
BRET-Based Biosensors to Measure Agonist Efficacies in Histamine H1 Receptor-Mediated G Protein Activation, Signaling and Interactions with GRKs and β-Arrestins


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
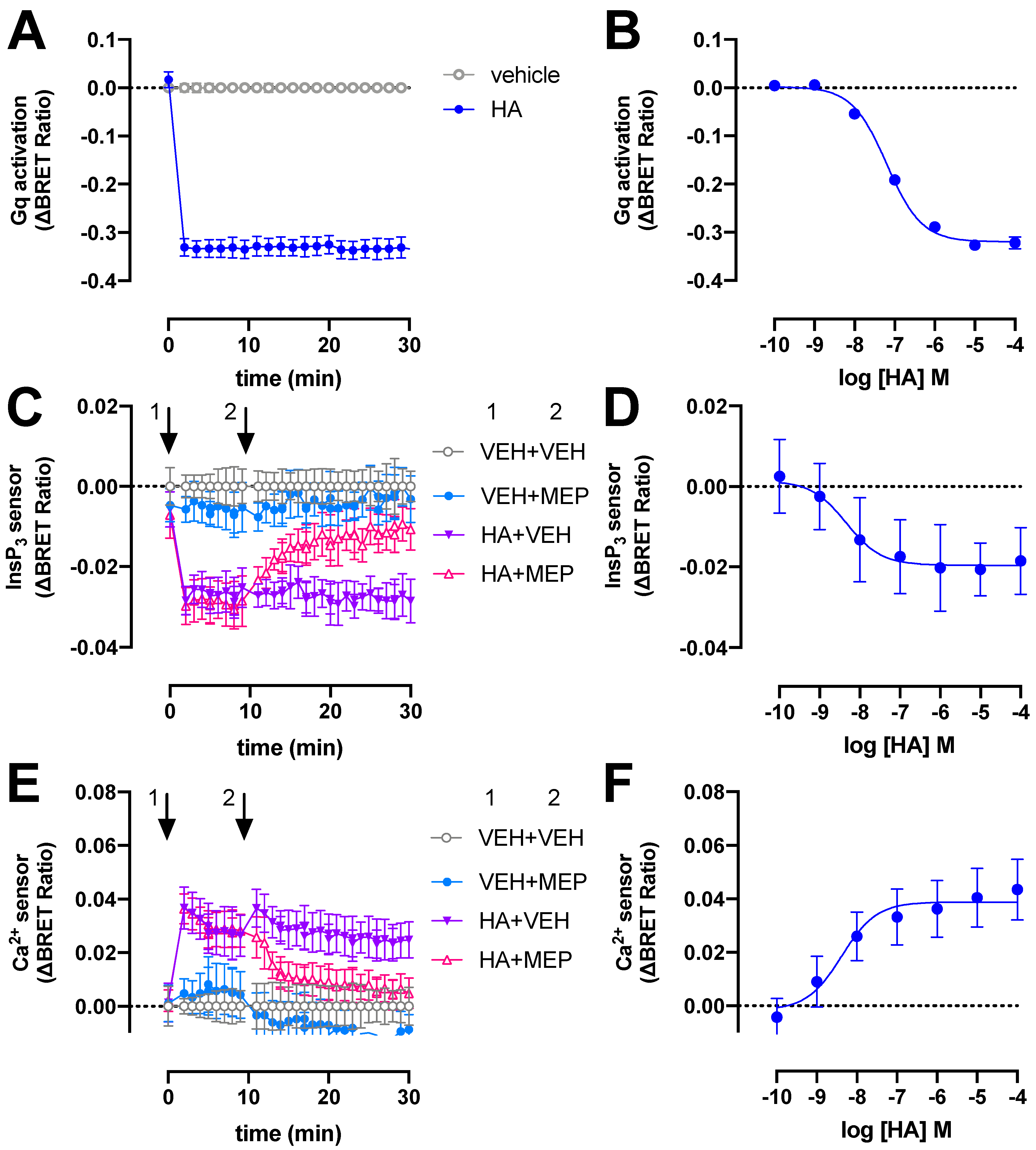
2.1. BRET-Based Detection of Gq Activation and Signaling by H1R
2.2. BRET-Based Detection of β-Arrestin1/2 Recruitment to H1R
2.3. BRET-Based Detection of GRKs Interaction with H1R
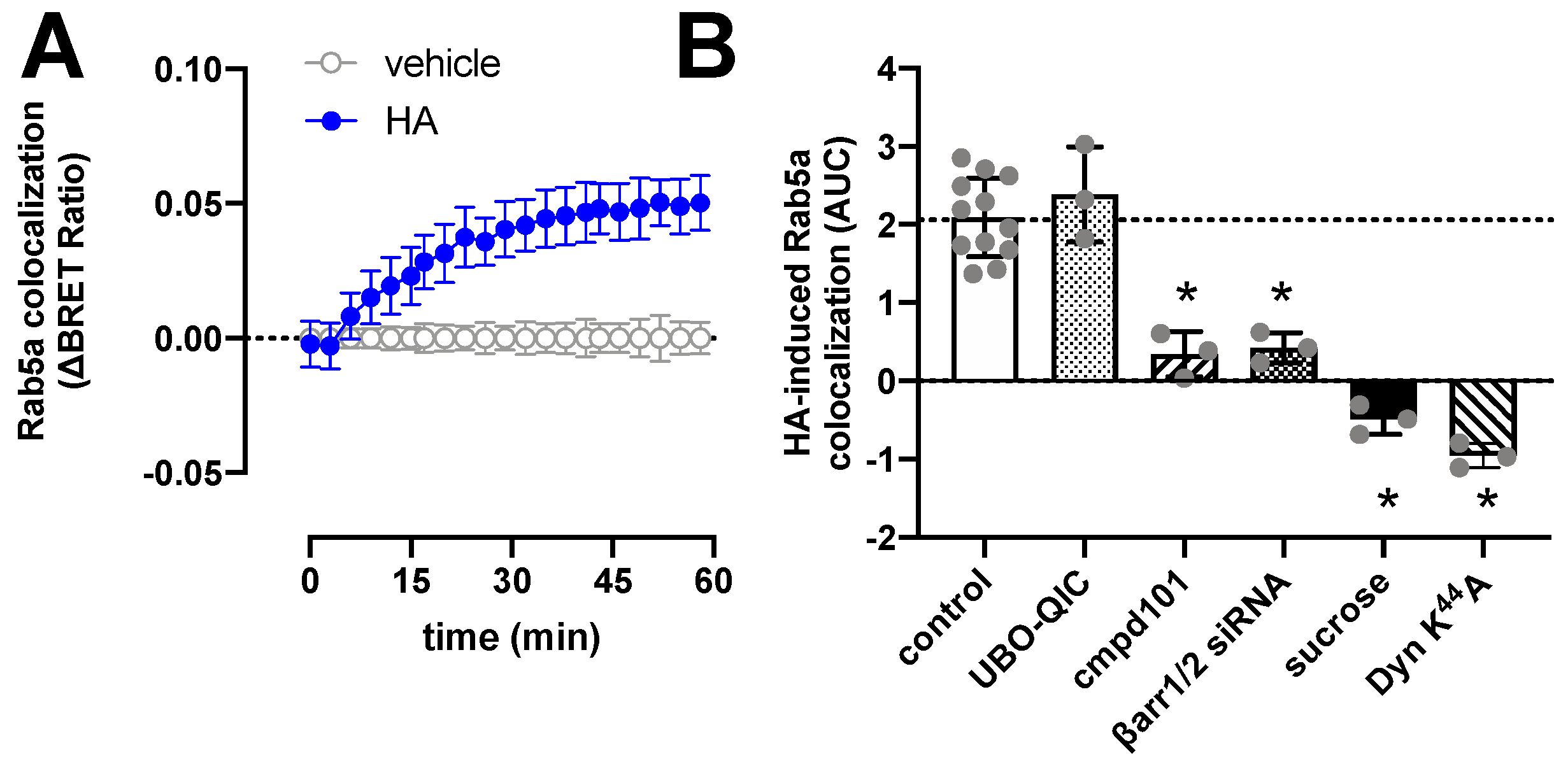
2.4. H1R Internalization Requires GRK2/3 and β-Arrestin1/2
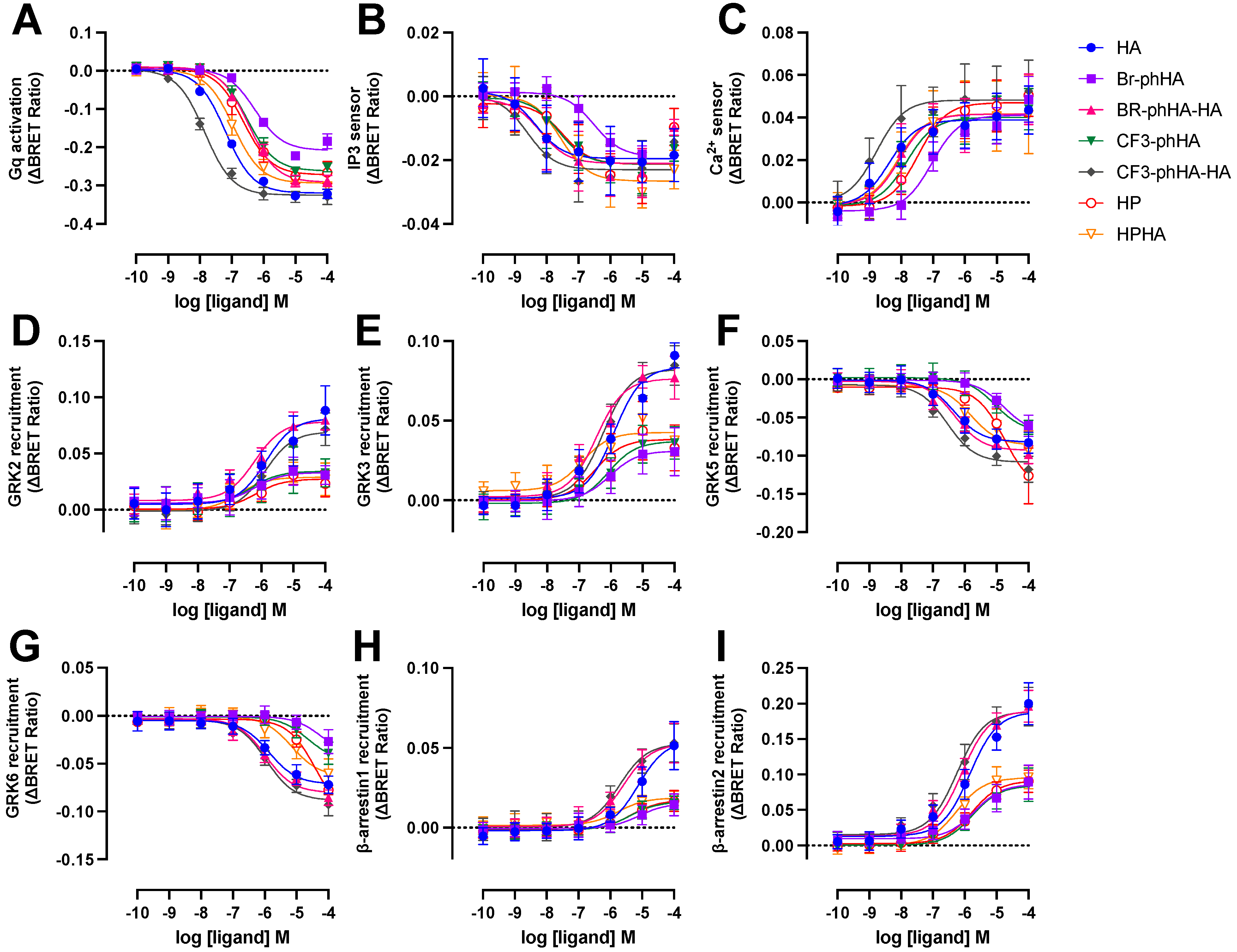
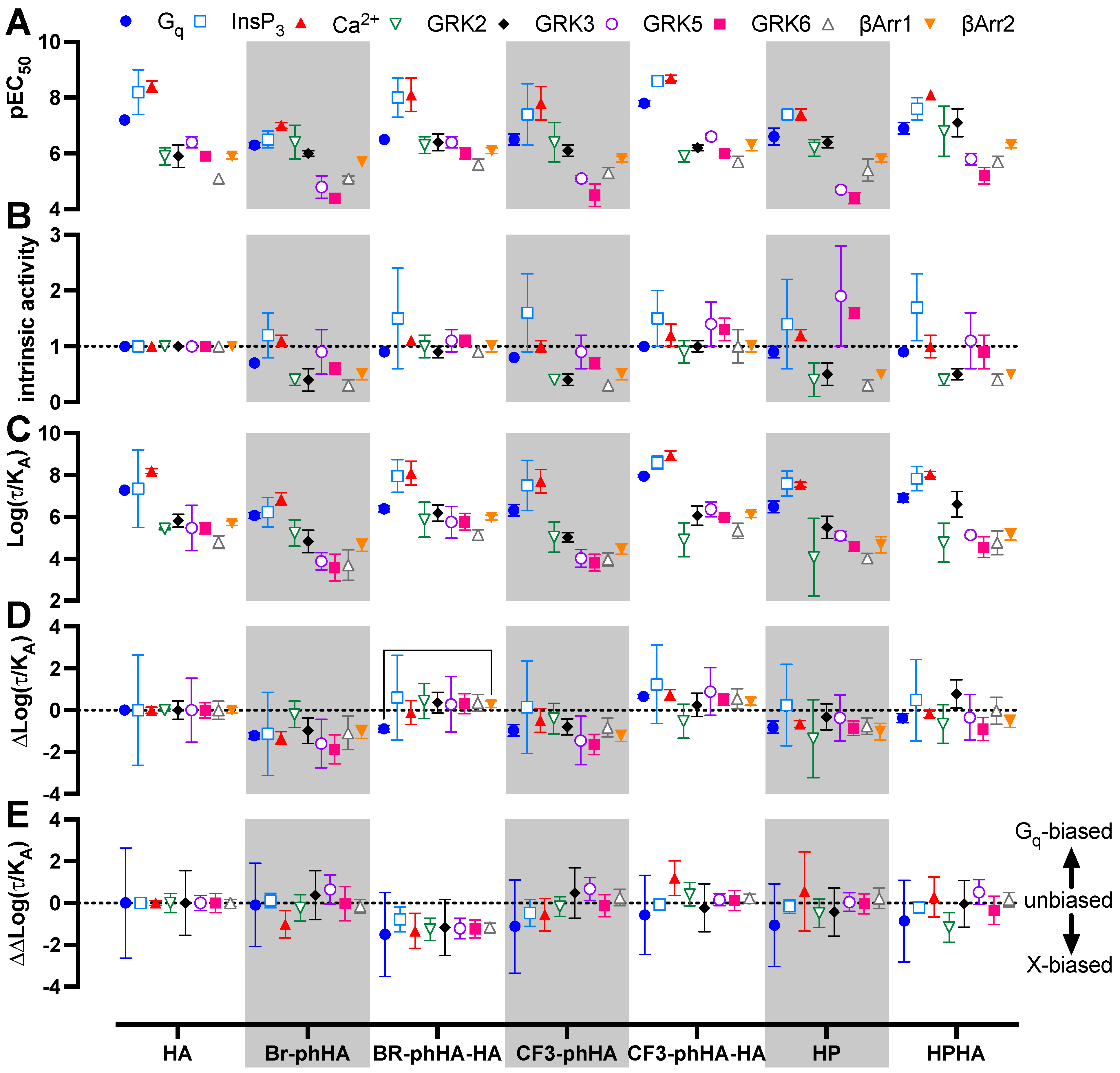
2.5. Histamine H1R Agonists Display Distinct Efficacies in BRET-Based Responses in HEK293T Cells
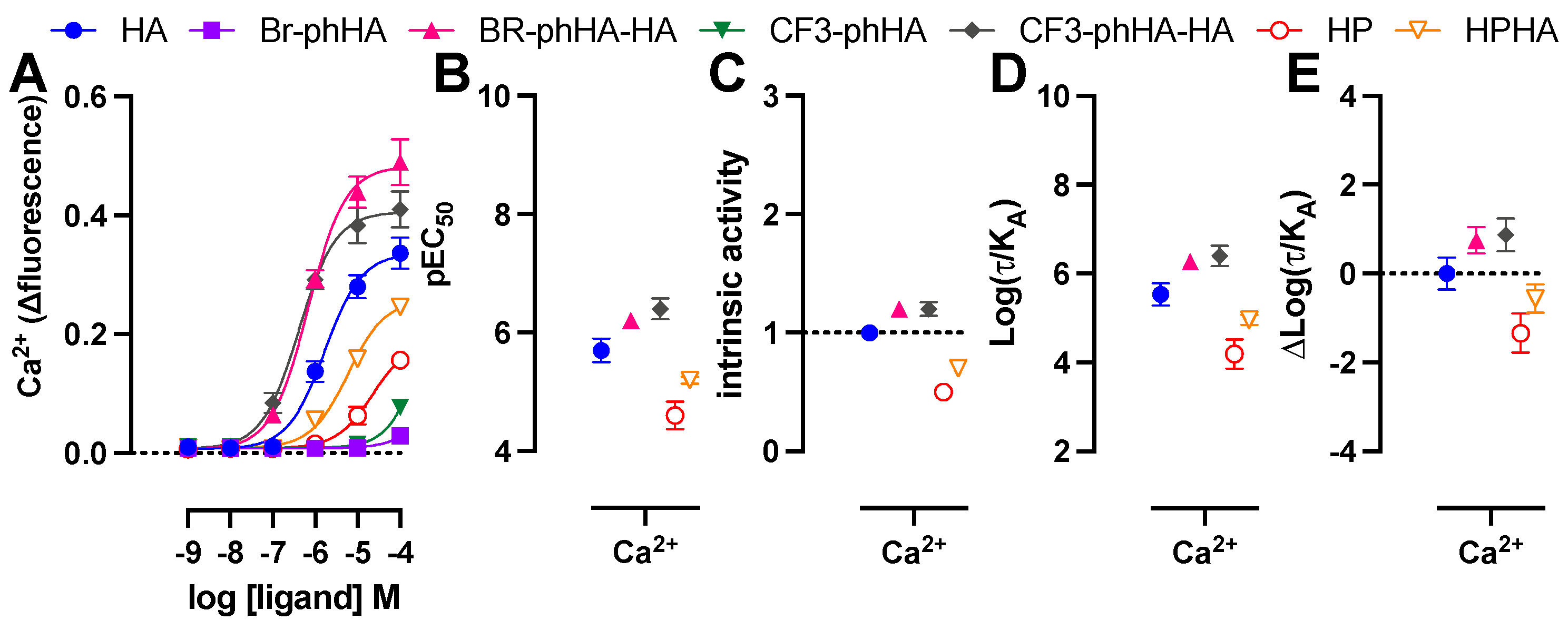
2.6. Histamine H1R Agonists Induced Ca2+ Mobilization in HeLa Cells Endogenously Expressing H1R
3. Materials and Methods
3.1. Materials
3.2. DNA Constructs
3.3. Cell Culture and Transfection
3.4. Radioligand Binding Experiments
3.5. Nuclear Factor Activated T-cells (NFAT)-Driven Reporter Gene Assay
3.6. Bioluminescence Resonance Energy Transfer (BRET) Assays
3.7. Western Blot
3.8. Intracellular Ca2+ Mobilization in HeLa Cells
3.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thurmond, R.L.; Gelfand, E.W.; Dunford, P.J. The role of histamine H1 and H4 receptors in allergic inflammation: The search for new antihistamines. Nat. Rev. Drug Discov. 2008, 7, 41–53. [Google Scholar] [CrossRef]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.S.; Stark, H.; Thurmond, R.L.; Haas, H.L. International union of basic and clinical pharmacology. XCVIII. Histamine receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [Green Version]
- Tiligada, E.; Ennis, M. Histamine pharmacology: From sir Henry Dale to the 21st century. Br. J. Pharmacol. 2020, 177, 469–489. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. GPCR signaling regulation: The role of GRKs and arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Peterson, Y.K.; Luttrell, L.M. The diverse roles of arrestin scaffolds in G protein-coupled receptor signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef] [Green Version]
- Bosma, R.; Moritani, R.; Leurs, R.; Vischer, H.F. BRET-based β-arrestin2 recruitment to the histamine H1 receptor for investigating antihistamine binding kinetics. Pharmacol. Res. 2016, 111, 679–687. [Google Scholar] [CrossRef]
