
Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy



Abstract
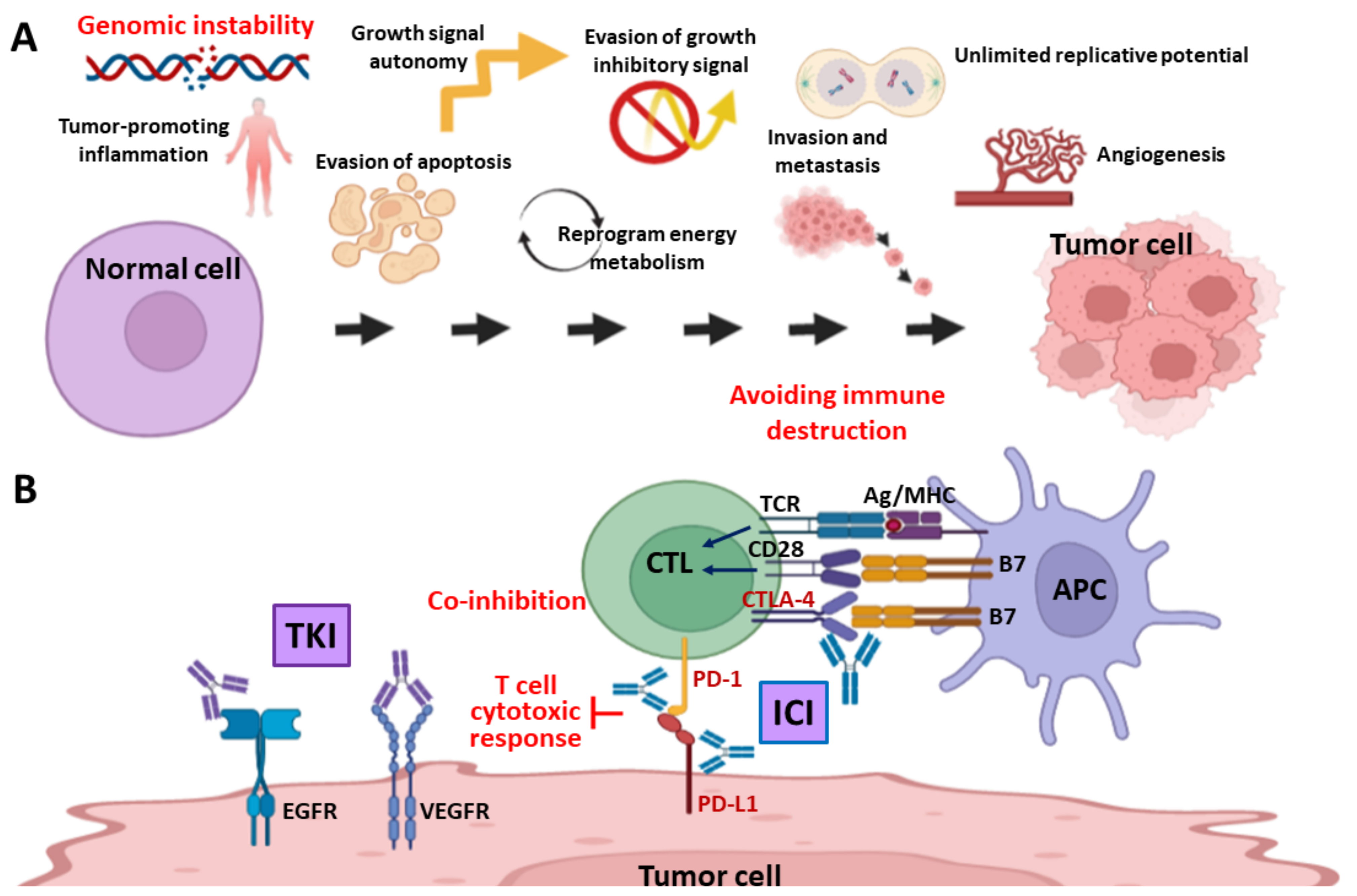
1. Introduction
2. DNA Damage Response (DDR) and DNA Repair Systems
2.1. Nucleotide Excise Repair (NER)
2.2. Base Excision Repair (BER)
2.3. Mismatch Repair (MMR)
2.4. Duble-Strand Break (DSB) Repair and the Cell Cycle Checkpoint
3. Targeting DDR—Induction of Synthetic Lethality in Cancer Cells
4. Targeting DDR—Stimulation of Immune Response through Generation of Cytosolic DNA
5. Targeting DDR and Immune Checkpoint—Contemporary Clinical Trials in Breast, Colorectal, and Pancreatic Cancers
5.1. Breast Cancer
5.1.1. Combination Therapy
MEDIOLA Trial (NCT02734004)
TOPACIO Trial (NCT02657889, KEYNOTE-162)
TALAVE Trial (NCT03964532)
NCT02849496

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | DDRi | Target | ICI | Target | Disease Setting | Biomarker | Trial ID | Phase | Comment/Reference |
|---|---|---|---|---|---|---|---|---|---|
| Combination | Olaparib | PARP | Durvalumab | PD-L1 | Metastatic HER2- BC 1 | Germline BRCAm 2 | NCT02734004 | II | MEDIOLA trial [109] |
| Niraparib | PARP | Pembrolizumab | PD-1 | aTNBC 3 or mTNBC 4 | BRCAm | NCT02657889 | II | TOPACIO trial [111] | |
| Talazoparib | PARP | Avelumab | PD-L1 | aBC 5 | NCT03964532 | I/II | TALAVE trial [112] | ||
| Olaparib | PARP | Atezolizurmab | PD-L1 | aHER2- BC or mHER2- BC | BRCAm, HRD 6 | NCT02849496 | II | ||
| Olaparib | PARP | - | - | ||||||
| Olaparib | PARP | Pembrolizumab | PD-1 | aTNBC or mTNBC | CT+ICI positive 7 | NCT04191135 | II/III | KEYLYNK-009 trial [113] | |
| v.s. CT (C+G) 8 | DNA | Pembrolizumab | PD-1 | ||||||
| Monotherapy DDRi or ICI | CT (nP) 9 | Atezolizurmab | PD-L1 | Untreated mTNBC | NCT02425891 | III | IMpassion 130 trial [114] | ||
| CT (nP) | v.s. Placebo | ||||||||
| Durvalumab | PD-L1 | NACT Resist TNBC 10 | NCT03740893 | II | Phoenix trial | ||||
| AZD6738 | ATR | ||||||||
| Olaparib | PARP | ||||||||
| Monotherapy DDRi or ICI (molecular selection) | Talazoparib | PARP | aBC and/or mBC 11 | Germline BRCAm | NCT01945775 | Approved | EMBRACA trial [115,116] | ||
| Olaparib | PARP | mBC | Germline BRCAm | NCT02000622 | Approved | OlympiAD trial [117] | |||
| Pembrolizumab | PD-1 | mBC, etc. | BRCAm, POLD1m 12, POLEm 13 | NCT03428802 | II |
KEYLYNK-009 Trial (NCT04191135)
5.1.2. Monotherapy
IMpassion130 Trial (NCT02425891)
PHOENIX Trial (NCT03740893)
5.1.3. Monotherapy in Biomarker-Selected Patients
EMBRACA Trial (NCT01945775)
OlympiAD Trial (NCT02000622)
NCT03428802
5.2. Colorectal Cancer (CRC)
5.2.1. Combination Therapy
DAPPER Trial (NCT03851614)
NCT02484404
5.2.2. Monotherapy in Biomarker-Selected Patients
KEYNOTE-177 Trial (NCT02563002)
NCT01876511
| Treatment | DDRi | Target | ICI | Target | TT/CT 1 | Disease Setting | Biomarker | Trial ID | Phase | Comment/Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Combination | Olaparib | PARP | Durvalumab | PD-L1 | Cediranib (VEGF) | CRC 2, PA 3, LMS 4 | MSS 5, pMMR 6 | NCT03851614 | II | DAPPER trial |
| Olaparib | PARP | Durvalumab | PD-L1 | Cediranib (VEGF) | Solid tumors | NCT02484404 | I/II | [15,127] | ||
