
Molecular Dynamics Simulations of Transmembrane Cyclic Peptide Nanotubes Using Classical Force Fields, Hydrogen Mass Repartitioning, and Hydrogen Isotope Exchange Methods: A Critical Comparison

 ,
,
Abstract
1. Introduction
2. Materials and Methods
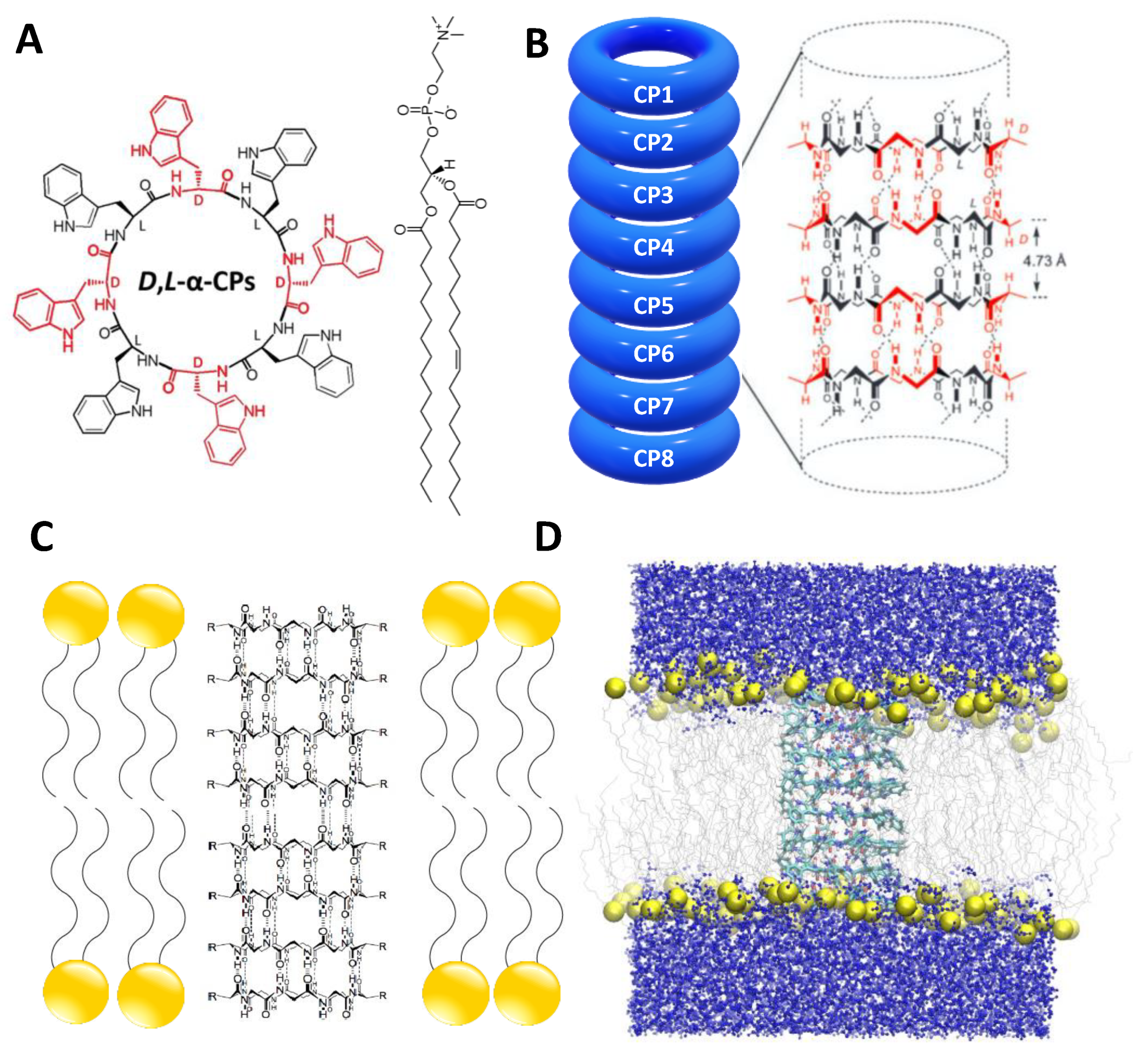
2.1. Simulation Systems and Parameters
2.2. Analysis of the Trajectories
3. Results and Discussion
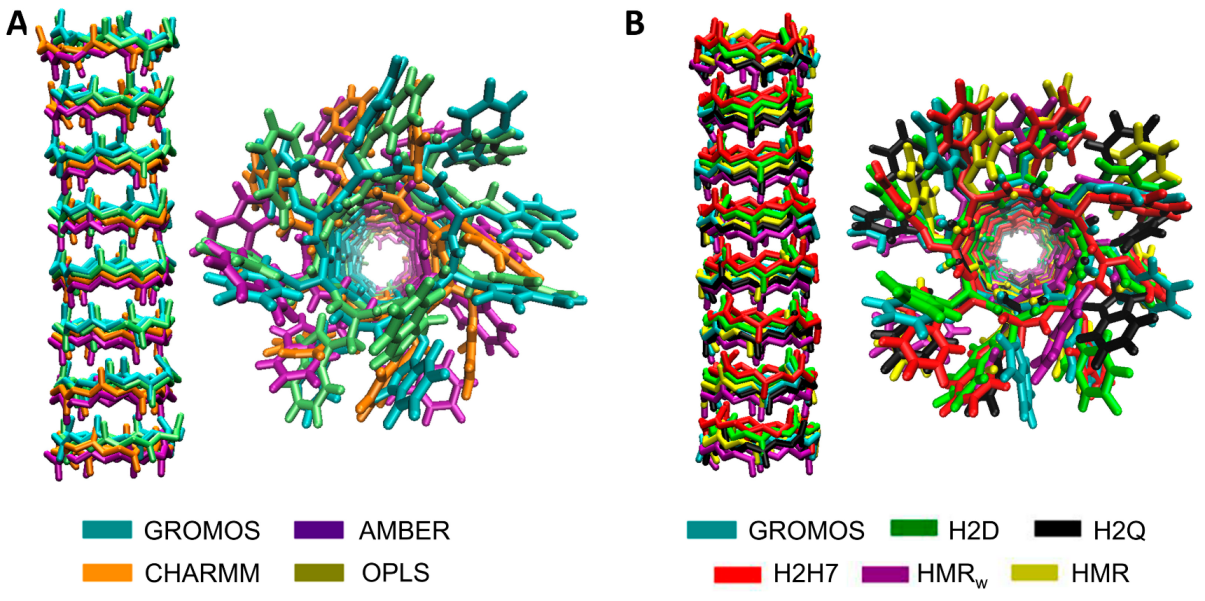
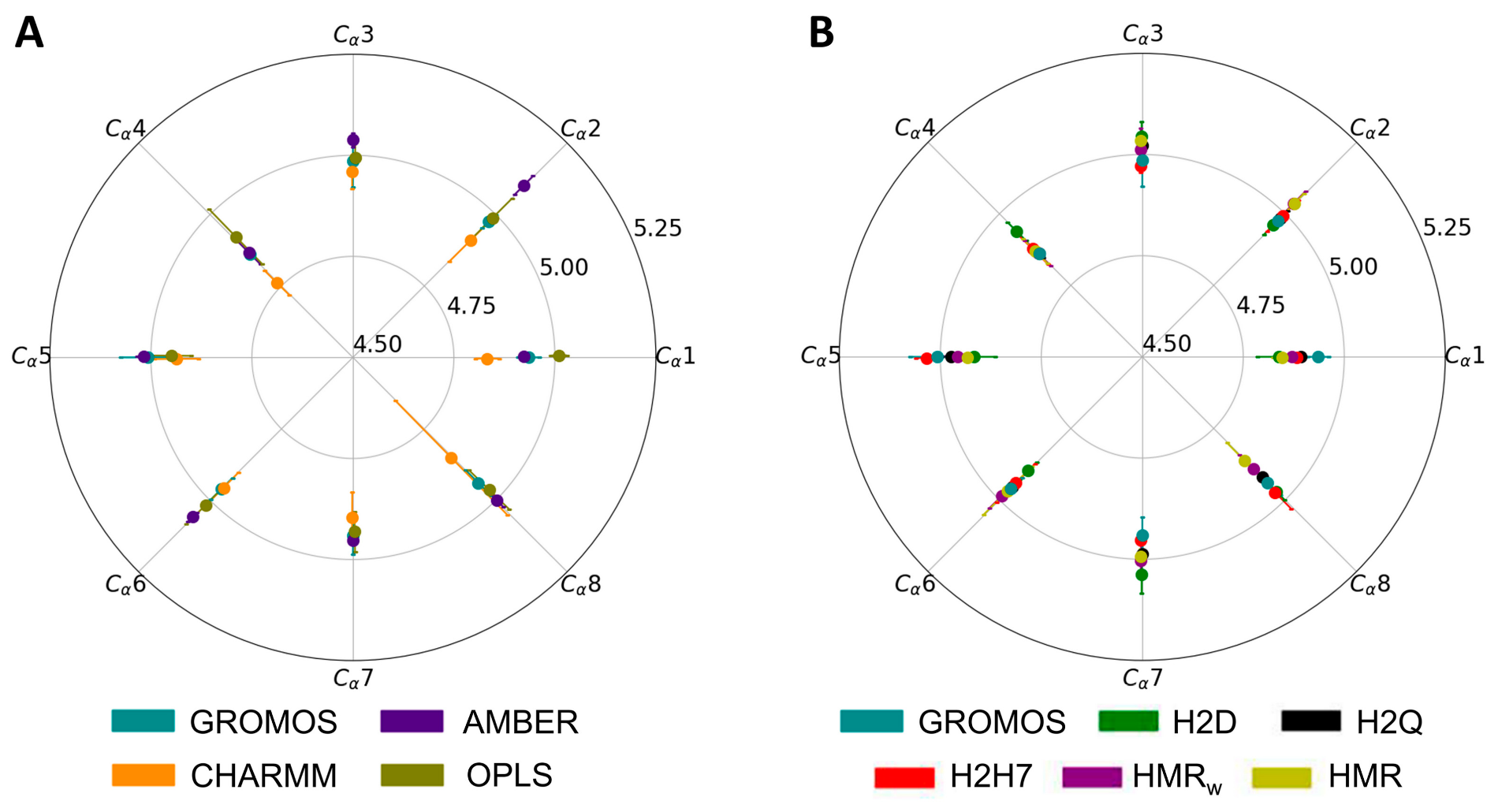
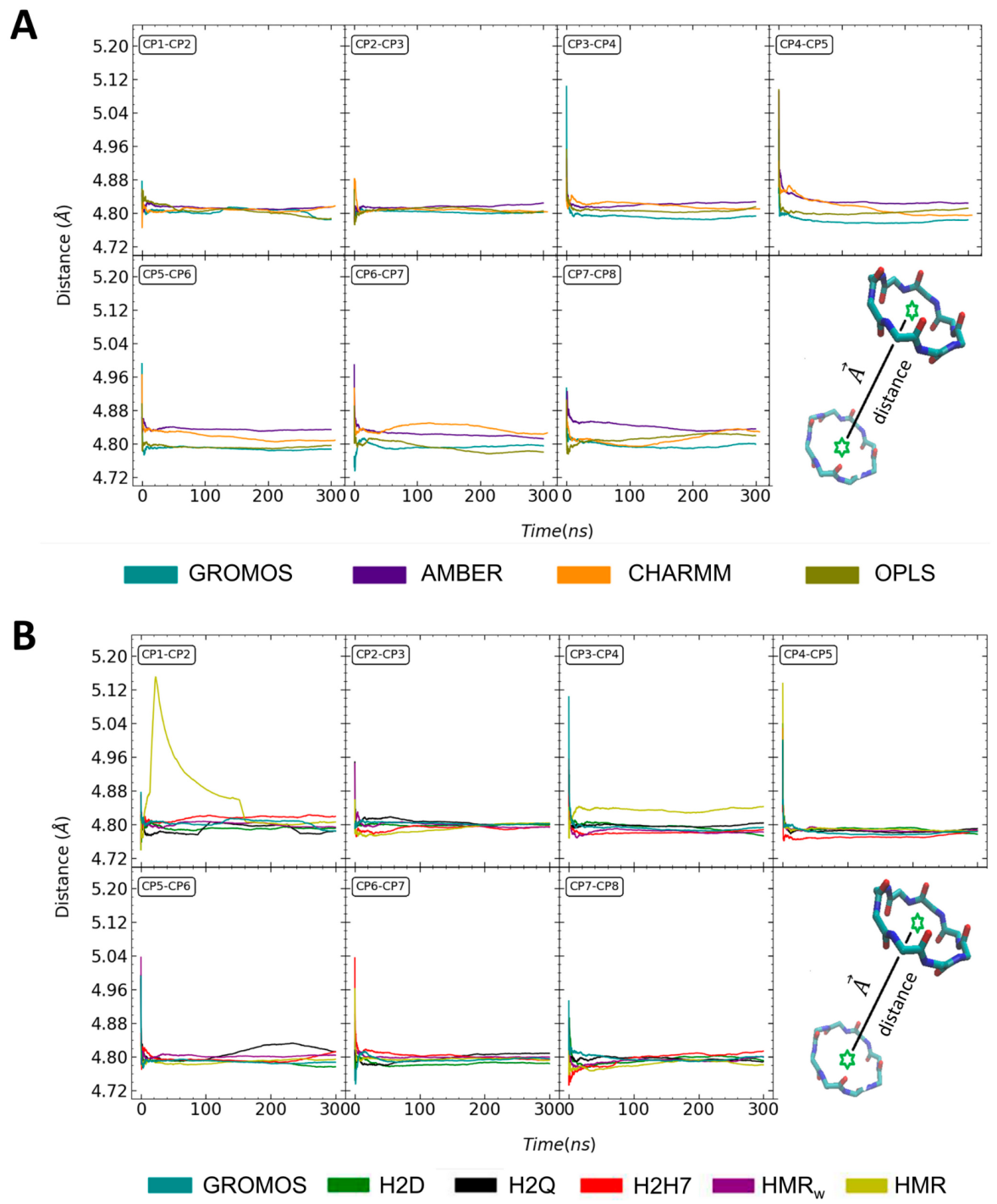
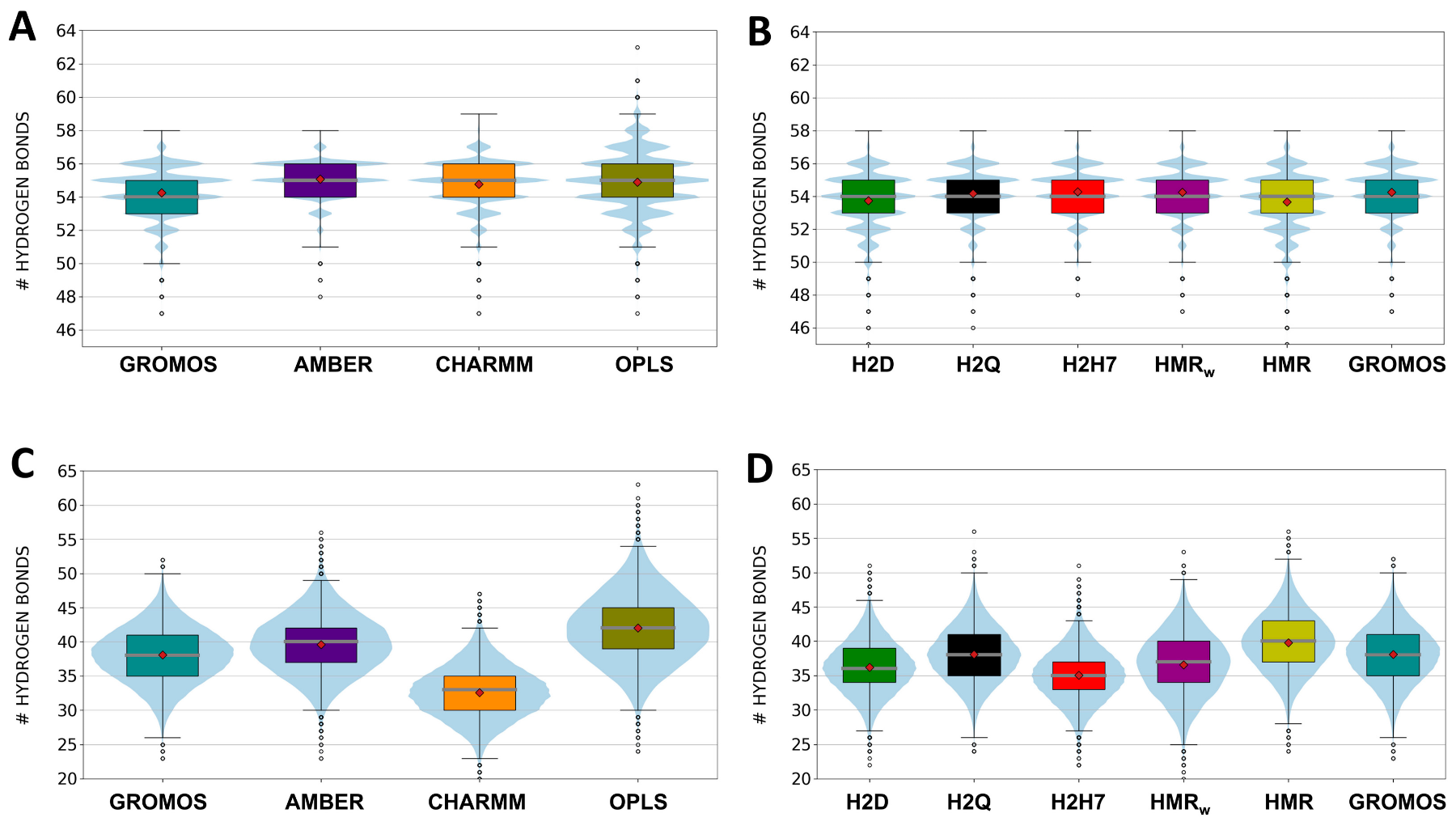
3.1. Force Field Dependence of the Structural Stability of the D,L-α-SCPN
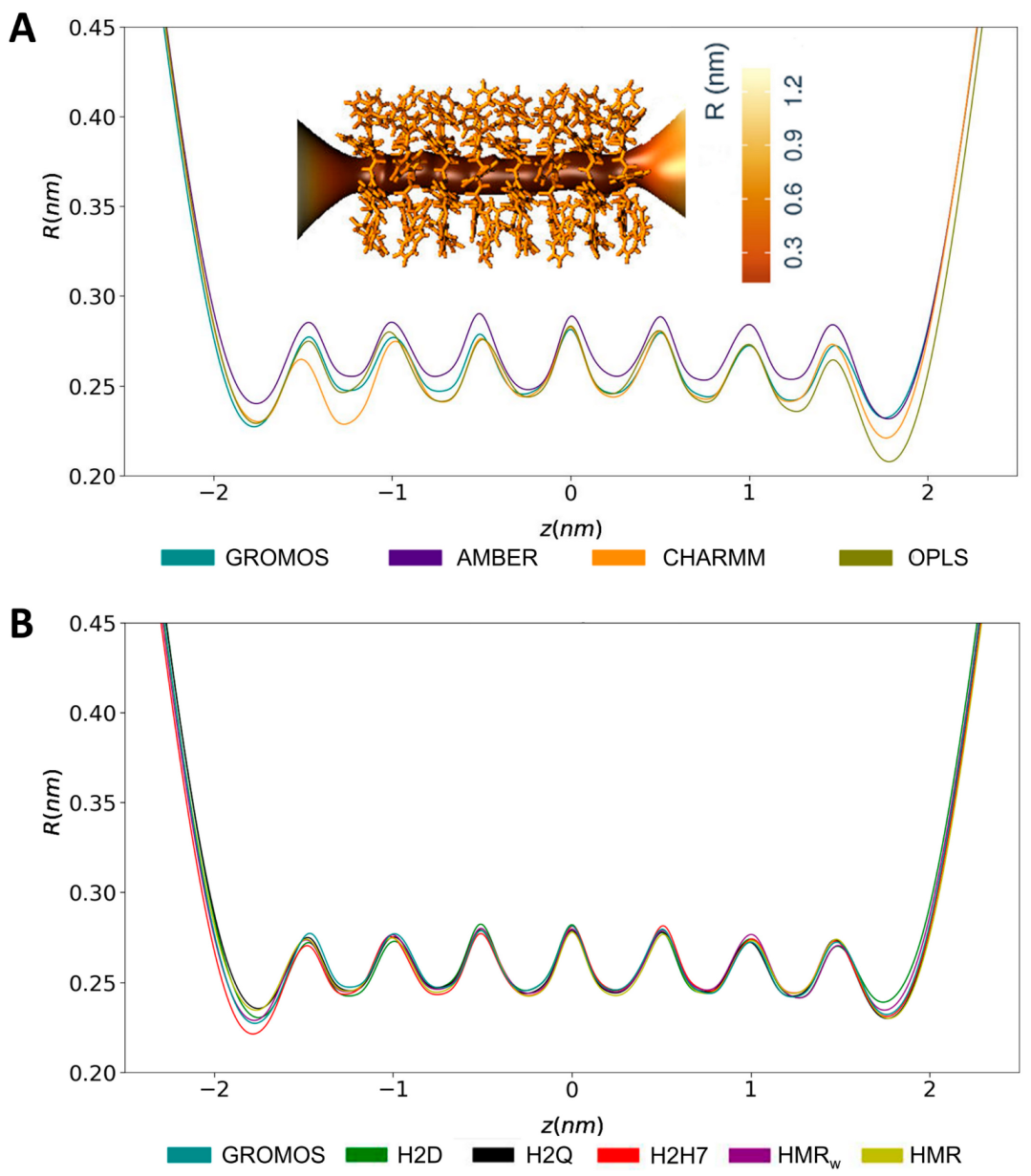
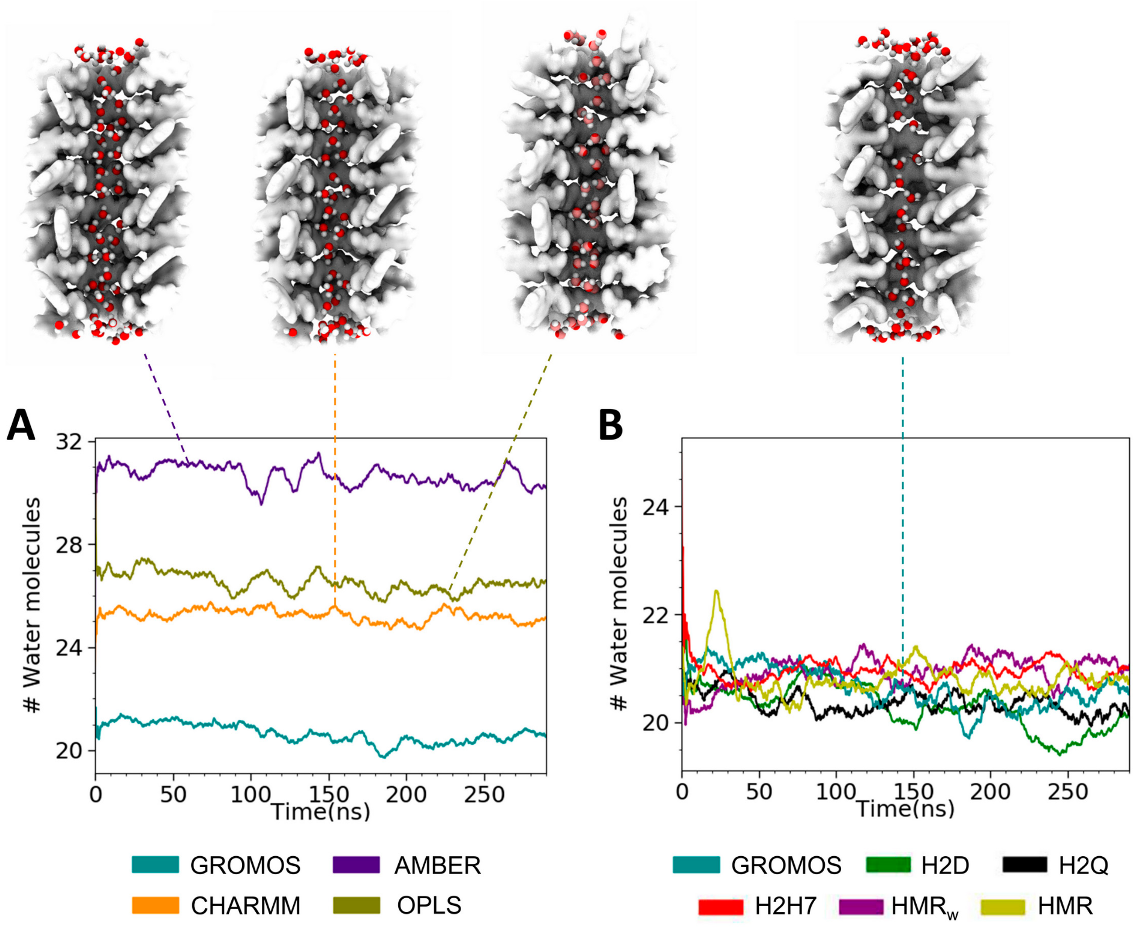
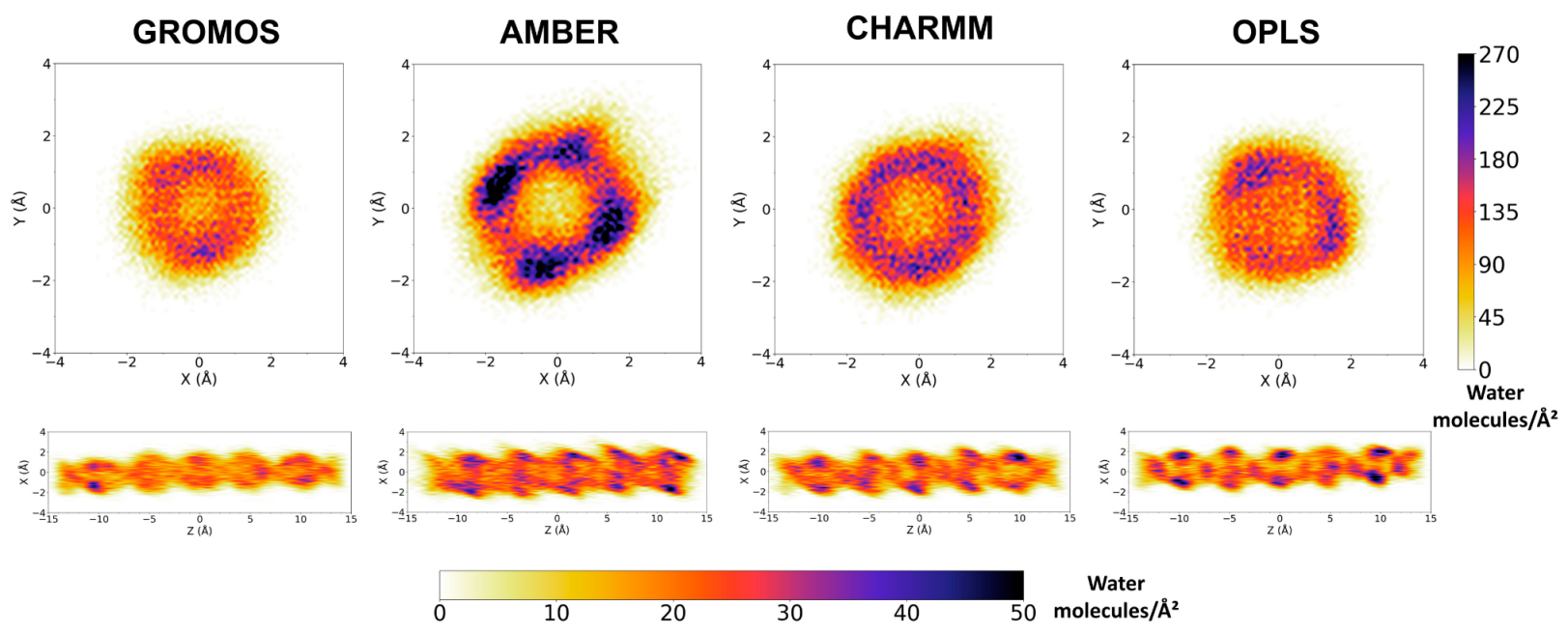
3.2. Force Field Dependence of the Structure of the Nanopores Formed by the D,L-α-SCPN
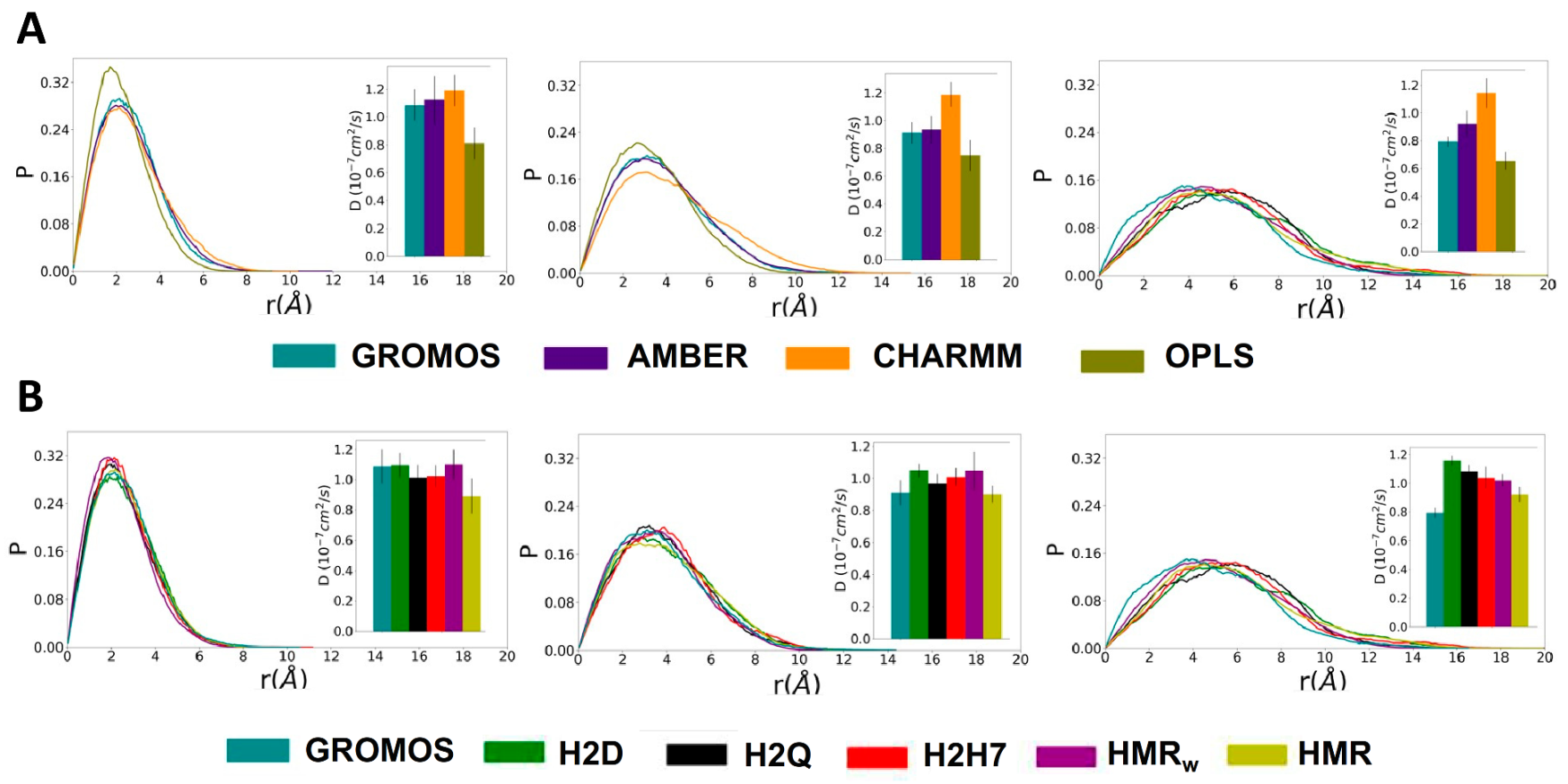
3.3. Force Field Dependence of D,L-α-SCPN Diffusion Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Claro, B.; Bastos, M.; Garcia-Fandino, R. Design and Applications of Cyclic Peptides. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 87–129. [Google Scholar] [CrossRef]
- Garcia-Fandiño, R.; Calvelo, M.; Granja, J.R. Pore- and Channel-Forming Peptides and Their Mimetics. In Comprehensive Supramolecular Chemistry II; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 4, pp. 539–573. [Google Scholar] [CrossRef]
- De Santis, P.; Morosetti, S.; Rizzo, R. Conformational Analysis of Regular Enantiomeric Sequences; UTC: Chattanooga, TN, USA, 1974; Volume 7. [Google Scholar]