- Ma, X.; Leurs, R.; Vischer, H.F. NanoLuc-based methods to measure β-arrestin2 recruitment to G protein-coupled receptors. Methods Mol. Biol. 2021, 2268, 233–248. [Google Scholar]
- Lieb, S.; Michaelis, S.; Plank, N.; Bernhardt, G.; Buschauer, A.; Wegener, J. Label-free analysis of GPCR-stimulation: The critical impact of cell adhesion. Pharmacol. Res. 2016, 108, 65–74. [Google Scholar] [CrossRef]
- Littmann, T.; Buschauer, A.; Bernhardt, G. Split luciferase-based assay for simultaneous analyses of the ligand concentration- and time-dependent recruitment of β-arrestin2. Anal. Biochem. 2019, 573, 8–16. [Google Scholar] [CrossRef]
- Brighton, P.J.; Rana, S.; Challiss, R.J.; Konje, J.C.; Willets, J.M. Arrestins differentially regulate histamine- and oxytocin-evoked phospholipase C and mitogen-activated protein kinase signalling in myometrial cells. Br. J. Pharmacol. 2011, 162, 1603–1617. [Google Scholar] [CrossRef] [Green Version]
- Iwata, K.; Luo, J.; Penn, R.B.; Benovic, J.L. Bimodal regulation of the human H1 histamine receptor by G protein-coupled receptor kinase 2. J. Biol. Chem. 2005, 280, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Willets, J.M.; Taylor, A.H.; Shaw, H.; Konje, J.C.; Challiss, R.A.J. Selective regulation of H1 histamine receptor signaling by G protein-coupled receptor kinase 2 in uterine smooth muscle cells. Mol. Endocrinol. 2008, 22, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Wootten, D.; Christopoulos, A.; Martí-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 27, 129. [Google Scholar] [CrossRef]
- Wingler, L.M.; Lefkowitz, R.J. Conformational basis of G protein-coupled receptor signaling versatility. Trends Cell Biol. 2020, 30, 736–747. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Masureel, M.; Hollingsworth, S.A.; Heydenreich, F.M.; Suomivuori, C.-M.; Brinton, C.; Townshend, R.J.L.; Bouvier, M.; Kobilka, B.K.; Dror, R.O. How GPCR phosphorylation patterns orchestrate arrestin-mediated signaling. Cell 2020, 183, 1813–1825.e18. [Google Scholar] [CrossRef]