| Monotherapy (ICI) | Pembrolizumab | PD-1 | CRC, solid tumor | MSI 7-H or dMMR, TMB 8-H | NCT02563002 | approved | KEYNOTE-177 trial | |||
| Pembrolizumab | PD-1 | CRC, MSI+ non-CRC | CRC (MSI or MSS) | NCT01876511 | II | [42,129] | ||||
| Nivolumab Ipilimumab | PD-1 CTLA-4 | Celecoxib (COX-2i) | mCRC 9 | MSI, MSS | NCT03026140 | II | NICHE trial [130] | |||
| Avelumab | PD-L1 | mCRC | MSI-H or POLEm 10 | NCT03150706 | II | |||||
| Durvalumab | PD-L1 | mCRC | MSI-H or POLEm | NCT03435107 | II | |||||
| Monothreapy (DDRi) | AZD1775 | WEE1 | Irinotecan (Top1) | mCRC | RAS, BRAFm 12 | NCT02906059 | I | |||
| Niraparib | PARP | Panitumumab (EGFR) | aCRC 11 and mCRC | RasWT or MSI-H or MSS | NCT03983993 | II | NIPAVect trial | |||
| Olaparib | PARP | Temozolomide | aCRC | MGMT 13 promoter hypermethylation | NCT04166435 | II | ||||
| Adavosertib | WEE1 | mCRC | RAS, TP53 | FOCUS4-C | II | [44] |
NICHE Trial (NCT03026140)
NCT03150706 and NCT03435107
NCT02906059
NIPAVect Trial (NCT03983993)
NCT04166435
FOCUS4-C Trial
5.3. Pancreatic Cancer
5.3.1. Combination Therapy
DAPPER Trial (NCT03851614)
NCT04548752
Parpvax Trial (NCT03404960)
5.3.2. Monotherapy with DDR Inhibitors or ICIs
The Match Screening Trial (NCT02465060)
5.3.3. DDR Inhibitors in Combination with Chemotherapy or Radiotherapy
NCT04514497
NCT04172532
NCT01296763
| Treatment | DDRi | Target | ICI | Target | CT/RT | Disease Setting | Biomarker | Trial ID | Phase | Comment/Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Combination DDRi+ICi | Olaparib | PARP | Durvalumab | PD-L1 | Cediranib (VEGFi) | PC 1, CRC 2,LMS 3 | MSS 4 | NCT03851614 | II | DAPPER trial |
| Olaparib | PARP | Pembrolizumab | PD-1 | mPC 5 | gBRCA1/2m | NCT04548752 | II | |||
| Niraparib | PARP | Nivolumab | PD-1 | After platinum-based CT | Progression Free PC | NCT03404960 | Ib | Parpvax trial | ||
| Ipilimumab | CTLA-4 | II | ||||||||
| Molecular Match | - | - | Nivolumab | PD-1 | PC, BC 6, CRC | MLH1, MSH2, | NCT02465060 | II | [137] | |
| Adavosertib | WEE1 | - | - | BRCA1/2 | NCI match trail | |||||
| Combination | Elimusertib | ATR | Irinotecan | aPC 7, SCLC 8 PD-NEC 9 | NCT04514497 | I | ||||
| DDRi+CT/RT | Elimusertib | ATR | Topotecan | |||||||
| Peposertib | DNA-PK | Hypofractionated RT | localized aPC | NCT04172532 | I/II | [70] | ||||
| Olaparib | PARP | Irinotecan, Cisplatin, Mitomycin | aPC | BRCA/FANC, HRD | NCT01296763 | I | [138] | |||
| Veliparib | PARP | Cisplatin, Gemcitabine | mPC | BRCA/PALB2 | NCT01585805 | II | [139] | |||
| Veliparib | PARP | FOLFOX 10 | mPC | BRCA1/2, PALBB2, FANC | NCT01489865 | I/II | [140] | |||
| Rucaparib | PARP | Irinotecan Fluorouracil, Leucovorin | mPC, CRC | BRCA1/2, PALB2 (HRD 11) | NCT03337087 | I/II | [141] | |||
| Olaparib Selumetinib | PARP MEK | FOLFIRI 12 | mPC | sBRCAm, KRAS or no mutation | NCT04348045 | MAZEPPA trial | ||||
| Monotherapy PARPi (Molecular Selection) | Olaparib | PARP | mPC | gBRCA1/2m | NCT02184195 | approved | POLO trial. [142] | |||
| Olaparib | PARP | PC, OC 13, BC Prostate Ca | gBRCA1/2m | NCT01078662 | II | [143] | ||||
| Rucaparib | PARP | PC, EC 14, OC, etc. | BRCA1/2, PALB2, RAD51C, RAD51D, BARD1, BRIP1, FANCA, NBN, RAD51, RAD51B | NCT04171700 | II | LODESTAR trial | ||||