- Ghadiri, M.R.; Granja, J.R.; Milligan, R.A.; McRee, D.E.; Khazanovich, N. Self-Assembling Organic Nanotubes Based on a Cyclic Peptide Architecture. Nature 1993, 366, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Bong, D.T.; Clark, T.D.; Granja, J.R.; Ghadiri, M.R. Self-Assembling Organic Nanotubes. Angew. Chem. Int. Ed. 2001, 40, 988–1011. [Google Scholar] [CrossRef]
- García-Fandiño, R.; Amorín, M.; Granja, J.R. Synthesis of Supramolecular Nanotubes. In Supramolecular Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2012. [Google Scholar] [CrossRef]
- Rodriguez-Vazquez, N.; Ozores, H.; Guerra, A.; Gonzalez-Freire, E.; Fuertes, A.; Panciera, M.; Priegue, J.; Outeiral, J.; Montenegro, J.; Garcia-Fandino, R.; et al. Membrane-Targeted Self-Assembling Cyclic Peptide Nanotubes. Curr. Top. Med. Chem. 2015, 14, 2647–2661. [Google Scholar] [CrossRef]
- Rodríguez-Vázquez, N.; Amorín, M.; Granja, J.R. Recent Advances in Controlling the Internal and External Properties of Self-Assembling Cyclic Peptide Nanotubes and Dimers. Org. Biomol. Chem. 2017, 15, 4490–4505. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, M.R.; Granja, J.R.; Buehler, L.K. Artificial Transmembrane Ion Channels from Self-Assembling Peptide Nanotubes. Nature 1994, 369, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Huang, L.; Sun, Z.; Barboiu, M. Self-Assembled Artificial Ion-Channels toward Natural Selection of Functions. Angew. Chem. Int. Ed. 2021, 60, 566–597. [Google Scholar] [CrossRef] [PubMed]
