barry, A.D.F.; Moll, T.; Harvey, C.; Klimentidis, Y.C.; Newell, M.; Zhang, S.; Snyder, M.P.; Cooper-Knock, J.; et al. Physical exercise is a risk factor for amyotrophic lateral sclerosis: Convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 2021, 68, 103397. [Google Scholar] [CrossRef]
- Alkadhi, K.A.; Dao, A.T. Exercise decreases BACE and APP levels in the hippocampus of a rat model of Alzheimer’s disease. Mol. Cell. Neurosci. 2018, 86, 25–29. [Google Scholar] [CrossRef]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef]
- Dao, A.T.; Zagaar, M.A.; Levine, A.T.; Salim, S.; Eriksen, J.L.; Alkadhi, K.A. Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr. Alzheimer Res. 2013, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Dao, A.T.; Zagaar, M.A.; Salim, S.; Eriksen, J.L.; Alkadhi, K.A. Regular exercise prevents non-cognitive disturbances in a rat model of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2014, 17, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef]
- Marwarha, G.; Raza, S.; Meiers, C.; Ghribi, O. Leptin attenuates BACE1 expression and amyloid-β genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys. Acta 2014, 1842, 1587–1595. [Google Scholar] [CrossRef]
- Dao, A.T.; Zagaar, M.A.; Alkadhi, K.A. Moderate Treadmill Exercise Protects Synaptic Plasticity of the Dentate Gyrus and Related Signaling Cascade in a Rat Model of Alzheimer’s Disease. Mol. Neurobiol. 2014, 52, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Dao, A.T.; Zagaar, M.A.; Levine, A.T.; Alkadhi, K.A. Comparison of the Effect of Exercise on Late-Phase LTP of the Dentate Gyrus and CA1 of Alzheimer’s Disease Model. Mol. Neurobiol. 2016, 53, 6859–6868. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, H.L.; Zhang, H.; Tong, X.J. Treadmill exercise enhances synaptic plasticity, but does not alter β-amyloid deposition in hippocampi of aged APP/PS1 transgenic mice. Neuroscience 2015, 298, 357–366. [Google Scholar] [CrossRef]
- Moore, K.M.; Girens, R.E.; Larson, S.K.; Jones, M.R.; Restivo, J.L.; Holtzman, D.M.; Cirrito, J.R.; Yuede, C.M.; Zimmerman, S.D.; Timson, B.F. A spectrum of exercise training reduces soluble Aβ in a dose-dependent manner in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 85, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, T.C.; Beleza, J.; Rizo-Roca, D.; SantosAlves, E.; Leal, C.; Martins, M.J.; Ascensao, A.; Magalhães, J. Physical exercise mitigates behavioral impairments in a rat model of sporadic Alzheimer’s disease. Behav. Brain Res. 2020, 379, 112358. [Google Scholar] [CrossRef]
- Jiang, L.; Ma, J.; Zhang, Y.; Zhou, C.-N.; Zhang, L.; Chao, F.-L.; Chen, L.-M.; Jiang, R.; Wu, H.; Tang, Y. Effect of running exercise on the number of the neurons in the hippocampus of young transgenic APP/PS1 mice. Brain Res. 2018, 1692, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Robison, L.S.; Popescu, D.; Anderson, M.E.; Francis, N.; Hatfield, J.; Sullivan, J.K.; Beigelman, S.I.; Xu, F.; Anderson, B.J.; Van Nostrand, W.E.; et al. Long-term voluntary wheel running does not alter vascular amyloid burden but reduces neuroinflammation in the Tg-SwDI mouse model of cerebral amyloid angiopathy. J. Neuroinflammation 2019, 16, 144. [Google Scholar] [CrossRef] [PubMed]
- Belaya, I.; Ivanova, M.; Sorvari, A.; Ilicic, M.; Loppi, S.; Koivisto, H.; Varricchio, A.; Tikkanen, H.; Walker, F.R.; Atalay, M.; et al. Astrocyte remodeling in the beneficial effects of long-term voluntary exercise in Alzheimer’s disease. J. Neuroinflammation 2020, 17, 271. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Andersson, E.; Manouchehrian, O.; Yang, Y.; Deierborg, T. Voluntary running does not reduce neuroinflammation or improve non-cognitive behavior in the 5xFAD mouse model of Alzheimer’s disease. Sci. Rep. 2020, 10, 1346. [Google Scholar] [CrossRef]