| Niraparib | PARP | PC | BRCA1/2, PALB2, CHEK2 ATM | NCT03601923 | II |
NCT01585805
NCT01489865
NCT03337087
MAZEPPA Trial (NCT04348045)
5.3.4. Monotherapy with PARP Inhibitor in Biomarker-Selected Patients
POLO Trial (NCT02184195)
NCT01078662
Lodestar Trial (NCT04171700)
NCT03601923
6. Discussion and Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Jonna, S.; Deneka, A.; Patel, J.; Abazeed, M.; Golemis, E.; Borghaei, H.; Boumber, Y. Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies. Cancers 2021, 13, 3164. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Nilsson, M.; Goldman, J.; Reck, M.; Nakagawa, K.; Kato, T.; Ares, L.P.; Frimodt-Moller, B.; Wolff, K.; Visseren-Grul, C.; et al. Dual EGFR-VEGF Pathway Inhibition: A Promising Strategy for Patients with EGFR-Mutant NSCLC. J. Thorac. Oncol. 2020, 16, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, A.J.; Bivona, T.G. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol. Med. 2019, 25, 185–197. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gourley, C.; Balmaña, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Dickson, K.-A.; Xie, T.; Evenhuis, C.; Ma, Y.; Marsh, D.J. PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 8506. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2018, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.-C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Brandi, G. Biochemical predictors of response to immune checkpoint inhibitors in unresectable hepatocellular carcinoma. Cancer Treat. Res. Commun. 2021, 27, 100328. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.D.; Rizzo, A.; Brandi, G. The DNA damage repair (DDR) pathway in biliary tract cancer (BTC): A new Pandora’s box? ESMO Open 2020, 5, e001042. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Tomimatsu, N.; Tahimic, C.G.T.; Otsuki, A.; Burma, S.; Fukuhara, A.; Sato, K.; Shiota, G.; Oshimura, M.; Chen, D.J.; Kurimasa, A. Ku70/80 Modulates ATM and ATR Signaling Pathways in Response to DNA Double Strand Breaks. J. Biol. Chem. 2007, 282, 10138–10145. [Google Scholar] [CrossRef]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shih, D.J.H.; Lin, S.-Y. Role of DNA repair defects in predicting immunotherapy response. Biomark. Res. 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, Z.; Tang, X.; Zhang, X.; Chen, Y.; Hu, T.; Zhang, H.; Guan, M.; Zhang, X.; Wu, Z. Pan-cancer analysis reveals homologous recombination deficiency score as a predictive marker for immunotherapy responders. Hum. Cell 2021, 35, 199–213. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; Bent, M.J.V.D.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Cressey, D. DNA repair sleuths win chemistry Nobel. Nature 2015, 526, 307–308. [Google Scholar] [CrossRef][Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, T.; Tsubata, Y.; Tanino, R.; Nakao, M.; Hotta, T.; Hamaguchi, M.; Hamaguchi, S.; Araki, A.; Isobe, T. ERCC1 Is a Predictive Biomarker for Non-small Cell Lung Cancer But Is Antibody-dependent. Anticancer Res. 2021, 41, 2653–2660. [Google Scholar] [CrossRef] [PubMed]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA Repair by ERCC1 in Non–Small-Cell Lung Cancer and Cisplatin-Based Adjuvant Chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015, 13, 118. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Kim, T.-M.; Laird, P.W.; Park, P.J. The Landscape of Microsatellite Instability in Colorectal and Endometrial Cancer Genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Elbæk, C.R.; Petrosius, V.; Sørensen, C.S. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat. Res. Mol. Mech. Mutagen. 2020, 819–820, 111694. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.-Y.; Obuse, C.; Nishi, R.; et al. BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Welcsh, P.L. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef]
- Tomasova, K.; Cumova, A.; Seborova, K.; Horák, J.; Koucka, K.; Vodickova, L.; Vaclavikova, R.; Vodicka, P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers 2020, 12, 1713. [Google Scholar] [CrossRef]
- Clague, J.; Wilhoite, G.; Adamson, A.; Bailis, A.; Weitzel, J.N.; Neuhausen, S.L. RAD51C Germline Mutations in Breast and Ovarian Cancer Cases from High-Risk Families. PLoS ONE 2011, 6, e25632. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic Sequencing Identifies PALB2 as a Pancreatic Cancer Susceptibility Gene. Science 2009, 324, 217. [Google Scholar] [CrossRef]
- Tischkowitz, M.D.; Sabbaghian, N.; Hamel, N.; Borgida, A.; Rosner, C.; Taherian, N.; Srivastava, A.; Holter, S.; Rothenmund, H.; Ghadirian, P.; et al. Analysis of the Gene Coding for the BRCA2-Interacting Protein PALB2 in Familial and Sporadic Pancreatic Cancer. Gastroenterology 2009, 137, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Slater, E.; Langer, P.; Niemczyk, E.; Strauch, K.; Butler, J.; Habbe, N.; Neoptolemos, J.; Greenhalf, W.; Bartsch, D. PALB2 mutations in European familial pancreatic cancer families. Clin. Genet. 2010, 78, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Macedo, G.S.; Alemar, B.; Ashton-Prolla, P. Reviewing the characteristics of BRCA and PALB2-related cancers in the precision medicine era. Genet. Mol. Biol. 2019, 42, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Byrum, A.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors fromBRCAMutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef]
- LaFargue, C.; Molin, G.Z.D.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef]
- Siddiqui, A.; Tumiati, M.; Joko, A.; Sandholm, J.; Roering, P.; Aakko, S.; Vainionpää, R.; Kaipio, K.; Huhtinen, K.; Kauppi, L.; et al. Targeting DNA Homologous Repair Proficiency With Concomitant Topoisomerase II and c-Abl Inhibition. Front. Oncol. 2021, 11, 733700. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Haines, E.; Nishida, Y.; Carr, M.I.; Montoya, R.H.; Ostermann, L.B.; Zhang, W.; Zenke, F.T.; Blaukat, A.; Andreeff, M.; Vassilev, L.T. DNA-PK inhibitor peposertib enhances p53-dependent cytotoxicity of DNA double-strand break inducing therapy in acute leukemia. Sci. Rep. 2021, 11, 12148. [Google Scholar] [CrossRef]
- McLornan, D.P.; List, A.; Mufti, G.J. Applying Synthetic Lethality for the Selective Targeting of Cancer. N. Engl. J. Med. 2014, 371, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2020, 26, e164–e172. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Arena, S.; Siena, S.; Bardelli, A.; Sartore-Bianchi, A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann. Oncol. 2020, 31, 1135–1147. [Google Scholar] [CrossRef]