- Butcher, A.J.; Prihandoko, R.; Kong, K.C.; McWilliams, P.; Edwards, J.M.; Bottrill, A.; Mistry, S.; Tobin, A.B. Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 2011, 286, 11506–11518. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yang, F.; Zhang, D.; Liu, Z.; Lin, A.; Liu, C.; Xiao, P.; Yu, X.; Sun, J.-P. Phosphorylation of G protein-coupled receptors: From the barcode hypothesis to the flute model. Mol. Pharmacol. 2017, 92, 201–210. [Google Scholar] [CrossRef]
- Zidar, D.A.; Violin, J.D.; Whalen, E.J.; Lefkowitz, R.J. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 9649–9654. [Google Scholar] [CrossRef] [Green Version]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.-Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Kenakin, T. Biased receptor signaling in drug discovery. Pharmacol. Rev. 2019, 71, 267–315. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Oliceridine: First approval. Drugs 2020, 80, 1739–1744. [Google Scholar] [CrossRef]
- Schihada, H.; Shekhani, R.; Schulte, G. Quantitative assessment of constitutive G protein-coupled receptor activity with BRET-based G protein biosensors. Sci. Signal. 2021, 14, eabf1653. [Google Scholar] [CrossRef]
- Gulyás, G.; Tóth, J.T.; Tóth, D.J.; Kurucz, I.; Hunyady, L.; Balla, T.; Várnai, P. Measurement of inositol 1,4,5-trisphosphate in living cells using an improved set of resonance energy transfer-based biosensors. PLoS ONE 2015, 10, e0125601. [Google Scholar] [CrossRef] [Green Version]
- Adjobo-Hermans, M.J.W.; Goedhart, J.; van Weeren, L.; Nijmeijer, S.; Manders, E.M.M.; Offermanns, S.; Gadella, T.W.J. Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol. 2011, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Van Unen, J.; Rashidfarrokhi, A.; Hoogendoorn, E.; Postma, M.; Gadella, T.W.J.; Goedhart, J. Quantitative single-cell analysis of signaling pathways activated immediately downstream of histamine receptor subtypes. Mol. Pharmacol. 2016, 90, 162–176. [Google Scholar] [CrossRef] [Green Version]
- Höring, C.; Seibel, U.; Tropmann, K.; Grätz, L.; Mönnich, D.; Pitzl, S.; Bernhardt, G.; Pockes, S.; Strasser, A. A dynamic, split-luciferase-based mini-G protein sensor to functionally characterize ligands at all four histamine receptor subtypes. Int. J. Mol. Sci. 2020, 21, 8440. [Google Scholar] [CrossRef]