- Hartgerink, J.D.; Granja, J.R.; Milligan, R.A.; Ghadiri, M.R. Self-Assembling Peptide Nanotubes. J. Am. Chem. Soc. 1996, 118, 43–50. [Google Scholar] [CrossRef]
- Zhu, J.; Cheng, J.; Liao, Z.; Lai, Z.; Liu, B. Investigation of Structures and Properties of Cyclic Peptide Nanotubes by Experiment and Molecular Dynamics. J. Comput.-Aided. Mol. Des. 2008, 22, 773–781. [Google Scholar] [CrossRef]
- Lynch, C.I.; Rao, S.; Sansom, M.S.P. Water in Nanopores and Biological Channels: A Molecular Simulation Perspective. Chem. Rev. 2020, 120, 10298–10335. [Google Scholar] [CrossRef]
- Dauber-Osguthorpe, P.; Hagler, A.T. Biomolecular Force Fields: Where Have We Been, Where Are We Now, Where Do We Need to Go and How Do We Get There? J. Comput.-Aided Mol. Des. 2019, 33, 133–203. [Google Scholar] [CrossRef]
- Lopes, P.E.M.; Guvench, O.; Mackerell, A.D. Current Status of Protein Force Fields for Molecular Dynamics Simulations. Methods Mol. Biol. 2015, 1215, 47–71. [Google Scholar] [CrossRef]
- Rosenberger, D.; Smith, J.S.; Garcia, A.E. Modeling of Peptides with Classical and Novel Machine Learning Force Fields: A Comparison. J. Phys. Chem. B 2021, 125, 3598–3612. [Google Scholar] [CrossRef]
- Beauchamp, K.A.; Lin, Y.S.; Das, R.; Pande, V.S. Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements. J. Chem. Theory Comput. 2012, 8, 1409–1414. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic Validation of Protein Force Fields against Experimental Data. PLoS ONE 2012, 7, e0032131. [Google Scholar] [CrossRef]
