
Type I–IV Halogen⋯Halogen Interactions: A Comparative Theoretical Study in Halobenzene⋯Halobenzene Homodimers

 ,
,  , ,
, ,  ,
, 

Abstract
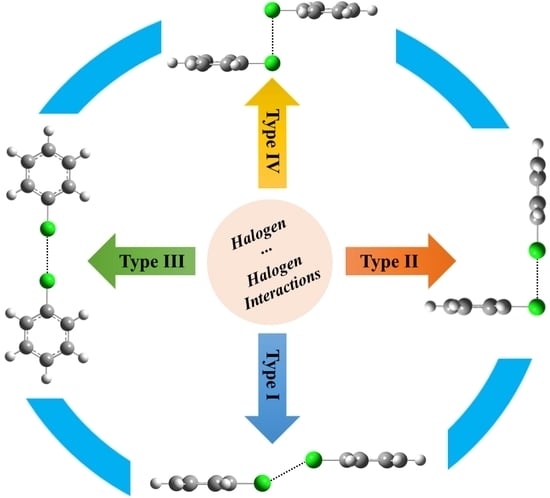
:
1. Introduction
2. Results
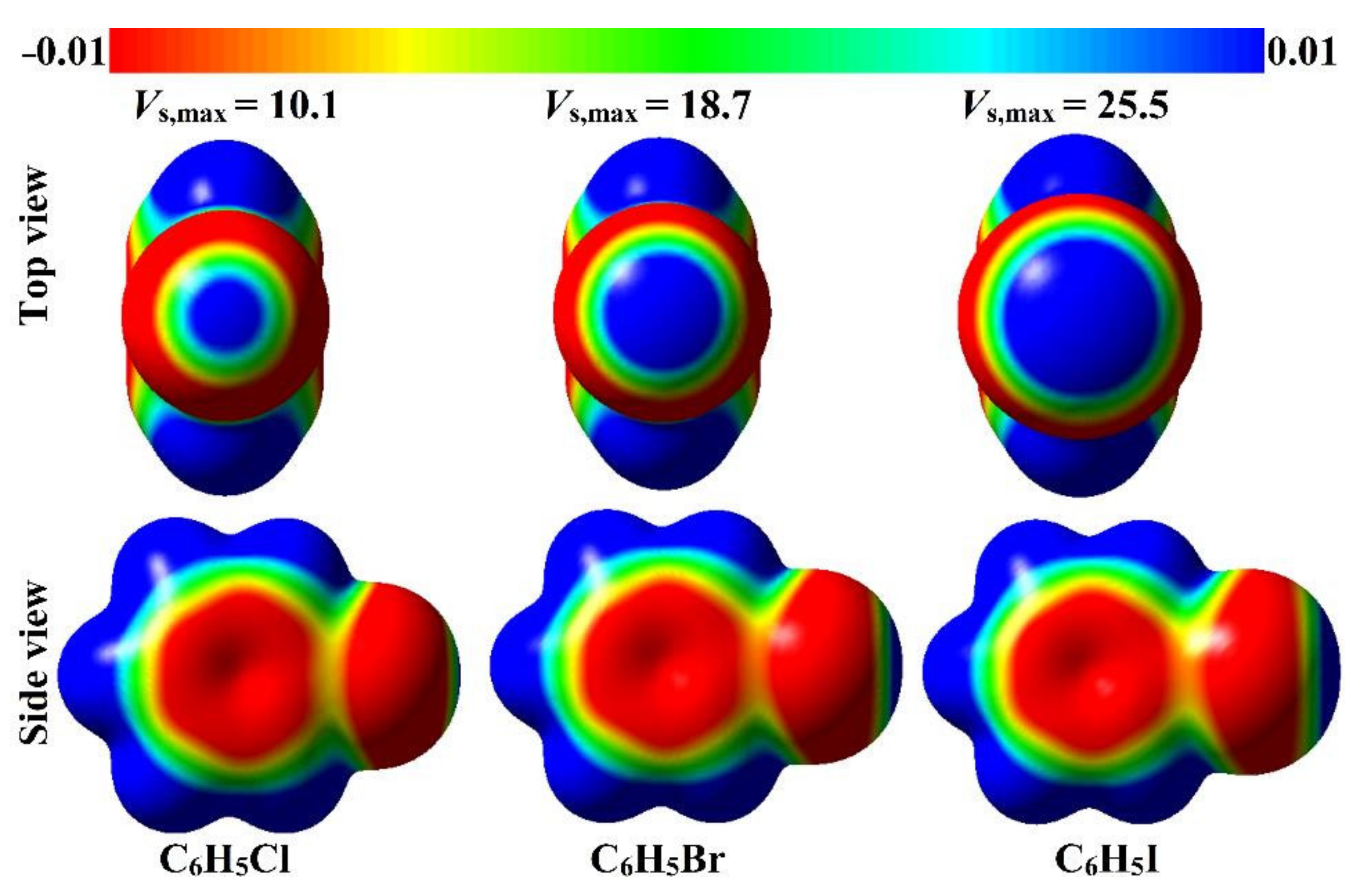
2.1. Electrostatic Potential (EP) Analysis
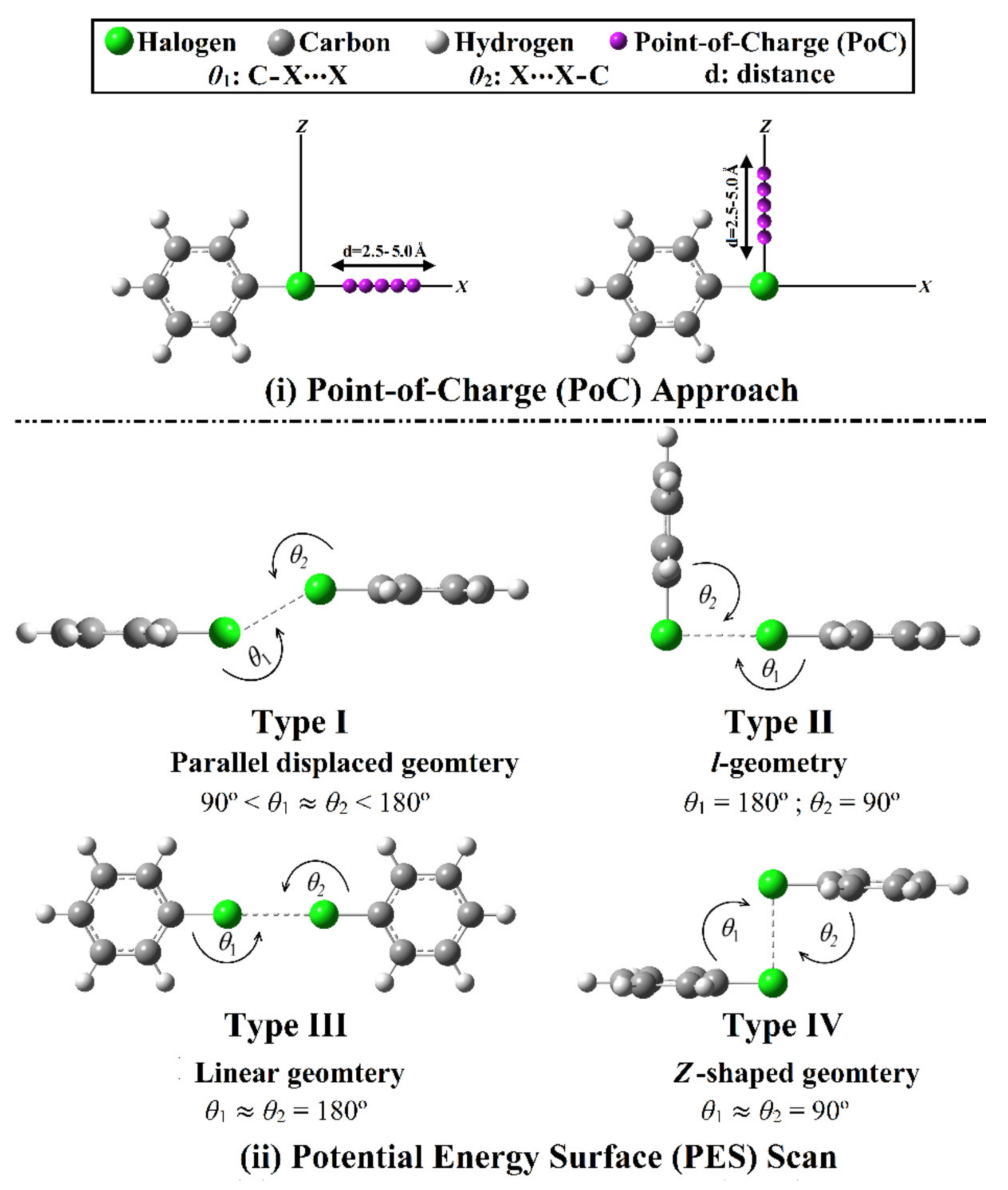
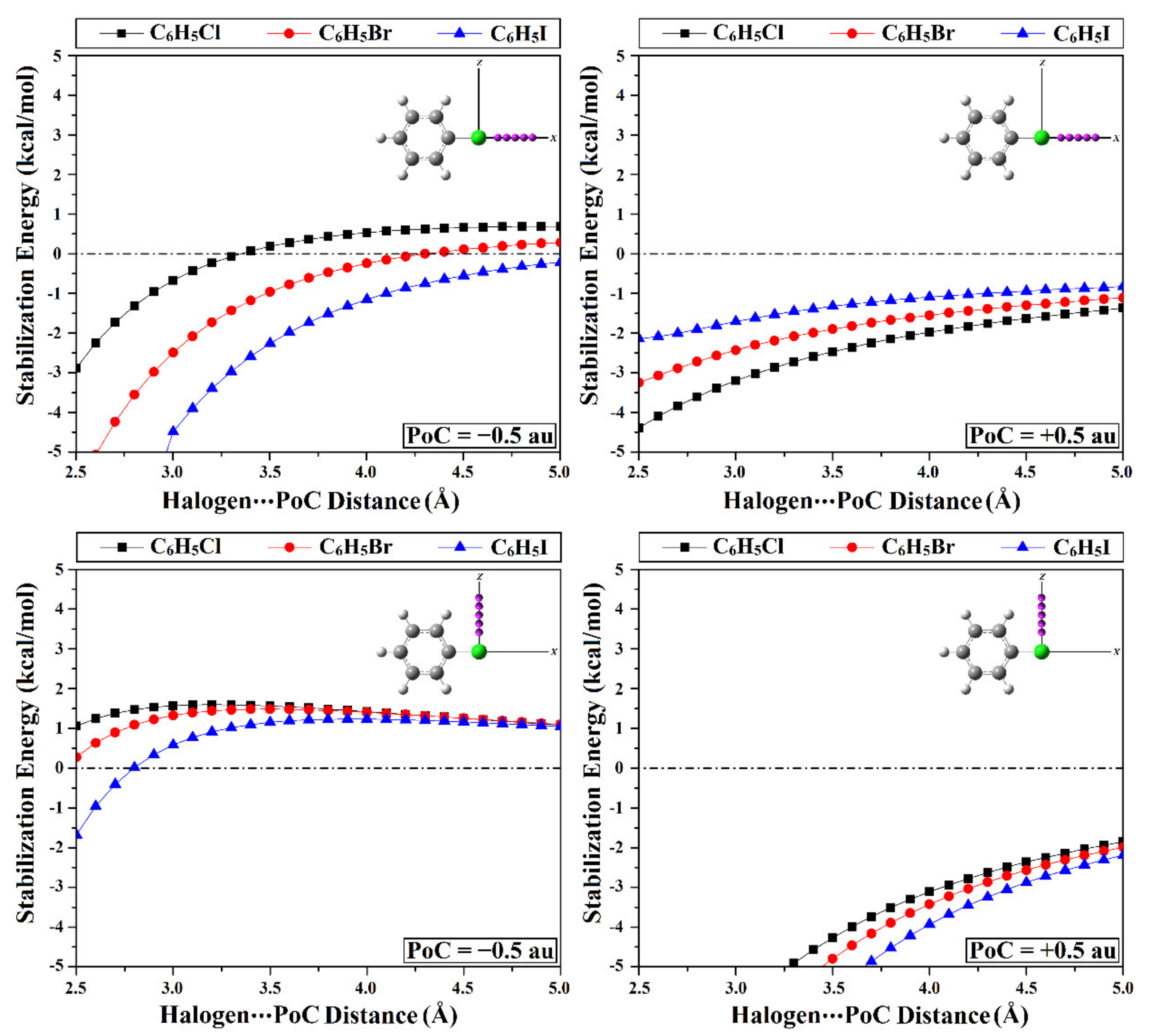
2.2. Point-of-Charge (PoC) Calculations
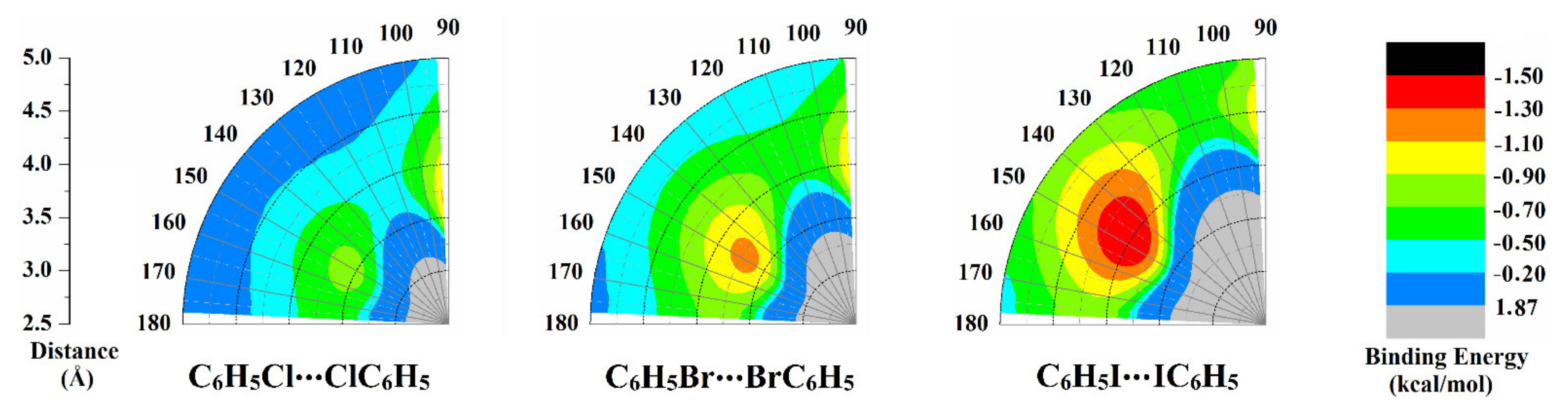
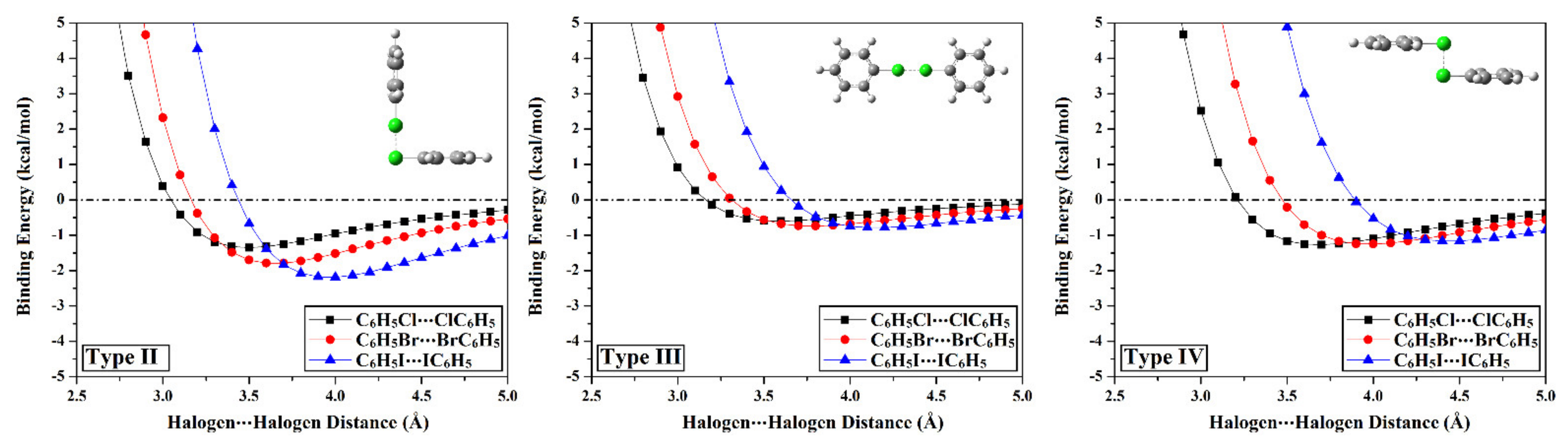
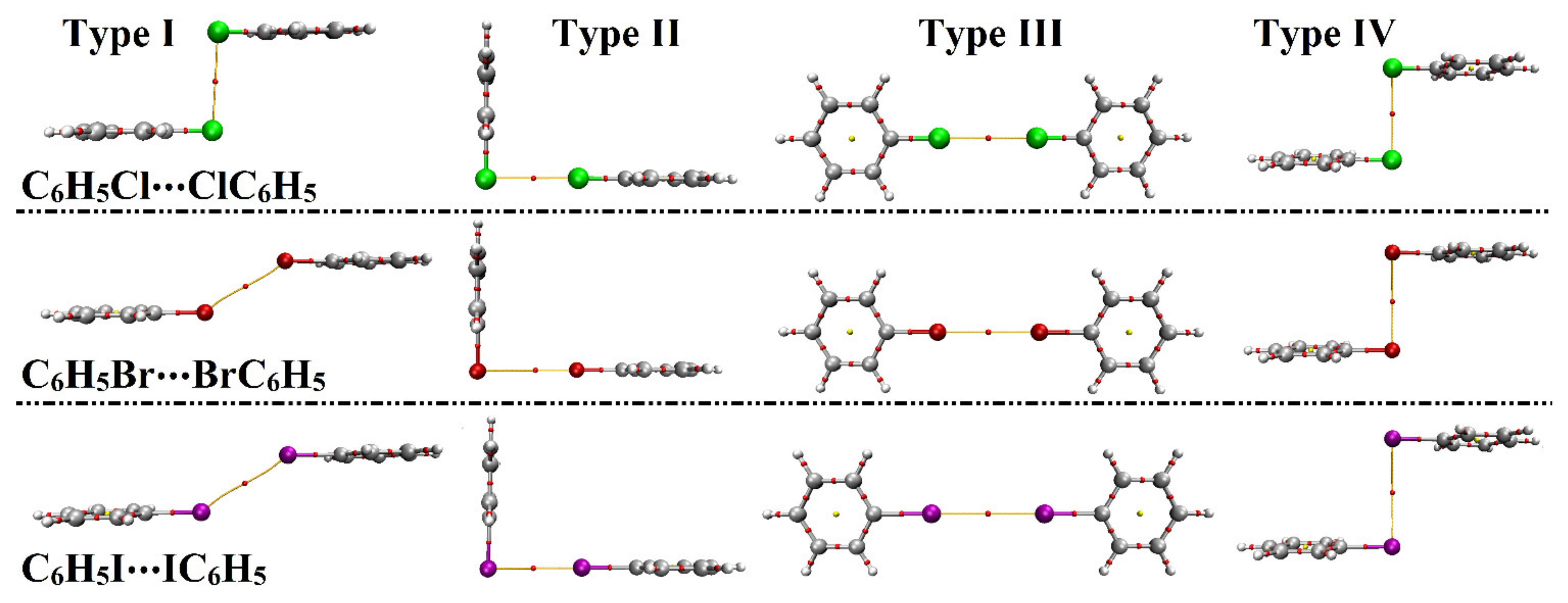
2.3. Potential Energy Surface (PES) Scan
2.4. Quantum Theory of Atom in Molecules (QTAIM) Analysis
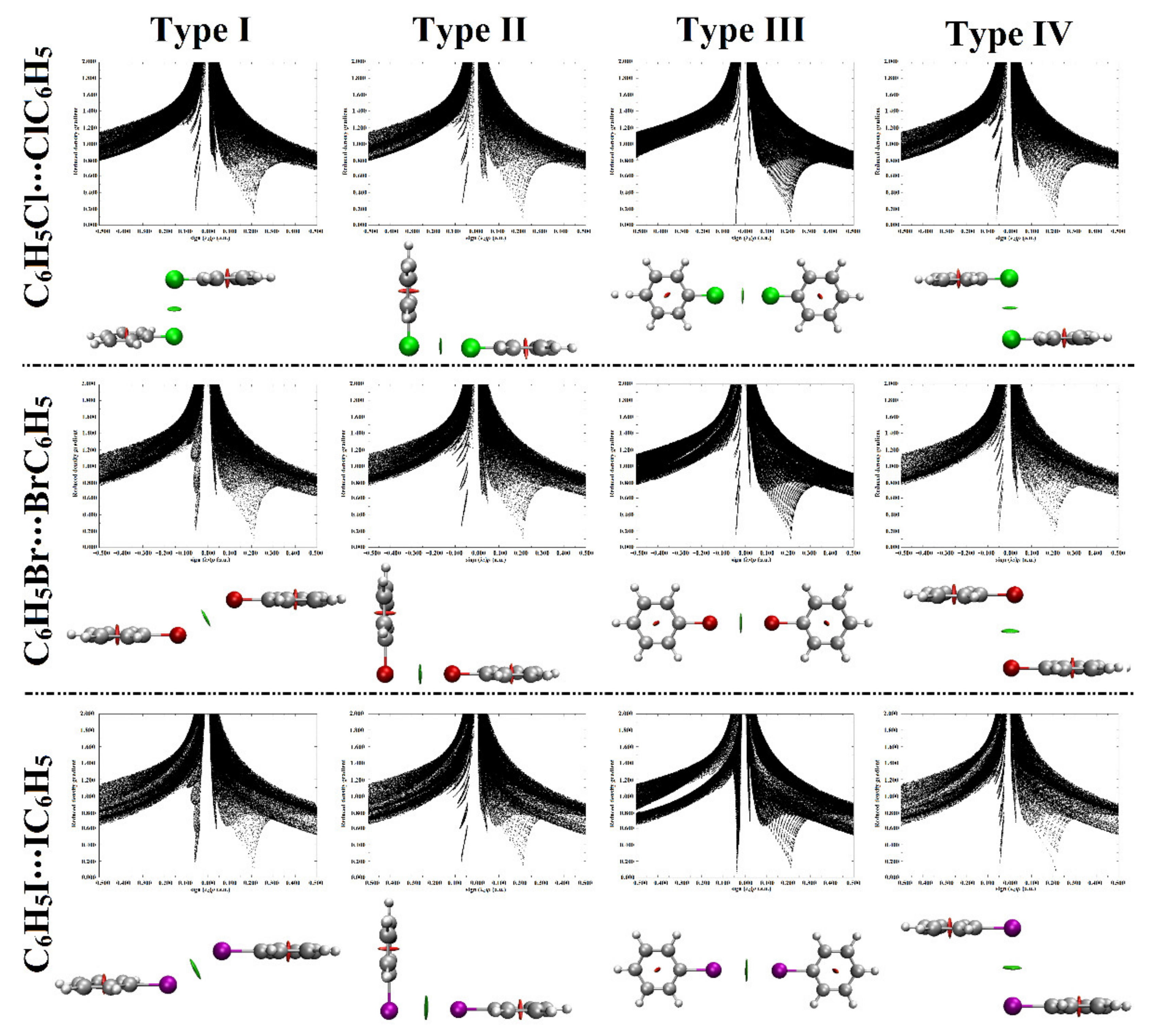
2.5. Noncovalent Interactions (NCI) Analysis
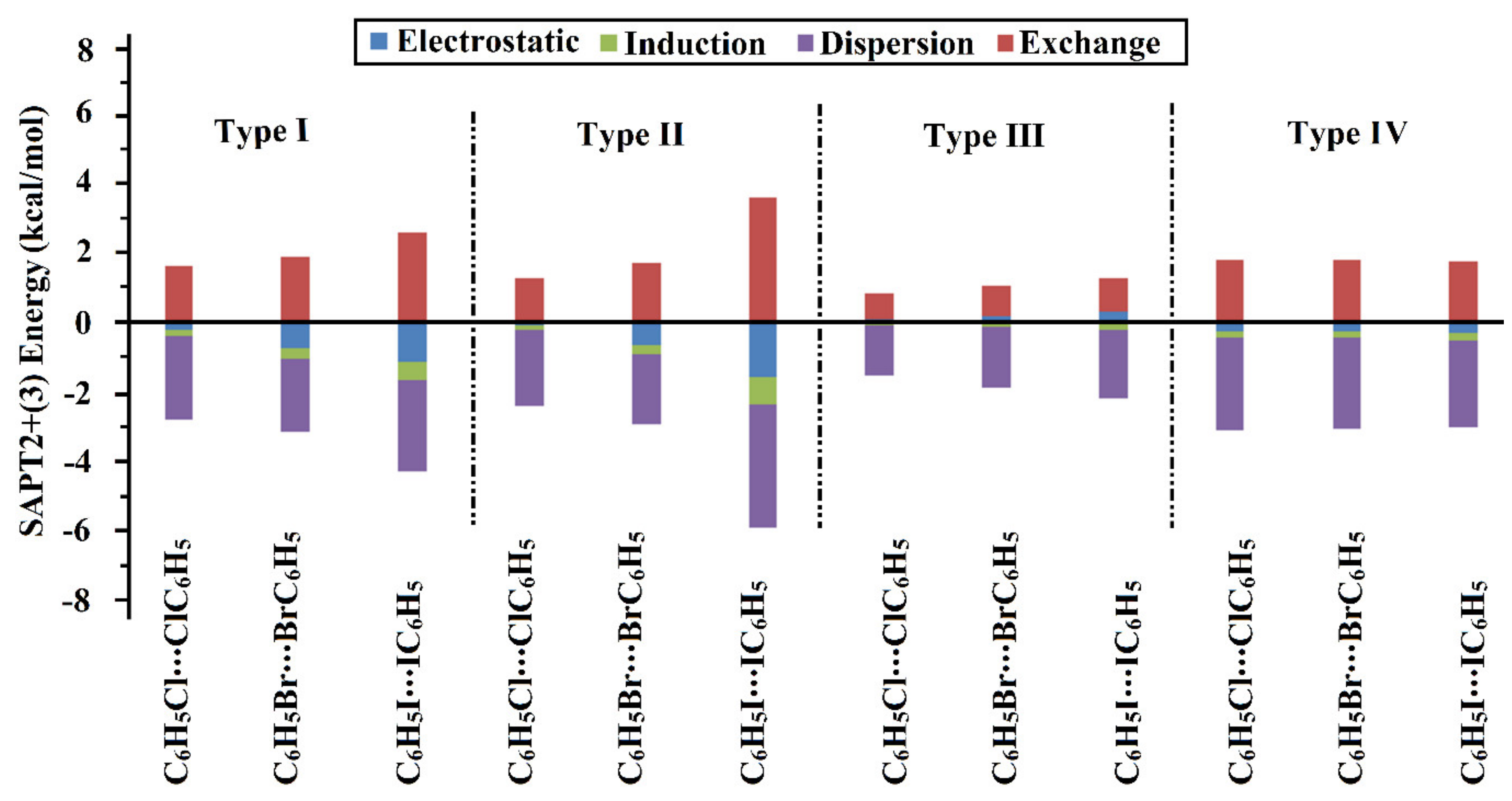
2.6. Symmetry-Adapted Perturbation Theory-Based Energy Decomposition Analysis (SAPT-EDA)
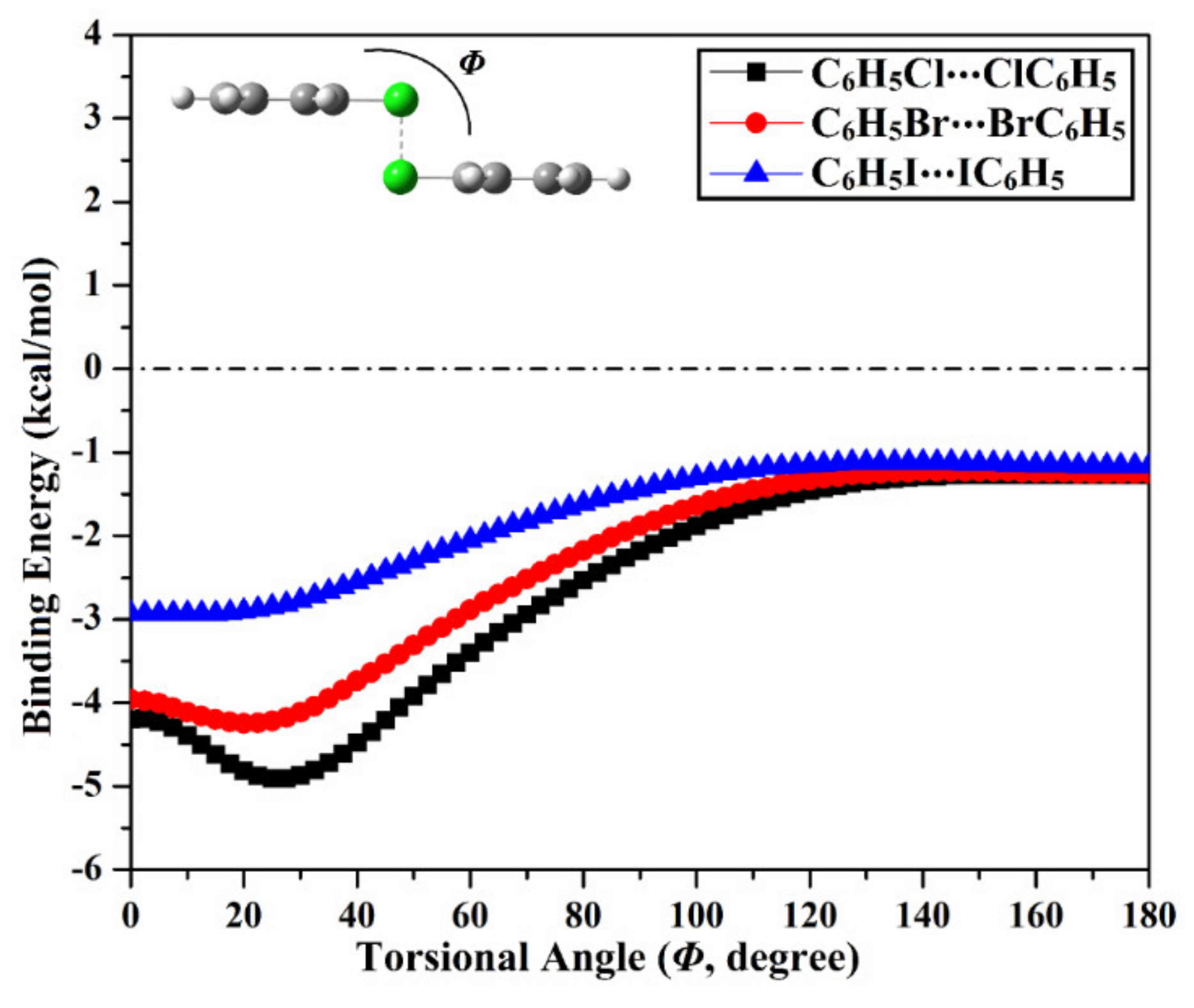
2.7. π⋯π. Contribution into Type IV Halogen⋯Halogen Interaction
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breugst, M.; Koenig, J.J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 2020, 5473–5487. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Beer, P.D. Sigma-Hole Interactions in Anion Recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Kriz, K.; Fanfrlik, J.; Lepsik, M. Chalcogen Bonding in Protein-Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. ChemPhysChem 2018, 19, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Scholfield, M.R.; Zanden, C.M.; Carter, M.; Ho, P.S. Halogen bonding (X-bonding): A biological perspective. Protein Sci. 2013, 22, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, G.; Robeyns, K.; Soubhye, J.; Wintjens, R.; Meyer, F. Halogen bonding in a multi-connected 1,2,2-triiodo-alkene involving geminal and/or vicinal iodines: A crystallographic and DFT study. CrystEngComm 2016, 18, 683–690. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A. Molecular mechanical perspective on halogen bonding. J. Mol. Model. 2012, 18, 4625–4638. [Google Scholar] [CrossRef] [PubMed]
- Mustoe, C.L.; Gunabalasingam, M.; Yu, D.; Patrick, B.O.; Kennepohl, P. Probing covalency in halogen bonds through donor K-edge X-ray absorption spectroscopy: Polyhalides as coordination complexes. Faraday Discuss. 2017, 203, 79–91. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The sigma-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Alkorta, I.; Sanchez-Sanz, G.; Elguero, J.; Del Bene, J.E. Influence of Hydrogen Bonds on the P...P Pnicogen Bond. J. Chem. Theory Comput. 2012, 8, 2320–2327. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Strength of the pnicogen bond in complexes involving group Va elements N, P, and As. J. Phys. Chem. A 2015, 119, 1642–1656. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The Origin of Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.A.; Kamel, A.A.K.; Soliman, M.E.S.; Moustafa, M.F.; El-Mageed, H.R.A.; Taha, F.; Mohamed, L.A.; Moussa, N.A.M. Effect of external electric field on tetrel bonding interactions in (FTF3...FH) complexes (T = C, Si, Ge, and Sn). ACS Omega 2021, 6, 25476–25485. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The sigma-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the sigma-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Nazare, M.; Will, D.W.; Matter, H.; Schreuder, H.; Ritter, K.; Urmann, M.; Essrich, M.; Bauer, A.; Wagner, M.; Czech, J.; et al. Probing the subpockets of factor Xa reveals two binding modes for inhibitors based on a 2-carboxyindole scaffold: A study combining structure-activity relationship and X-ray crystallography. J. Med. Chem. 2005, 48, 4511–4525. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, Y.; Zhu, W. Nonbonding interactions of organic halogens in biological systems: Implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [Google Scholar] [CrossRef]
- Zhou, P.; Zou, J.; Tian, F.; Shang, Z. Fluorine bonding—How does it work in protein-ligand interactions? J. Chem. Inf. Model. 2009, 49, 2344–2355. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Metal-bound halogen atoms in crystal engineering. Chem. Commun. 2013, 49, 1783–1785. [Google Scholar] [CrossRef] [PubMed]
- Teyssandier, J.; Mali, K.S.; De Feyter, S. Halogen Bonding in Two-Dimensional Crystal Engineering. ChemistryOpen 2020, 9, 225–241. [Google Scholar] [CrossRef]
- Ding, X.; Tuikka, M.; Haukka, M. Halogen bonding in crystal engineering. In Recent Advances in Crystallography; IntechOpen: London, UK, 2012; Volume 262, pp. 143–168. [Google Scholar]
- Bruckmann, A.; Pena, M.A.; Bolm, C. Organocatalysis through halogen-bond activation. Synlett 2008, 2008, 900–902. [Google Scholar] [CrossRef]
- Kniep, F.; Jungbauer, S.H.; Zhang, Q.; Walter, S.M.; Schindler, S.; Schnapperelle, I.; Herdtweck, E.; Huber, S.M. Organocatalysis by neutral multidentate halogen-bond donors. Angew. Chem. Int. Ed. Engl. 2013, 52, 7028–7032. [Google Scholar] [CrossRef] [PubMed]
- Mahlanen, R.; Jalkanen, J.P.; Pakkanen, T.A. Potential energy surfaces of CF4, CCl4 and CBr4 dimers. Chem. Phys. 2005, 313, 271–277. [Google Scholar] [CrossRef]