- Nehmé, R.; Carpenter, B.; Singhal, A.; Strege, A.; Edwards, P.C.; White, C.F.; Du, H.; Grisshammer, R.; Tate, C.G. Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS ONE 2017, 12, e0175642. [Google Scholar] [CrossRef]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N.; et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 2019, 177, 1933–1947.e25. [Google Scholar] [CrossRef]
- Olsen, R.H.J.; DiBerto, J.F.; English, J.G.; Glaudin, A.M.; Krumm, B.E.; Slocum, S.T.; Che, T.; Gavin, A.C.; McCorvy, J.D.; Roth, B.L.; et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16, 841–849. [Google Scholar] [CrossRef]
- Lieb, S.; Littmann, T.; Plank, N.; Felixberger, J.; Tanaka, M.; Schäfer, T.; Krief, S.; Elz, S.; Friedland, K.; Bernhardt, G.; et al. Label-free versus conventional cellular assays: Functional investigations on the human histamine H1 receptor. Pharmacol. Res. 2016, 114, 13–26. [Google Scholar] [CrossRef]
- Schrage, R.; Schmitz, A.-L.; Gaffal, E.; Annala, S.; Kehraus, S.; Wenzel, D.; Büllesbach, K.M.; Bald, T.; Inoue, A.; Shinjo, Y.; et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 2015, 6, 10156. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J.G. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Verweij, E.W.E.; Al Araaj, B.; Prabhata, W.R.; Prihandoko, R.; Nijmeijer, S.; Tobin, A.B.; Leurs, R.; Vischer, H.F. Differential role of serines and threonines in intracellular loop 3 and C-terminal tail of the histamine H4 receptor in β-arrestin and G protein-coupled receptor kinase interaction, internalization, and signaling. ACS Pharmacol. Transl. Sci. 2020, 3, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miess, E.; Gondin, A.B.; Yousuf, A.; Steinborn, R.; Mösslein, N.; Yang, Y.; Göldner, M.; Ruland, J.G.; Bünemann, M.; Krasel, C.; et al. Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid μ-opioid receptor desensitization. Sci. Signal. 2018, 11, eaas9609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, J.D.; Sanderson, H.S.; Cooke, A.E.; Ostovar, M.; Tsisanova, E.; Withey, S.L.; Chavkin, C.; Husbands, S.M.; Kelly, E.; Henderson, G.; et al. Role of G protein-coupled receptor kinases 2 and 3 in μ-opioid receptor desensitization and internalization. Mol. Pharmacol. 2015, 88, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Pack, T.F.; Orlen, M.I.; Ray, C.; Peterson, S.M.; Caron, M.G. The dopamine D2 receptor can directly recruit and activate GRK2 without G protein activation. J. Biol. Chem. 2018, 293, 6161–6171. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Hayata-Takano, A.; Moriguchi, K.; Nakazawa, T.; Ago, Y.; Kasai, A.; Seiriki, K.; Shintani, N.; Hashimoto, H. β-arrestin1 and 2 differentially regulate PACAP-induced PAC1 receptor signaling and trafficking. PLoS ONE 2018, 13, e0196946. [Google Scholar] [CrossRef] [PubMed]
- Drube, J.; Haider, R.S.; Matthees, E.S.F.; Reichel, M.; Zeiner, J.; Fritzwanker, S.; Ziegler, C.; Barz, S.; Klement, L.; Filor, J.; et al. GPCR kinase knockout cells reveal the impact of individual GRKs on arrestin binding and GPCR regulation. Nat. Commun. 2022, 13, 540. [Google Scholar] [CrossRef]
- Zarca, A.; Perez, C.; van den Bor, J.; Bebelman, J.P.; Heuninck, J.; de Jonker, R.J.F.; Durroux, T.; Vischer, H.F.; Siderius, M.; Smit, M.J. Differential involvement of ACKR3 C-tail in β-arrestin recruitment, trafficking and internalization. Cells 2021, 10, 618. [Google Scholar] [CrossRef]
- Møller, T.C.; Pedersen, M.F.; van Senten, J.R.; Seiersen, S.D.; Mathiesen, J.M.; Bouvier, M.; Bräuner-Osborne, H. Dissecting the roles of GRK2 and GRK3 in μ-opioid receptor internalization and β-arrestin2 recruitment using CRISPR/Cas9-edited HEK293 cells. Sci. Rep. 2020, 10, 17395. [Google Scholar] [CrossRef]
- Hasbi, A.; Devost, D.; Laporte, S.A.; Zingg, H.H. Real-time detection of interactions between the human oxytocin receptor and G protein-coupled receptor kinase-2. Mol. Endocrinol. 2004, 18, 1277–1286. [Google Scholar] [CrossRef] [Green Version]
- Matti, C.; Salnikov, A.; Artinger, M.; D’Agostino, G.; Kindinger, I.; Uguccioni, M.; Thelen, M.; Legler, D.F. ACKR4 recruits GRK3 prior to β-arrestins but can scavenge chemokines in the absence of β-arrestins. Front. Immunol. 2020, 11, 720. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.M.; Jorgensen, R.; Clark, R.B. Phosphorylation of the β2-adrenergic receptor in plasma membranes by intrinsic GRK5. Biochemistry 2007, 46, 14438–14449. [Google Scholar] [CrossRef]