- Lange, O.F.; Van Der Spoel, D.; De Groot, B.L. Scrutinizing Molecular Mechanics Force Fields on the Submicrosecond Timescale with NMR Data. Biophys. J. 2010, 99, 647–655. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Paissoni, C.; Nardelli, F.; Zanella, S.; Curnis, F.; Belvisi, L.; Musco, G.; Ghitti, M. A Critical Assessment of Force Field Accuracy against NMR Data for Cyclic Peptides Containing β-Amino Acids. Phys. Chem. Chem. Phys. 2018, 20, 15807–15816. [Google Scholar] [CrossRef]
- McHugh, S.M.; Rogers, J.R.; Solomon, S.A.; Yu, H.; Lin, Y.S. Computational Methods to Design Cyclic Peptides. Curr. Opin. Chem. Biol. 2016, 34, 95–102. [Google Scholar] [CrossRef]
- Yu, H.; Lin, Y.S. Toward Structure Prediction of Cyclic Peptides. Phys. Chem. Chem. Phys. 2015, 17, 4210–4219. [Google Scholar] [CrossRef]
- Lin, H.; Fan, J.; Weng, P.; Si, X.; Zhao, X. Molecular Dynamics Simulations on the Behaviors of Hydrophilic/Hydrophobic Cyclic Peptide Nanotubes at the Water/Hexane Interface. J. Phys. Chem. A 2017, 121, 6863–6873. [Google Scholar] [CrossRef]
- Maroli, N.; Kolandaivel, P. Comparative Study of Stability and Transport of Molecules through Cyclic Peptide Nanotube and Aquaporin: A Molecular Dynamics Simulation Approach. J. Biomol. Struct. Dyn. 2020, 38, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Tarek, M.; Maigret, B.; Chipot, C. Molecular Dynamics Investigation of an Oriented Cyclic Peptide Nanotube in DMPC Bilayers. Biophys. J. 2003, 85, 2287–2298. [Google Scholar] [CrossRef][Green Version]