- Das, S.; Cardin, D. Targeting DNA Damage Repair Pathways in Pancreatic Adenocarcinoma. Curr. Treat. Options Oncol. 2020, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liu, P. Cytosolic DNA sensing by cGAS: Regulation, function, and human diseases. Signal Transduct. Target. Ther. 2021, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Parker, B.; Rautela, B.S.P.J.; Hertzog, P. Antitumour actions of interferons: Implications for cancer therapy. Nat. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 2020, 39, 96–108.e6. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Storozynsky, Q.; Hitt, M.M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef]
- Cao, X.; Liang, Y.; Hu, Z.; Li, H.; Yang, J.; Hsu, E.J.; Zhu, J.; Zhou, J.; Fu, Y.-X. Next generation of tumor-activating type I IFN enhances anti-tumor immune responses to overcome therapy resistance. Nat. Commun. 2021, 12, 5866. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. Cancer Clin. Trials 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; DeMaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.H.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunol. Immunother. 2014, 63, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 81. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Song, Y.; Chu, Q.; Wu, K. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 35. [Google Scholar] [CrossRef]
- Aval, L.M.; Pease, J.E.; Sharma, R.; Pinato, D.J. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J. Clin. Med. 2020, 9, 3323. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, K.; Xiong, H.; Huang, Y.; Chen, X.; Zhou, Y.; Qin, W.; Su, J.; Chen, R.; Qiu, H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Provides a New Combination Therapy in Pancreatic Cancer. Front. Immunol. 2021, 12, 762989. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Kaestner, V.; Mailankody, S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type—FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018, 4, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-Inducible Protein 10 (IP-10; CXCL10)-Deficient Mice Reveal a Role for IP-10 in Effector T Cell Generation and Trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef]
- Shi, Z.; Shen, J.; Qiu, J.; Zhao, Q.; Hua, K.; Wang, H. CXCL10 potentiates immune checkpoint blockade therapy in homologous recombination-deficient tumors. Theranostics 2021, 11, 7175–7187. [Google Scholar] [CrossRef]
- Reisländer, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef]