- Latajka, Z.; Berski, S. Density functional study of the H3N□Cl2 system—The importance of Hartree-Fock exchange in density functional methods. J. Mol. Struc. THEOCHEM 1996, 371, 11–16. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, C.-Y.; You, X.-Z. Systematic Theoretical Study of Structures and Bondings of the Charge-Transfer Complexes of Ammonia with HX, XY, and X2 (X and Y are Halogens). J. Phys. Chem. A 1997, 101, 2879–2885. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Charge-transfer complexes between dihalogen compounds and electron donors. J. Phys. Chem. A 1998, 102, 9278–9285. [Google Scholar] [CrossRef]
- Price, S.L.; Stone, A.J.; Lucas, J.; Rowland, R.S.; Thornley, A.E. The Nature of -Cl.cntdot..cntdot..cntdot.Cl- Intermolecular Interactions. J. Am. Chem. Soc. 2002, 116, 4910–4918. [Google Scholar] [CrossRef]
- Bui, T.T.; Dahaoui, S.; Lecomte, C.; Desiraju, G.R.; Espinosa, E. The nature of halogen...halogen interactions: A model derived from experimental charge-density analysis. Angew. Chem. Int. Ed. Engl. 2009, 48, 3838–3841. [Google Scholar] [CrossRef]
- Voth, A.R.; Khuu, P.; Oishi, K.; Ho, P.S. Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem. 2009, 1, 74–79. [Google Scholar] [CrossRef]
- Jones, G.R.; Burbank, R.D.; Bartlett, N. Crystal structure of the 1:1 molecular addition compound xenon difluoride-iodine pentafluoride, XeF2.IF5. Inorg. Chem. 2002, 9, 2264–2268. [Google Scholar] [CrossRef]