- Hwang, H.; Schatz, G.C.; Ratner, M.A. Steered Molecular Dynamics Studies of the Potential of Mean Force of a Na+ or K+ Ion in a Cyclic Peptide Nanotube. J. Phys. Chem. B 2006, 110, 26448–26460. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fan, J.; Tang, M.; Zhou, W. Molecular Dynamics Simulation for the Structure of the Water Chain in a Transmembrane Peptide Nanotube. J. Phys. Chem. A 2010, 114, 2376–2383. [Google Scholar] [CrossRef]
- Vijayaraj, R.; Van Damme, S.; Bultinck, P.; Subramanian, V. Structure and Stability of Cyclic Peptide Based Nanotubes: A Molecular Dynamics Study of the Influence of Amino Acid Composition. Phys. Chem. Chem. Phys. 2012, 14, 15135–15144. [Google Scholar] [CrossRef]
- Bertran, O.; Curcó, D.; Zanuy, D.; Alemán, C. Atomistic Organization and Characterization of Tube-like Assemblies Comprising Peptide-Polymer Conjugates: Computer Simulation Studies. Faraday Discuss. 2013, 166, 59–82. [Google Scholar] [CrossRef]
- Vijayakumar, V.; Vijayaraj, R.; Peters, G.H. In Silico Study of Amphiphilic Nanotubes Based on Cyclic Peptides in Polar and Non-Polar Solvent. J. Mol. Model. 2016, 22, 264. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous Parallelization and Acceleration of Molecular Dynamics Simulations in GROMACS. J. Chem. Phys. 2020, 153, 44130. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Stone, J.E.; Hardy, D.J.; Ufimtsev, I.S.; Schulten, K. GPU-Accelerated Molecular Modeling Coming of Age. J. Mol. Graph. Model. 