
Molecular Mechanisms Involved in Hypoxia-Induced Alterations in Bone Remodeling


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bone Remodeling and Cells Involved
2.1. Osteocytes
2.2. Osteoclasts
2.3. Osteoblasts
3. Hypoxia: Local Effects on Bone Metabolism Cells
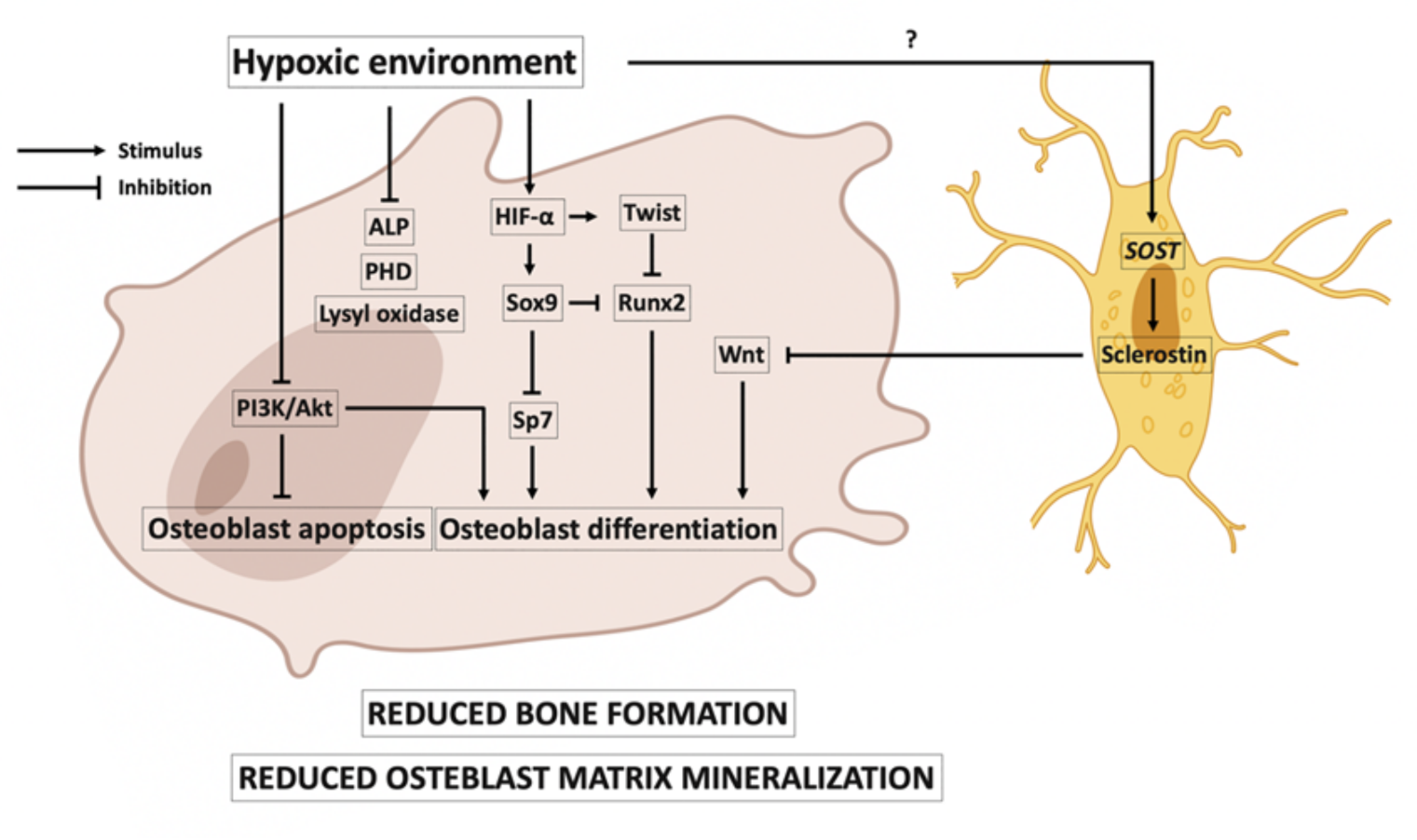
3.1. Hypoxia and Osteoblasts
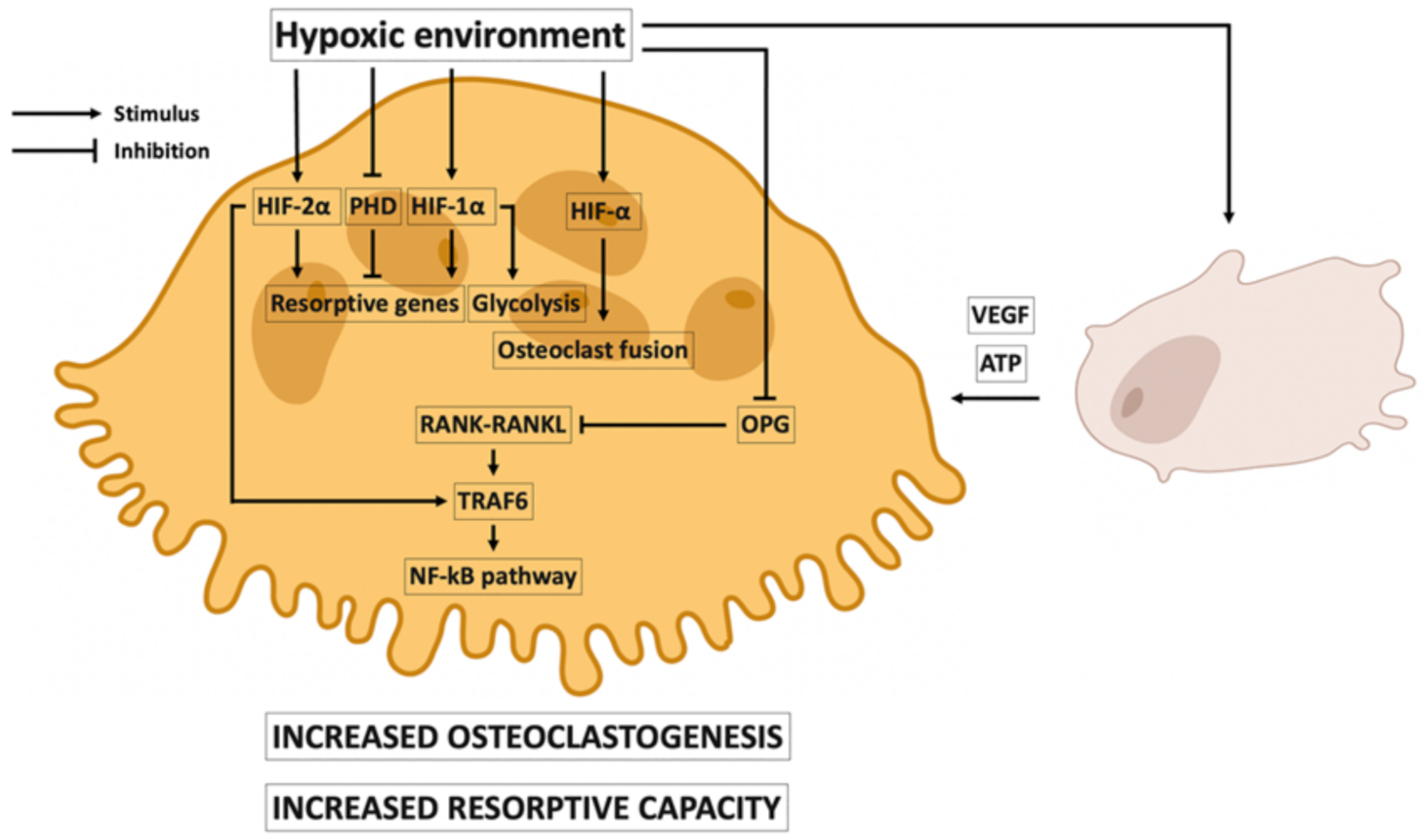
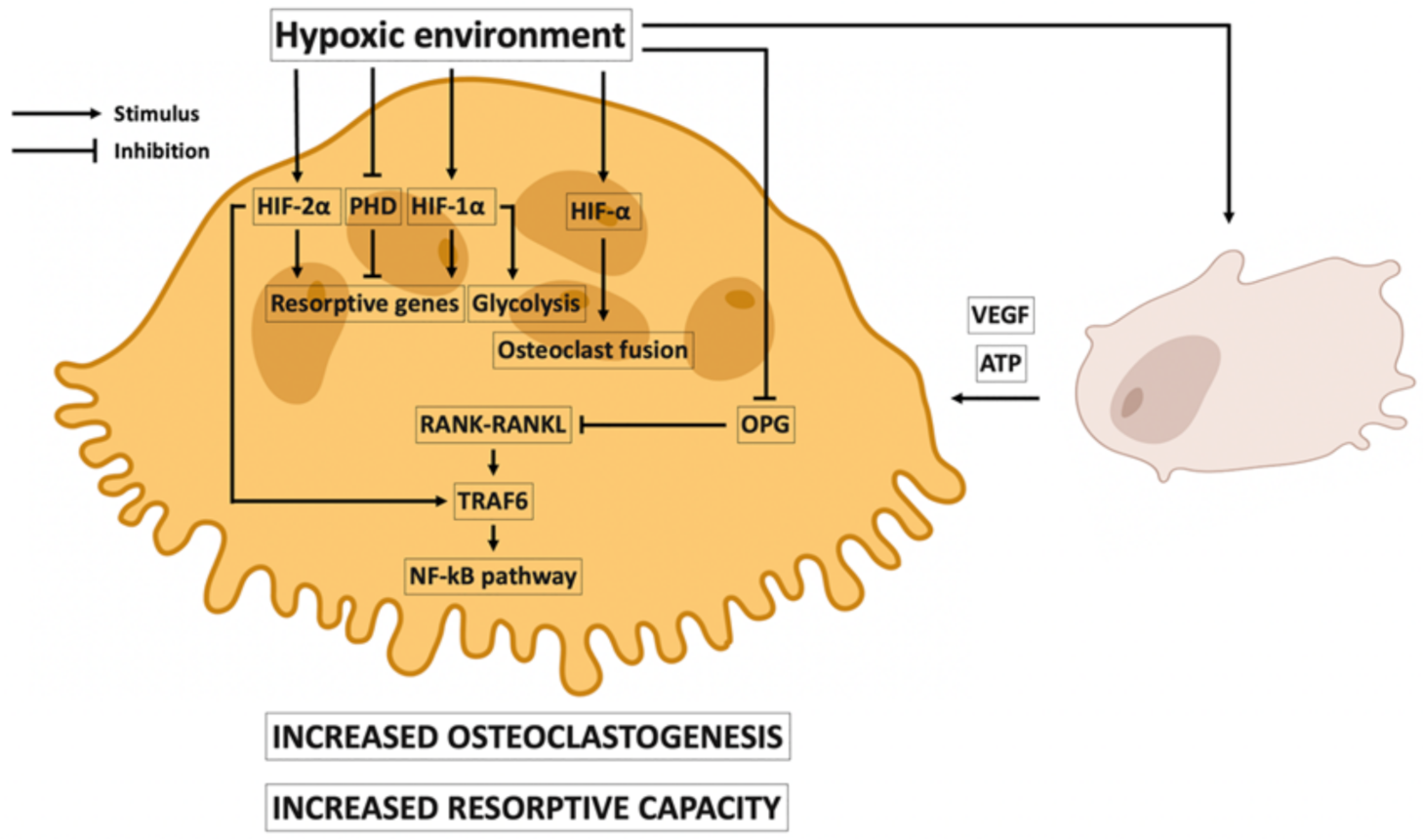
3.2. Local Effects of Hypoxia on Osteoclasts
4. Hypoxia, Erythropoietin, and Bone Remodeling
5. Vitamin D Metabolism, Inflammation, Hypoxia, and Bone
6. Hypoxia and Bone Remodeling in Clinical Studies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ralston, S.H. Bone structure and metabolism. Medicine 2013, 41, 581–585. [Google Scholar] [CrossRef]
- Ninomiya, J.T.; Tracy, R.P.; Calore, J.D.; Gendreau, M.A.; Kelm, R.J.; Mann, K.G. Heterogeneity of human bone. J. Bone Miner. Res. 1990, 5, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.S.; Poundarik, A.A.; Cabral, J.M.S.; da Silva, C.L.; Vashishth, D. Biomimetic matrices for rapidly forming mineralized bone tissue based on stem cell-mediated osteogenesis. Sci. Rep. 2018, 8, 14388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3 (Suppl. 3), S131–S139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florencio-Silva, R.; da Silva Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, e421746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Malhotra, A.S.; Pal, K.; Chatterjee, T.; Ghosh, D.; Haldar, K.; Verma, S.K.; Kumar, S.; Sharma, Y.K.; Sawhney, R.C. Determination of bone mass using multisite quantitative ultrasound and biochemical markers of bone turnover during residency at extreme altitude: A longitudinal study. High Alt. Med. Biol. 2013, 14, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Malhotra, A.S.; Pal, K.; Kumar, R.; Bajaj, R.; Verma, S.K.; Ghosh, D.; Sharma, Y.K.; Sawhney, R.C. Alterations in different indices of skeletal health after prolonged residency at high altitude. High Alt. Med. Biol. 2014, 15, 170–175. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.A.; Pollock, R.D.; Stroud, M.; Lambert, R.J.; Kumar, A.; Atkinson, R.A.; Green, D.; Anton-Solanas, A.; Edwards, L.; Harridge, S. Human physiological and metabolic responses to an attempted winter crossing of Antarctica: The effects of prolonged hypobaric hypoxia. Physiol. Rep. 2018, 6, e13613. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. BoneKEy Rep. 2014, 3, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell … and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; O’Brien, C.A. Osteocyte RANKL: New insights into the control of bone remodeling. J. Bone Miner. Res. 2012, 27, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonewald, L.F.; Johnson, M.L. Osteocytes, mechanosensing and Wnt signaling. Bone 2008, 42, 606–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Schoppet, M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA 2004, 292, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Bollerslev, J.; Everts, V.; Karsdal, M.A. Osteoclast activity and subtypes as a function of physiology and pathology--implications for future treatments of osteoporosis. Endocr. Rev. 2011, 32, 31–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2011, 13, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Komori, T.; Suda, T. Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehogs, and Cbfa1. Endocr. Rev. 2000, 21, 393–411. [Google Scholar] [CrossRef]
- Dirckx, N.; Moorer, M.C.; Clemens, T.L.; Riddle, R.C. The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol. 2019, 15, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Corrado, A.; Cantatore, F.P. Osteoblast physiology in normal and pathological conditions. Cell Tissue Res. 2011, 343, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J. Fibroblast growth factor signaling controlling bone formation: An update. Gene 2012, 498, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Michigami, T.; Ozono, K. Wnt signaling in bone metabolism. J. Bone Miner. Metab. 2009, 27, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Andre, P.; Ye, L.; Yang, Y.-Z. The Hedgehog signalling pathway in bone formation. Int. J. Oral Sci. 2015, 7, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-L.; Shen, H.; Liu, A.; Dong, S.-S.; Zhang, L.; Deng, F.-Y.; Zhao, Q.; Deng, H.W. A road map for understanding molecular and genetic determinants of osteoporosis. Nat. Rev. Endocrinol. 