- Xia, J.; Li, B.; Yin, L.; Zhao, N.; Yan, Q.; Xu, B. Treadmill exercise decreases β-amyloid burden in APP/PS1 transgenic mice involving regulation of the unfolded protein response. Neurosci. Lett. 2019, 703, 125–131. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Y.; Wang, Y.; Song, L.; Zhang, R.; Du, Y. Long-term treadmill exercise attenuates Aβ burdens and astrocyte activation in APP/PS1 mouse model of Alzheimer’s disease. Neurosci. Lett. 2018, 666, 70–77. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study Design | Participants | Result | Refs. |
|---|---|---|---|
| Systematic review and meta-analysis of observational prospective studies and randomised controlled trials | Participants from 243 observational prospective studies and 153 randomised controlled trials | 21 most important evidence-based suggestions for life-course practices to prevent AD were identified and divided by the level of evidence (into levels A and B) and strength of suggestion (into class I and class III). PA was classified among the 21 most important evidence-based suggestions for life-course practices to prevent AD into level B, class I. | [231] |
| Randomised controlled trial | Men and women (n = 106) randomised in control (n = 51) and treated groups (n = 55), aged 60 years or older, with MCI or subjective memory complaints (SMC) and at least one CVR factor (physical inactivity, obesity, hypertension, heart disease, type II diabetes, smoking, hypercholesterolemia) | After 24-months, the home based PA programme improved CVF and leg strength; cognition was not evaluated. | [232] |
| Meta-analysis of randomised control trials | 673 subjects with AD in 13 randomized controlled trials, treated to different quantities of physical activity and exercise interventions, were included. | PA and PE improved cognition of older adults with AD. High frequency PA and PE interventions did not have a greater effect on cognition compared to low frequency interventions. | [233] |
| Large prospective observational study | Two prospective studies. First study on 197,685 long-distance skiers to compare the incidence of vascular dementia (VAD) or AD to matched individuals from the general population (n = 197,684 participants) during 21 years of follow-up. Second study evaluated the association between self-reported PA and the incidence of VAD and AD in 20,639 participants. | PA in midlife reduced the incidence of VAD. There was no significant association between PA and the risk of subsequent development of AD. | [234] |
| Randomised controlled trail | 198 male and female patients, average age 71 and 70 for control and exercise groups, with clinically diagnosed AD by the NINCDS-ADRDA criteria and an MMSE >19. | 16 weeks of moderate-to-high PE attenuated plasma INFγ in APO ε4 patients with AD. | [211] |
| Randomised trial | 1260 people, age (60–77 years), at risk for dementia were randomized 1:1 to multidomain intervention (diet, PE, cognition (evaluated by the Mini–Mental State Examination, and vascular risk management) and regular health advice). | The 2-year study intervention improved overall cognitive performance (measured with an extended Neuropsychological Test Battery (NTB) and was beneficial regardless of participants’ sex, age, income, cognition, body mass index, blood pressure, cholesterol, fasting glucose, overall cardiovascular risk, and cardiovascular comorbidity | [235] |
| Randomised controlled trail | 494 male and female participants with dementia (measured by the Alzheimer’s disease assessment scale-cognitive subscale), average age 77 years. | 12 months of moderate to high intensity aerobic and strength PE training programme did not attenuate cognitive impairment nor improve activities of daily living in people with mild to moderate dementia. Physical fitness was improved. | [236] |
| Meta-analysis of randomised controlled trilas | Older adults with MCI or dementia from 10 randomized controlled trials that evaluated the effect of a combined cognitive-physical intervention on cognition. | Combined cognitive-physical interventions were equally beneficial to older adults with MCI or with dementia: there was a small-to-medium positive effect on global cognitive function and a moderate-to-large positive effect for activities of daily living. | [237] |