2010, 29, 116–125. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Claro, B.; González-Freire, E.; Calvelo, M.; Bessa, L.J.; Goormaghtigh, E.; Amorín, M.; Granja, J.R.; Garcia-Fandiño, R.; Bastos, M. Membrane Targeting Antimicrobial Cyclic Peptide Nanotubes—An Experimental and Computational Study. Colloids Surf. B Biointerfaces 2020, 196, 111349. [Google Scholar] [CrossRef]
- Khalfa, A.; Tarek, M. On the Antibacterial Action of Cyclic Peptides: Insights from Coarse-Grained MD Simulations. J. Phys. Chem. B 2010, 114, 2676–2684. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Yang, Y.I.; Shao, Q.; Zhang, J.; Yang, L.; Gao, Y.Q. Enhanced Sampling in Molecular Dynamics. J. Chem. Phys. 2019, 151, 70902. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Feenstra, K.A.; Hess, B.; Berendsen, H.J.C. Improving Efficiency of Large Time-Scale Molecular Dynamics Simulations of Hydrogen-Rich Systems. J. Comput. Chem. 1999, 20, 786–798. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Beyer, H.; Tukey, J.W. Exploratory Data Analysis. Addison-Wesley Publishing Company Reading, Mass.—Menlo Park, Cal., London, Amsterdam, Don Mills, Ontario, Sydney 1977, XVI, 688 S. Biom. J. 1981, 23, 413–414. [Google Scholar] [CrossRef]
- Garrido, P.F.; Calvelo, M.; Garcia-Fandiño, R.; Piñeiro, Á. Rings, Hexagons, Petals, and Dipolar Moment Sink-Sources: The Fanciful Behavior of Water around Cyclodextrin Complexes. Biomolecules 2020, 10, 431. [Google Scholar] [CrossRef]