2020, 16, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Zepeda, A.B.; Pessoa, A.; Castillo, R.L.; Figueroa, C.A.; Pulgar, V.M.; Farías, J.G. Cellular and molecular mechanisms in the hypoxic tissue: Role of HIF-1 and ROS. Cell Biochem. Funct. 2013, 31, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Déry, M.-A.C.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Kaluz, S.; Kaluzová, M.; Stanbridge, E.J. Rational design of minimal hypoxia-inducible enhancers. Biochem. Biophys. Res. Commun. 2008, 370, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, V.; Davis, D.A.; Haque, M.; Huang, L.E.; Yarchoan, R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005, 65, 3299–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.S.; Rameshwar, P.; Chang, V.; Bandari, P. Oxygen saturation in the bone marrow of healthy volunteers. Blood 2002, 99, 394. [Google Scholar] [CrossRef] [PubMed]
- Arnett, T.R. Acidosis, hypoxia and bone. Arch. Biochem. Biophys. 2010, 503, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Marenzana, M.; Arnett, T.R. The Key Role of the Blood Supply to Bone. Bone Res. 2013, 1, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utting, J.C.; Robins, S.P.; Brandao-Burch, A.; Orriss, I.R.; Behar, J.; Arnett, T.R. Hypoxia inhibits the growth, differentiation and bone-forming capacity of rat osteoblasts. Exp. Cell Res. 2006, 312, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.; Sun, D.; Xin, J.; Wang, L.; Huang, D.; Wu, W.; Xian, C.J. Short-Term Hypoxia Accelerates Bone Loss in Ovariectomized Rats by Suppressing Osteoblastogenesis but Enhancing Osteoclastogenesis. Med. Sci. Monit. 2016, 22, 2962–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T. Regulation of osteoblast differentiation by Runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49. [Google Scholar] [PubMed]
- Ontiveros, C.; Irwin, R.; Wiseman, R.W.; McCabe, L.R. Hypoxia suppresses runx2 independent of modeled microgravity. J. Cell. Physiol. 2004, 200, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-H.; Wu, K.-J. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle 2008, 7, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Merceron, C.; Ranganathan, K.; Wang, E.; Tata, Z.; Makkapati, S.; Khan, M.P.; Mangiavini, L.; Yao, A.Q.; Castellini, L.; Levi, B.; et al. Hypoxia-inducible factor 2α is a negative regulator of osteoblastogenesis and bone mass accrual. Bone Res. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Yang, S.; Zhang, T.; Sun, H.; Wang, Y.; Xue, H.; Zhou, D. Hypoxia enhances glucocorticoid-induced apoptosis and cell cycle arrest via the PI3K/Akt signaling pathway in osteoblastic cells. J. Bone Miner. Metab. 2015, 33, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Kubota, T.; Wang, W.; Ohata, Y.; Miura, K.; Kitaoka, T.; Okuzaki, D.; Namba, N.; Michigami, T.; Kitabatake, Y.; et al. Successful induction of sclerostin in human-derived fibroblasts by 4 transcription factors and its regulation by parathyroid hormone, hypoxia, and prostaglandin E2. Bone 2016, 85, 91–98. [Google Scholar] [CrossRef]
- Genetos, D.C.; Toupadakis, C.A.; Raheja, L.F.; Wong, A.; Papanicolaou, S.E.; Fyhrie, D.P.; Loots, G.; Yellowley, C.E. Hypoxia decreases sclerostin expression and increases Wnt signaling in osteoblasts. J. Cell. Biochem. 2010, 110, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Myllyharju, J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 2003, 22, 15–24. [Google Scholar] [CrossRef]
- Schipani, E.; Maes, C.; Carmeliet, G.; Semenza, G.L. Regulation of osteogenesis-angiogenesis coupling by HIFs and VEGF. J. Bone Miner. Res. 2009, 24, 1347–1353. [Google Scholar] [CrossRef]
- Orriss, I.R.; Knight, G.E.; Utting, J.C.; Taylor, S.E.B.; Burnstock, G.; Arnett, T.R. Hypoxia stimulates vesicular ATP release from rat osteoblasts. J. Cell. Physiol. 2009, 220, 155–162. [Google Scholar] [CrossRef]
- Arnett, T.R.; Gibbons, D.C.; Utting, J.C.; Orriss, I.R.; Hoebertz, A.; Rosendaal, M.; Meghji, S. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J. Cell. Physiol. 2003, 196, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Utting, J.C.; Flanagan, A.M.; Brandao-Burch, A.; Orriss, I.R.; Arnett, T.R. Hypoxia stimulates osteoclast formation from human peripheral blood. Cell Biochem. Funct. 2010, 28, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J. Distinct roles for the hypoxia-inducible transcription factors HIF-1α and HIF-2α in human osteoclast formation and function. Sci. Rep. 2020, 10, 21072. [Google Scholar] [CrossRef] [PubMed]