- Jorgensen, R.; Holliday, N.D.; Hansen, J.L.; Vrecl, M.; Heding, A.; Schwartz, T.W.; Elling, C.E. Characterization of G-protein coupled receptor kinase interaction with the neurokinin-1 receptor using bioluminescence resonance energy transfer. Mol. Pharmacol. 2008, 73, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Jensen, D.D.; Godfrey, C.B.; Niklas, C.; Canals, M.; Kocan, M.; Poole, D.P.; Murphy, J.E.; Alemi, F.; Cottrell, G.S.; Korbmacher, C.; et al. The bile acid receptor TGR5 does not interact with β-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J. Biol. Chem. 2013, 288, 22942–22960. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Lieu, T.M.; Barlow, N.; Sostegni, S.; Haerteis, S.; Korbmacher, C.; Liedtke, W.; Jimenez-Vargas, N.N.; Vanner, S.J.; Bunnett, N.W. Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J. Biol. Chem. 2015, 290, 13875–13887. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, N.T.; Mohan, M.L.; Gupta, M.K.; Martelli, E.E.; Hussain, A.K.; Qin, Y.; Chandrasekharan, U.M.; Young, D.; Feldman, A.M.; Sen, S.; et al. Gβγ-independent recruitment of G-protein coupled receptor kinase 2 drives tumor necrosis factor α-induced cardiac β-adrenergic receptor dysfunction. Circulation 2013, 128, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Hishinuma, S.; Nozawa, H.; Akatsu, C.; Shoji, M. C-terminal of human histamine H1 receptors regulates their agonist-induced clathrin-mediated internalization and G-protein signaling. J. Neurochem. 2016, 139, 552–565. [Google Scholar] [CrossRef] [Green Version]
- Bruysters, M.; Pertz, H.H.; Teunissen, A.; Bakker, R.A.; Gillard, M.; Chatelain, P.; Schunack, W.; Timmerman, H.; Leurs, R. Mutational analysis of the histamine H1-receptor binding pocket of histaprodifens. Eur. J. Pharmacol. 2004, 487, 55–63. [Google Scholar] [CrossRef]
- Littmann, T.; Ozawa, T.; Hoffmann, C.; Buschauer, A.; Bernhardt, G. A split luciferase-based probe for quantitative proximal determination of Gαq signalling in live cells. Sci. Rep. 2018, 8, 17179. [Google Scholar] [CrossRef]
- Strasser, A.; Wittmann, H.-J.; Kunze, M.; Elz, S.; Seifert, R. Molecular basis for the selective interaction of synthetic agonists with the human histamine H1-receptor compared with the guinea pig H1-receptor. Mol. Pharmacol. 2009, 75, 454–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasser, A.; Striegl, B.; Wittmann, H.