- Gong, E.A.Z.; Yang, Y.; Zhang, J.; Guo, W. Evaluation of 30 DNA damage response and 6 mismatch repair gene mutations as biomarkers for immunotherapy outcomes across multiple solid tumor types. Cancer Biol. Med. 2021, 18, 1080. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N.J. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Juliá, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front. Immunol. 2018, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, P.; Pilat, M.J.P.; Santa-Maria, C.A.; Connolly, R.M.; Roesch, E.E.; Afghahi, A.; Han, H.S.; Nanda, R.; Wulf, G.M.; Assad, H.; et al. Trial in progress: A phase II open-label, randomized study of PARP inhibition (olaparib) either alone or in combination with anti-PD-L1 therapy (atezolizumab) in homologous DNA repair (HDR) deficient, locally advanced or metastatic non-HER2-positive breast cancer. J. Clin. Oncol. 2020, 38, TPS1102. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Ettl, J.; Quek, R.; Lee, K.-H.; Rugo, H.; Hurvitz, S.; Gonçalves, A.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.; Martin, M.; et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: Patient-reported outcomes from the EMBRACA phase III trial. Ann. Oncol. 2018, 29, 1939–1947. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Adams, S.; Diéras, V.; Barrios, C.; Winer, E.; Schneeweiss, A.; Iwata, H.; Loi, S.; Patel, S.; Henschel, V.; Chui, S.; et al. Patient-reported outcomes from the phase III IMpassion130 trial of atezolizumab plus nab-paclitaxel in metastatic triple-negative breast cancer. Ann. Oncol. 2020, 31, 582–589. [Google Scholar] [CrossRef]
- Wang, H.; Ma, H.; Sové, R.J.; Emens, L.A.; Popel, A.S. Quantitative systems pharmacology model predictions for efficacy of atezolizumab and nab-paclitaxel in triple-negative breast cancer. J. Immunother. Cancer 2021, 9, e002100. [Google Scholar] [CrossRef]
- Litton, J.; Hurvitz, S.; Mina, L.; Rugo, H.; Lee, K.-H.; Gonçalves, A.; Diab, S.; Woodward, N.; Goodwin, A.; Yerushalmi, R.; et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: Final overall survival results from the EMBRACA trial. Ann. Oncol. 2020, 31, 1526–1535. [Google Scholar] [CrossRef]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2015, 140, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, Q.; Wang, Y.-N.; Jin, Y.; He, M.-M.; Liu, Z.-X.; Xu, R.-H. Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes across Multiple Cancer Types. JAMA Oncol. 2019, 5, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Seyedin, S.N.; Hasibuzzaman, M.; Pham, V.; Petronek, M.S.; Callaghan, C.; Kalen, A.L.; Mapuskar, K.A.; Mott, S.L.; Spitz, D.R.; Allen, B.G.; et al. Combination Therapy with Radiation and PARP Inhibition Enhances Responsiveness to Anti-PD-1 Therapy in Colorectal Tumor Models. Int. J. Radiat. Oncol. 2020, 108, 81–92. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Nichols, E.; Cimino-Mathews, A.; Peer, C.; Cao, L.; Lee, M.-J.; Kohn, E.C.; Annunziata, C.M.; Lipkowitz, S.; Trepel, J.B.; et al. A phase I study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1–3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J. Immunother. Cancer 2019, 7, 197. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Van Den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef]