- Hulley, P.A.; Bishop, T.; Vernet, A.; Schneider, J.E.; Edwards, J.R.; Athanasou, N.A.; Knowles, H.J. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2. J. Pathol. 2017, 242, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J. Hypoxia, hypoxia-inducible factor (HIF) and bone homeostasis: Focus on osteoclast-mediated bone resorption. Trends Cell Mol. Biol. 2015, 10, 91–104. [Google Scholar]
- Lee, S.Y.; Park, K.H.; Yu, H.-G.; Kook, E.; Song, W.-H.; Lee, G.; Koh, J.T.; Shin, H.I.; Choi, J.Y.; Huh, Y.H.; et al. Controlling hypoxia-inducible factor-2α is critical for maintaining bone homeostasis in mice. Bone Res. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- ShIrakura, M.; Tanimoto, K.; Eguchi, H.; Miyauchi, M.; Nakamura, H.; Hiyama, K.; Tanimoto, K.; Tanka, E.; Takata, T.; Tanne, K. Activation of the Hypoxia-Inducible Factor-1 in Overloaded Temporomandibular Joint, and Induction of Osteoclastogenesis. Biochemical and Biophysical Research Communications. 2010. Available online: https://www.scienceopen.com/document?vid=4da73899-94ed-4bce-8d0c-74e38794cb82 (accessed on 18 February 2022).
- Kanemoto, S.; Kobayashi, Y.; Yamashita, T.; Miyamoto, T.; Cui, M.; Asada, R.; Cui, X.; Hino, K.; Kaneko, M.; Takai, T.; et al. Luman is involved in osteoclastogenesis through the regulation of DC-STAMP expression, stability and localization. J. Cell Sci. 2015, 128, 4353–4365. [Google Scholar] [CrossRef] [Green Version]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589 Pt 6, 1251–1258. [Google Scholar] [CrossRef]
- Oikonomidou, P.R.; Casu, C.; Yang, Z.; Crielaard, B.; Shim, J.H.; Rivella, S.; Vogiatzi, M.G. Polycythemia is associated with bone loss and reduced osteoblast activity in mice. Osteoporos. Int. 2016, 27, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Farmer, S.; Horváth-Puhó, E.; Vestergaard, H.; Hermann, A.P.; Frederiksen, H. Chronic myeloproliferative neoplasms and risk of osteoporotic fractures; a nationwide population-based cohort study. Br. J. Haematol. 2013, 163, 603–610. [Google Scholar] [CrossRef]