-J.; Seifert, R. Pharmacological profile of histaprodifens at four recombinant histamine H1 receptor species isoforms. J. Pharmacol. Exp. Ther. 2008, 324, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namkung, Y.; Radresa, O.; Armando, S.; Devost, D.; Beautrait, A.; le Gouill, C.; Laporte, S.A. Quantifying biased signaling in GPCRs using BRET-based biosensors. Methods 2016, 92, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.; Watson, C.; Muniz-Medina, V.; Christopoulos, A.; Novick, S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 2012, 3, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Westhuizen, E.T.; Breton, B.; Christopoulos, A.; Bouvier, M. Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: Implications for drug taxonomy. Mol. Pharmacol. 2014, 85, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Bosma, R.; Witt, G.; Vaas, L.A.I.; Josimovic, I.; Gribbon, P.; Vischer, H.F.; Gul, S.; Leurs, R. The target residence time of antihistamines determines their antagonism of the G protein-coupled histamine H1 receptor. Front. Pharmacol. 2017, 8, 667. [Google Scholar] [CrossRef] [PubMed]
- Klein Herenbrink, C.; Sykes, D.A.; Donthamsetti, P.; Canals, M.; Coudrat, T.; Shonberg, J.; Scammells, P.J.; Capuano, B.; Sexton, P.M.; Charlton, S.J.; et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 2016, 7, 10842. [Google Scholar] [CrossRef]
- Leschke, C.; Elz, S.; Garbarg, M.; Schunack, W. Synthesis and histamine H1 receptor agonist activity of a series of 2-phenylhistamines, 2-heteroarylhistamines, and analogues. J. Med. Chem. 1995, 38, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Elz, S.; Kramer, K.; Pertz, H.H.; Detert, H.; ter Laak, A.M.; Kühne, R.; Schunack, W. Histaprodifens: Synthesis, pharmacological in vitro evaluation, and molecular modeling of a new class of highly active and selective histamine H(1)-receptor agonists. J. Med. Chem. 2000, 43, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Menghin, S.; Pertz, H.H.; Kramer, K.; Seifert, R.; Schunack, W.; Elz, S. N(alpha)-imidazolylalkyl and pyridylalkyl derivatives of histaprodifen: Synthesis and in vitro evaluation of highly potent histamine H(1)-receptor agonists. J. Med. Chem. 2003, 46, 5458–5470. [Google Scholar] [CrossRef] [PubMed]