| Pilot, randomised controlled trail | 76 male and female participants over 55 years of age, mean age 73, with MCI or dementia with etiologic diagnosis of probable AD based on clinical and cognitive test results. | 26 weeks of supervised PE improved cardiorespiratory fitness associated with a modest improvement in functional ability (measured by the Disability Assessment for Dementia). The was no measurable improvement in memory, executive function, or depressive symptoms. Improved cardiorespiratory fitness was positively correlated with change in memory performance and reduced bilateral hippocampal atrophy. | [230] |
| Randomised controlled trail |
40 male and female patients with AD, age 65–75, were divided into control (no training intervention) and treadmill aerobic exercise group. Both groups were evaluated for TNF-α, interleukin-6 IL-6, Rosenberg Self-Esteem Scale, Beck Depression Inventory, Profile of Mood States and SF-36 health quality of life before and at the end of the study. | 2-moths of PE improved quality of life, attenuated systemic inflammation markers and psychological wellbeing in patients with AD. | [210] |
| Animal Model, Age at Start of Experiment | PA or PE Design | Result, Compared to Sedentary Animal Models of AD and CAA | Refs. |
|---|---|---|---|
| CAAam (Tg-SwDI male and female M); C57BL/6 WT male and female M, age 4 months | Voluntary wheel running PA, wheel availability 1–12 h/day, 5 days/week, 8 consecutive months | PA improved motor function, reduced anxiety-like behaviour and attenuated neuroinflammation markers TNFα and IL6 but not vascular amyloid β accumulation. | [257] |
| FADam, 5xFAD male M; WT JAXC57BL/6J male M, age 6 weeks | Voluntary wheel running PA, wheel availability 24 h/day, 7 days/week, 6 consecutive months | PA mitigated Aβ pathology related cognitive deficits in spatial learning, memory and exploration activity with a temporal association to increased hippocampal glial fibrillary acid protein (GFAP) immunoreactivity and the number of GFAP-positive astrocytes, increased astrocytic brain-derived neurotrophic factor and restoration of postsynaptic protein PSD-95. Voluntary PE did not attenuate brain neuroinflammation markers. | [258] |
| FADam, 5xFAD female M; age 9–12 weeks | Voluntary wheel running PA, wheel availability 24 h/day, 7 days/week, 4 consecutive weeks | PA did not attenuate neuroinflammation markers (total amount of neuroglia in hippocampus, cytokine levels, levels of NLRP3), nor improve motor learning or reduce insoluble Aβ brain content. | [259] |
| FADam, APP/PS1 male and female M, age 12 months | MT PE, 20 min/day, 5 days/week, 4 consecutive months | PE mitigated Aβ pathology related cognitive deficits in spatial cognition with a temporal association to increases in spinophilin-immunoreactive puncta numbers in hippocampal areas. The effect of PE on neuroinflammation markers was not evaluated. | [24] |
| FADam, 5xFAD female M, age 9–12 weeks | Voluntary wheel running PA, wheel availability 24 h/day, 7 days/week, for 24 consecutive weeks. | PA did not mitigate Aβ pathology related cognitive deficits in object or working memory, nor synaptic proteins PSD-95 and synaptophysin contents, Aβ brain content or hippocampal Aβ42 concentration. The effect of PE on neuroinflammation markers was not evaluated. | [234] |
| FADam, APP/PS1 male M; WT C57BL/6 male M, age 3 months | MT PE, 45 min per day, 5 days/week, 3 consecutive months | PE mitigated Aβ pathology related deficits in cognition associated hippocampus, with reduced Aβ plaques and soluble Aβ forms, decreased β-site amyloid precursor protein-cleaving enzyme 1 and presenilin-1 expression, downregulated expression of GRP78, and inhibited activation of PERK, eIF2α, and ATF4. The effects of PE on neuroinflammation markers and animals’ cognitive behaviour were not evaluated. | [260] |