- Kim, J.; Jung, Y.; Sun, H.; Joseph, J.; Mishra, A.; Shiozawa, Y.; Wang, J.; Krebsbach, P.H.; Taichman, R.S. Erythropoietin mediated bone formation is regulated by mTOR signaling. J. Cell. Biochem. 2012, 113, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Rölfing, J.H.D.; Baatrup, A.; Stiehler, M.; Jensen, J.; Lysdahl, H.; Bünger, C. The osteogenic effect of erythropoietin on human mesenchymal stromal cells is dose-dependent and involves non-hematopoietic receptors and multiple intracellular signaling pathways. Stem Cell Rev. Rep. 2014, 10, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Zhang, F.; He, Q.; Tsang, W.P.; Lu, L.; Li, Q.; Wu, Z.; Qiu, G.; Zhou, G.; Wan, C. EPO promotes bone repair through enhanced cartilaginous callus formation and angiogenesis. PLoS ONE 2014, 9, e102010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggold, J.T.; Rankin, E.B. Erythropoiesis, EPO, macrophages, and bone. Bone 2019, 119, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, Z.; Song, J.; Wang, J.; Lee, C.H.; Sud, S.; et al. Erythropoietin couples hematopoiesis with bone formation. PLoS ONE 2010, 5, e10853. [Google Scholar] [CrossRef] [PubMed]
- Deshet-Unger, N.; Hiram-Bab, S.; Haim-Ohana, Y.; Mittelman, M.; Gabet, Y.; Neumann, D. Erythropoietin treatment in murine multiple myeloma: Immune gain and bone loss. Sci. Rep. 2016, 6, 30998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiram-Bab, S.; Neumann, D.; Gabet, Y. Erythropoietin in bone—Controversies and consensus. Cytokine 2017, 89, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yoshiko, Y.; Yamamoto, R.; Minamizaki, T.; Kozai, K.; Tanne, K.; Aubin, J.E.; Maeda, N. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J. Bone Miner. Res. 2008, 23, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Rauner, M.; Franke, K.; Murray, M.; Singh, R.P.; Hiram-Bab, S.; Platzbecker, U.; Gassmann, M.; Socolovsky, M.; Neumann, D.; Gabet, Y.; et al. Increased EPO Levels Are Associated with Bone Loss in Mice Lacking PHD2 in EPO-Producing Cells. J. Bone Miner. Res. 2016, 31, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Hiram-Bab, S.; Liron, T.; Deshet-Unger, N.; Mittelman, M.; Gassmann, M.; Rauner, M.; Franke, K.; Wielockx, B.; Neumann, D.; Gabet, Y. Erythropoietin directly stimulates osteoclast precursors and induces bone loss. FASEB J. 2015, 29, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.-C.; Jurutka, P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.-C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, M.J.; Ghosh, S. Signal transduction through NF-kappa B. Immunol. Today 1998, 19, 80–88. [Google Scholar]