- De Laeter, J.R.; Böhlke, J.K.; De Bièvre, P.; Hidaka, H.; Peiser, H.S.; Rosman, K.J.R.; Taylor, P.D.P. Atomic Weights of the Elements: Review 2000 (IUPAC Technical Report). Pure Appl. Chem. 2003, 75, 683–800. [Google Scholar] [CrossRef]
- Korsheninnikov, A.A.; Nikolskii, E.Y.; Kuzmin, E.A.; Ozawa, A.; Morimoto, K.; Tokanai, F.; Kanungo, R.; Tanihata, I.; Timofeyuk, N.K.; Golovkov, M.S.; et al. Experimental Evidence for the Existence of [Formula Presented] and for a Specific Structure of [Formula Presented]. Phys. Rev. Lett. 2003, 90, 082501. [Google Scholar] [CrossRef]
- Silk, M.R.; Newman, J.; Ratcliffe, J.C.; White, J.F.; Caradoc-Davies, T.; Price, J.R.; Perrier, S.; Thompson, P.E.; Chalmers, D.K. Parallel and Antiparallel Cyclic d/l Peptide Nanotubes. Chem. Commun. 2017, 53, 6613–6616. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D. Temperature and Size Dependence for Monte Carlo Simulations of TIP4P Water. Mol. Phys. 1985, 56, 1381–1392. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar] [CrossRef]
- Domański, J.; Stansfeld, P.J.; Sansom, M.S.P.; Beckstein, O. Lipidbook: A Public Repository for Force-Field Parameters Used in Membrane Simulations. J. Membr. Biol. 2010, 236, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Jämbeck, J.P.M.; Lyubartsev, A.P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Nencini, R. Simulation Data for CHARMM36 POPC Bilayer, 100 Lipids/Leaflet, 310K, GROMACS 5.1.4. 2018. Available online: https://doi.org/10.5281/ZENODO.1198158 (accessed on 10 February 2022).