- Mur, P.; García-Mulero, S.; del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Calegari, A.M.; Inno, A.; Monterisi, S.; Orlandi, A.; Santini, D.; Basso, M.; Cassano, A.; Martini, M.; Cenci, T.; De Pascalis, I.; et al. A phase 2 study of temozolomide in pretreated metastatic colorectal cancer with MGMT promoter methylation. Br. J. Cancer 2017, 116, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Yurgelun, M.B.; Goggins, M.G. Genetics of Familial and Sporadic Pancreatic Cancer. Gastroenterology 2019, 156, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Seeber, A.; Zimmer, K.; Kocher, F.; Puccini, A.; Xiu, J.; Nabhan, C.; Elliott, A.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; et al. Molecular characteristics of BRCA1/2 and PALB2 mutations in pancreatic ductal adenocarcinoma. ESMO Open 2020, 5, e000942. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Gray, R.J.; Chen, A.P.; Li, S.; McShane, L.M.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Iafrate, A.J.; Sklar, J.; et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 2020, 38, 3883–3894. [Google Scholar] [CrossRef]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A., III; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Lowery, M.A.; Kelsen, D.P.; Capanu, M.; Smith, S.C.; Lee, J.W.; Stadler, Z.K.; Moore, M.J.; Kindler, H.L.; Golan, T.; Segal, A.; et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur. J. Cancer 2017, 89, 19–26. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Hwang, J.J.; He, A.R.; Smaglo, B.G.; Kim, S.S.; Weinberg, B.A.; Weiner, L.M.; Marshall, J.L.; Brody, J.R. A Phase I/II Study of Veliparib (ABT-888) in Combination with 5-Fluorouracil and Oxaliplatin in Patients with Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5092–5101. [Google Scholar] [CrossRef]
- Takeuchi, S.; Doi, M.; Ikari, N.; Yamamoto, M.; Furukawa, T. Mutations in BRCA1, BRCA2, and PALB2, and a panel of 50 cancer-associated genes in pancreatic ductal adenocarcinoma. Sci. Rep. 2018, 8, 8105. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Keam, S.J. Selumetinib: First Approval. Drugs 2020, 80, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, N.; Tan, D. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Saner, F.A.M.; Herschtal, A.; Nelson, B.H.; DeFazio, A.; Goode, E.L.; Ramus, S.J.; Pandey, A.; Beach, J.A.; Fereday, S.; Berchuck, A.; et al. Going to extremes: Determinants of extraordinary response and survival in patients with cancer. Nat. Cancer 2019, 19, 339–348. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; LaFleur, M.; Nguyen, T.; Chen, S.; Chakravarthy, A.; Conway, J.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-L.; Chang, Y.-T.; Hong, Z.-Y.; Lin, C.-S. Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy. Int. J. Mol. Sci. 2022, 23, 3238. https://doi.org/10.3390/ijms23063238
Huang J-L, Chang Y-T, Hong Z-Y, Lin C-S. Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy. International Journal of Molecular Sciences. 2022; 23(6):3238. https://doi.org/10.3390/ijms23063238
Chicago/Turabian StyleHuang, Jau-Ling, Yu-Tzu Chang, Zhen-Yang Hong, and Chang-Shen Lin. 2022. "Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy" International Journal of Molecular Sciences 23, no. 6: 3238. https://doi.org/10.3390/ijms23063238
APA StyleHuang, J.-L., Chang, Y.-T., Hong, Z.-Y., & Lin, C.-S. (2022). Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy. International Journal of Molecular Sciences, 23(6), 3238. https://doi.org/10.3390/ijms23063238