- Won, S.; Sayeed, I.; Peterson, B.L.; Wali, B.; Kahn, J.S.; Stein, D.G. Vitamin D prevents hypoxia/reoxygenation-induced blood-brain barrier disruption via vitamin D receptor-mediated NF-kB signaling pathways. PLoS ONE 2015, 10, e0122821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D receptor inhibits nuclear factor κB activation by interacting with IκB kinase β protein. J. Biol. Chem. 2013, 288, 19450–19458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Wang, L.; Li, M.; Xu, N.; Yu, F.; Yang, F.; Li, R.; Zhang, F.; Zhao, B.; Du, J. Vitamin D/VDR signaling inhibits LPS-induced IFNγ and IL-1β in Oral epithelia by regulating hypoxia-inducible factor-1α signaling pathway. Cell Commun. Signal. 2019, 17, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Du, J.; Zhang, Z.; Liu, T.; Shi, Y.; Ge, X.; Li, Y.C. MicroRNA-346 Mediates Tumor Necrosis Factor-α-induced Down-Regulation of Gut Epithelial Vitamin D Receptor in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2014, 20, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Miyauchi, Y.; Yoshida, S.; Morita, M.; Kobayashi, T.; Kanagawa, H.; Katsuyama, E.; Fujie, A.; Hao, W.; Tando, T.; et al. The Vitamin D Analogue ED71 but Not 1,25(OH)2D3 Targets HIF1α Protein in Osteoclasts. PLoS ONE 2014, 9, e111845. [Google Scholar] [CrossRef] [PubMed]
- Rittweger, J.; Debevec, T.; Frings-Meuthen, P.; Lau, P.; Mittag, U.; Ganse, B.; Ferstl, P.G.; Simpson, E.J.; Macdonald, I.A.; Eiken, O.; et al. On the combined effects of normobaric hypoxia and bed rest upon bone and mineral metabolism: Results from the PlanHab study. Bone 2016, 91, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchavova, M.; Zikan, V.; Michalska, D.; Raska, I.; Kubena, A.A.; Stepan, J.J. The effect of timing of teriparatide treatment on the circadian rhythm of bone turnover in postmenopausal osteoporosis. Eur. J. Endocrinol. 2011, 164, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiyama, H.; Okazaki, R.; Inoue, D.; Ochiai, H.; Shiina, K.; Takata, Y.; Hashimoto, H.; Yamashina, A. Link between obstructive sleep apnea and increased bone resorption in men. Osteoporos. Int. 2008, 19, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Eimar, H.; Saltaji, H.; Ghorashi, S.; Isfeld, D.; MacLean, J.E.; Gozal, D.; Graf, D.; Flores-Mir, C. Association between sleep apnea and low bone mass in adults: A systematic review and meta-analysis. Osteoporos. Int. 2017, 28, 1835–1852. [Google Scholar] [CrossRef] [PubMed]