- Vögler, O.; Bogatkewitsch, G.S.; Wriske, C.; Krummenerl, P.; Jakobs, K.H.; van Koppen, C.J. Receptor subtype-specific regulation of muscarinic acetylcholine receptor sequestration by dynamin. Distinct sequestration of m2 receptors. J. Biol. Chem. 1998, 273, 12155–12160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.-H.; Kuravi, S.; Lambert, N.A. Internalization dissociates β2-adrenergic receptors. PLoS ONE 2011, 6, e17361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Segura, M.A.; Zarzycka, B.; Vischer, H.F.; Leurs, R. Analysis of missense variants in the human histamine receptor family reveals increased constitutive activity of E4106.30 × 30K variant in the histamine H1 receptor. Int. J. Mol. Sci. 2021, 22, 3702. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Verweij, E.W.E.; Siderius, M.; Leurs, R.; Vischer, H.F. Identification of TSPAN4 as novel histamine H4 receptor interactor. Biomolecules 2021, 11, 1127. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verweij, E.W.E.; Bosma, R.; Gao, M.; van den Bor, J.; Al Araaj, B.; de Munnik, S.M.; Ma, X.; Leurs, R.; Vischer, H.F. BRET-Based Biosensors to Measure Agonist Efficacies in Histamine H1 Receptor-Mediated G Protein Activation, Signaling and Interactions with GRKs and β-Arrestins. Int. J. Mol. Sci. 2022, 23, 3184. https://doi.org/10.3390/ijms23063184
Verweij EWE, Bosma R, Gao M, van den Bor J, Al Araaj B, de Munnik SM, Ma X, Leurs R, Vischer HF. BRET-Based Biosensors to Measure Agonist Efficacies in Histamine H1 Receptor-Mediated G Protein Activation, Signaling and Interactions with GRKs and β-Arrestins. International Journal of Molecular Sciences. 2022; 23(6):3184. https://doi.org/10.3390/ijms23063184
Chicago/Turabian StyleVerweij, Eléonore W. E., Reggie Bosma, Meichun Gao, Jelle van den Bor, Betty Al Araaj, Sabrina M. de Munnik, Xiaoyuan Ma, Rob Leurs, and Henry F. Vischer. 2022. "BRET-Based Biosensors to Measure Agonist Efficacies in Histamine H1 Receptor-Mediated G Protein Activation, Signaling and Interactions with GRKs and β-Arrestins" International Journal of Molecular Sciences 23, no. 6: 3184. https://doi.org/10.3390/ijms23063184
APA StyleVerweij, E. W. E., Bosma, R., Gao, M., van den Bor, J., Al Araaj, B., de Munnik, S. M., Ma, X., Leurs, R., & Vischer, H. F. (2022). BRET-Based Biosensors to Measure Agonist Efficacies in Histamine H1 Receptor-Mediated G Protein Activation, Signaling and Interactions with GRKs and β-Arrestins. International Journal of Molecular Sciences, 23(6), 3184. https://doi.org/10.3390/ijms23063184