- Mykhailo, G.; Samuli, O.O.H. POPC_Ulmschneider_OPLS_NaCl_015Mol. 2015. Available online: https://doi.org/10.5281/ZENODO.30892 (accessed on 10 February 2022).
- Ulmschneider, J.P.; Ulmschneider, M.B. United Atom Lipid Parameters for Combination with the Optimized Potentials for Liquid Simulations All-Atom Force Field. J. Chem. Theory Comput. 2009, 5, 1803–1813. [Google Scholar] [CrossRef]
- Poger, D.; Van Gunsteren, W.F.; Mark, A.E. A New Force Field for Simulating Phosphatidylcholine Bilayers. J. Comput. Chem. 2010, 31, 1117–1125. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Piggot, T.J.; Piñeiro, Á.; Khalid, S. Molecular Dynamics Simulations of Phosphatidylcholine Membranes: A Comparative Force Field Study. J. Chem. Theory Comput. 2012, 8, 4593–4609. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Klesse, G.; Rao, S.; Sansom, M.S.P.; Tucker, S.J. CHAP: A Versatile Tool for the Structural and Functional Annotation of Ion Channel Pores. J. Mol. Biol. 2019, 431, 3353–3365. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domá nski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Mckinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 56–61. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Garcia-Fandiño, R.; Piñeiro, Á.; Trick, J.L.; Sansom, M.S.P. Lipid Bilayer Membrane Perturbation by Embedded Nanopores: A Simulation Study. ACS Nano 2016, 10, 3693–3701. [Google Scholar] [CrossRef]
- Tiangtrong, P.; Thamwattana, N.; Baowan, D. Modelling Water Molecules inside Cyclic Peptide Nanotubes. Appl. Nanosci. 2016, 6, 345–357. [Google Scholar] [CrossRef]
- Hernandez-Pascacio, J.; Piñeiro, Á.; Ruso, J.M.; Hassan, N.; Campbell, R.A.; Campos-Terán, J.; Costas, M. Complex Behavior of Aqueous α-Cyclodextrin Solutions. Interfacial Morphologies Resulting from Bulk Aggregation. Langmuir 2016, 32, 6682–6690. [Google Scholar] [CrossRef]
- Luviano, A.S.; Hernández-Pascacio, J.; Ondo, D.; Campbell, R.A.; Piñeiro, Á.; Campos-Terán, J.; Costas, M. Highly Viscoelastic Films at the Water/Air Interface: α-Cyclodextrin with Anionic Surfactants. J. Colloid Interface Sci. 2020, 565, 601–613. [Google Scholar] [CrossRef]
- Siu, S.W.I.; Vácha, R.; Jungwirth, P.; Böckmann, R.A. Biomolecular Simulations of Membranes: Physical Properties from Different Force Fields. J. Chem. Phys. 2008, 128, 125103. [Google Scholar] [CrossRef]
- Sandoval-Perez, A.; Pluhackova, K.; Böckmann, R.A. Critical Comparison of Biomembrane Force Fields: Protein–Lipid Interactions at the Membrane Interface. J. Chem. Theory Comput. 2017, 13, 2310–2321. [Google Scholar] [CrossRef]
- Guvench, O.; MacKerell, A.D. Comparison of Protein Force Fields for Molecular Dynamics Simulations. Methods Mol. Biol. 2008, 443, 63–88. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMBER | CHARMM | OPLS | GROMOS | H2D | H2Q | H2H7 | HMR | HMRw | |
|---|---|---|---|---|---|---|---|---|---|
| Time step/fs | 2 | 2 | 2 | 2 | 5 | 6 | 6 | 6 | 7 |
| Water model | TIP3P | TIP3P | TIP4P | SPC | SPC | SPC | SPC | SPC | SPC |
| Polar H/Da | 1.008 | 1.008 | 1.008 | 1.008 | 2.014 | 4.026 | 7.053 | 4.032 | 4.032 |
| Polar C/Da | 12.010 | 12.011 | 12.011 | 12.011 | 12.011 | 12.011 | 12.011 | 8.987 | 8.987 |
| Water O/Da | 16.000 | 16.000 | 16.000 | 15.999 | 15.999 | 15.999 | 15.999 | 15.999 | 9.951 |
| Water H/Da | 1.008 | 1.008 | 1.008 | 1.008 | 1.008 | 1.008 | 1.008 | 1.008 | 4.032 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conde, D.; Garrido, P.F.; Calvelo, M.; Piñeiro, Á.; Garcia-Fandino, R. Molecular Dynamics Simulations of Transmembrane Cyclic Peptide Nanotubes Using Classical Force Fields, Hydrogen Mass Repartitioning, and Hydrogen Isotope Exchange Methods: A Critical Comparison. Int. J. Mol. Sci. 2022, 23, 3158. https://doi.org/10.3390/ijms23063158
Conde D, Garrido PF, Calvelo M, Piñeiro Á, Garcia-Fandino R. Molecular Dynamics Simulations of Transmembrane Cyclic Peptide Nanotubes Using Classical Force Fields, Hydrogen Mass Repartitioning, and Hydrogen Isotope Exchange Methods: A Critical Comparison. International Journal of Molecular Sciences. 2022; 23(6):3158. https://doi.org/10.3390/ijms23063158
Chicago/Turabian StyleConde, Daniel, Pablo F. Garrido, Martín Calvelo, Ángel Piñeiro, and Rebeca Garcia-Fandino. 2022. "Molecular Dynamics Simulations of Transmembrane Cyclic Peptide Nanotubes Using Classical Force Fields, Hydrogen Mass Repartitioning, and Hydrogen Isotope Exchange Methods: A Critical Comparison" International Journal of Molecular Sciences 23, no. 6: 3158. https://doi.org/10.3390/ijms23063158
APA StyleConde, D., Garrido, P. F., Calvelo, M., Piñeiro, Á., & Garcia-Fandino, R. (2022). Molecular Dynamics Simulations of Transmembrane Cyclic Peptide Nanotubes Using Classical Force Fields, Hydrogen Mass Repartitioning, and Hydrogen Isotope Exchange Methods: A Critical Comparison. International Journal of Molecular Sciences, 23(6), 3158. https://doi.org/10.3390/ijms23063158