| FADam, APP/PS1 male M; WT C57BL/6 male M, age 6 months | MT PE, 20 min per day, 5 days/week, 4 consecutive months | PE mitigated Aβ pathology related cognitive deficits in special learning and memory abilities with a temporal association to increased hippocampal volumes and increased number of hippocampal neurons. The effect of PE on neuroinflammation markers was not evaluated. | [256] |
| FADam, APP/PS1 M; WT C57BL/6 M, age 5 months | MT PE, 30 min per day, 6 days/week, 5 consecutive months | PE mitigated Aβ pathology in cognition asociated hippocampus and neocortex (attenuated Aβ area fraction, plaque number and size and decreased levels of insulin-degrading enzyme and receptor for advanced glycation end products). Also, PE increased neuronal density, attenuated activation of astrocytes and decreased β-site amyloid precursor protein cleaving enzyme 1 and presenilin 1 levels. The activity of non-amyloidogenic APP pathway was increased. The effect of PE on animals’ cognitive behaviour was not evaluated. Controlled PE had a possible inhibitory effect on neuroinflammation by supressing any numerical and morphological conversions of microglia and by reducing the total number of astrocytes and the number of astrocytes associated with Aβ pathology. | [261] |
| LOADam, icvi. of streptozotocin, Wistar male R, age 6 weeks | MT PE, 1 h/day, 5 days/week, 24 weeks (8 weeks before icvi and consecutive 12 weeks after) | PE mitigated Aβ pathology related cognitive deficits in spatial cognition and willingness to explore with a temporal association to positive changes in MITO oxygen consumption endpoints of synaptosomal and non-synaptosomal brain mitochondria. The effect of PE on neuroinflammation markers was not evaluated. | [255] |
| LOADam, icvi. of Aβ42 peptide, Wistar male R, age 7 weeks | MT PE, two, 15 min sessions/day in weeks 1 and 2, increased to 3 sessions/day in week 3 and 4 sessions/day in week 4 | PE prevented Aβ pathology associated increase in levels of APP, BACE-1 and Aβ proteins in hippocampal areas (associated with cognitive functions). The effects of PE on neuroinflammation markers and animals’ cognitive behaviour were not evaluated. | [245] |
| LOADam, ihi. of Aβ42 peptide, C57BL/6N male M, age 8 weeks | MT PE, 30/day, 7 consecutive days | PE mitigated Aβ pathology related cognitive deficits in object recognition and spatial cognition with a temporal association to hippocampal increased adult neurogenesis, decreased inflammatory cytokine levels and decreased astroglial cell density. Also, PE partly normalised MAPK signalling (i.e., attenuated JNK and P38 phosphorylation). | [89] |
| LOADam, icvi. of Aβ42 peptide, Swiss Albino male M, age 3 months | swimming PE with weights attached to the proximal portions of animal’s tail, duration progressively increased from 20 to 60 min/day, 5 days/week, 8 consecutive weeks | PE mitigated Aβ pathology related cognitive deficits (memory impairment and depressive/anxiety-like behaviour) with a temporal association to inhibition of inflammation/indoleamine-2,3-dioxygenase activation and up-regulation of neurotrophic factors in brain. | [196] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribarič, S. Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 3245. https://doi.org/10.3390/ijms23063245
Ribarič S. Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients. International Journal of Molecular Sciences. 2022; 23(6):3245. https://doi.org/10.3390/ijms23063245
Chicago/Turabian StyleRibarič, Samo. 2022. "Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients" International Journal of Molecular Sciences 23, no. 6: 3245. https://doi.org/10.3390/ijms23063245
APA StyleRibarič, S. (2022). Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients. International Journal of Molecular Sciences, 23(6), 3245. https://doi.org/10.3390/ijms23063245