- Sforza, E.; Thomas, T.; Barthélémy, J.-C.; Collet, P.; Roche, F. Obstructive Sleep Apnea is Associated with Preserved Bone Mineral Density in Healthy Elderly Subjects. Sleep 2013, 36, 1509–1515. [Google Scholar] [PubMed] [Green Version]
- Cauley, J.A.; Blackwell, T.L.; Redline, S.; Ensrud, K.E.; Ancoli-Israel, S.; Fink, H.A.; Orwoll, E.S.; Stone, K.L. Hypoxia During Sleep and the Risk of Falls and Fractures in Older Men: The Osteoporotic Fracture in Men Sleep Study. J. Am. Geriatr. Soc. 2014, 62, 1853–1859. [Google Scholar] [CrossRef]
- Choi, S.B.; Lyu, I.S.; Lee, W.; Kim, D.W. Increased fragility fracture risk in Korean women who snore: A 10-year population-based prospective cohort study. BMC Musculoskelet. Disord. 2017, 18, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usategui-Martín, R.; Rigual, R.; Ruiz-Mambrilla, M.; Fernández-Gómez, J.-M.; Dueñas, A.; Pérez-Castrillón, J.L. Molecular Mechanisms Involved in Hypoxia-Induced Alterations in Bone Remodeling. Int. J. Mol. Sci. 2022, 23, 3233. https://doi.org/10.3390/ijms23063233
Usategui-Martín R, Rigual R, Ruiz-Mambrilla M, Fernández-Gómez J-M, Dueñas A, Pérez-Castrillón JL. Molecular Mechanisms Involved in Hypoxia-Induced Alterations in Bone Remodeling. International Journal of Molecular Sciences. 2022; 23(6):3233. https://doi.org/10.3390/ijms23063233
Chicago/Turabian StyleUsategui-Martín, Ricardo, Ricardo Rigual, Marta Ruiz-Mambrilla, José-María Fernández-Gómez, Antonio Dueñas, and José Luis Pérez-Castrillón. 2022. "Molecular Mechanisms Involved in Hypoxia-Induced Alterations in Bone Remodeling" International Journal of Molecular Sciences 23, no. 6: 3233. https://doi.org/10.3390/ijms23063233
APA StyleUsategui-Martín, R., Rigual, R., Ruiz-Mambrilla, M., Fernández-Gómez, J.-M., Dueñas, A., & Pérez-Castrillón, J. L. (2022). Molecular Mechanisms Involved in Hypoxia-Induced Alterations in Bone Remodeling. International Journal of Molecular Sciences, 23(6), 3233. https://doi.org/10.3390/ijms23063233