- Maddox, H.; McCullough, J.D. The Crystal and Molecular Structure of the Iodine Complex of 1-Oxa-4-selenacyclohexane, C4H8OSe.I2. Inorg. Chem. 2002, 5, 522–526. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Hasb, A.A.M. Polarization plays the key role in halogen bonding: A point-of-charge-based quantum mechanical study. Theor. Chem. Acc. 2019, 138, 2–13. [Google Scholar] [CrossRef]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the fluorine in molecules dispersive? Is molecular electrostatic potential a valid property to explore fluorine-centered non-covalent interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen bonding: A halogen-centered noncovalent interaction yet to be understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Zhou, J.; Wang, Q.; Jena, P. Like charges attract? J. Phys. Chem. Lett. 2016, 7, 2689–2695. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Do surfaces of positive electrostatic potential on different halogen derivatives in molecules attract? like attracting like! J. Comput. Chem. 2018, 39, 343–350. [Google Scholar] [CrossRef]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Nature of halogen-centered intermolecular interactions in crystal growth and design: Fluorine-centered interactions in dimers in crystalline hexafluoropropylene as a prototype. J. Comput. Chem. 2019, 40, 1836–1860. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Revealing factors influencing the fluorine-centered non-covalent interactions in some fluorine-substituted molecular complexes: Insights from first-principles studies. ChemPhysChem 2018, 19, 1486–1499. [Google Scholar] [CrossRef]
- Echeverría, J.; Velásquez, J.D.; Alvarez, S. Understanding the interplay of dispersion, charge transfer, and electrostatics in noncovalent interactions: The case of bromine–carbonyl short contacts. Cryst. Growth Des. 2020, 20, 7180–7187. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Type II halogen...halogen contacts are halogen bonds. IUCrJ 2014, 1, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Moussa, N.A.M. Unconventional type III halogen...halogen interactions: A quantum mechanical elucidation of sigma-hole...sigma-hole and di-sigma-hole interactions. ACS Omega 2020, 5, 21824–21835. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Parthasarathy, R. The nature of halogen...halogen interactions: Are short halogen contacts due to specific attractive forces or due to close packing of nonspherical atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Shehata, M.N.I.; Soliman, M.E.S.; Moustafa, M.F.; El-Mageed, H.R.A.; Moussa, N.A.M. Unusual Chalcogen∙∙∙Chalcogen Interactions in Like∙∙∙Like and Unlike Y=C=Y∙∙∙Y=C=Y Complexes (Y = O, S, and Se). Phys. Chem. Chem. Phys. 2022, 24, 3386–3399. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Weiner, P.K.; Langridge, R.; Blaney, J.M.; Schaefer, R.; Kollman, P.A. Electrostatic potential molecular surfaces. Proc. Natl. Acad. Sci. USA 1982, 79, 3754–3758. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.A.; Mohamed, Y.A.M.; Abd Elhafez, H.S.M.; Shehata, M.N.I.; Soliman, M.E.S.; Ahmed, M.N.; Abd El-Mageed, H.R.; Moussa, N.A.M. R(*)-hole interactions of group IV-VII radical-containing molecules: A comparative study. J. Mol. Graph. Model. 2022, 111, 108097. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Rady, A.S.S.M.; Al-Fahemi, J.H.; Telb, E.M.Z.; Ahmed, S.A.; Shawky, A.M.; Moussa, N.A.M. ±π-Hole Interactions: A Comparative Investigation Based on Boron-Containing Molecules. ChemistrySelect 2020, 5, 13223–13231. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Rady, A.-S.S.M.; Soliman, M.E.S.; Moustafa, M.F.; El-Mageed, H.R.A.; Moussa, N.A.M. π-Hole interactions of group III–VI elements with π-systems and Lewis bases: A comparative study. Struct. Chem. 2022, 33, 9–21. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Mohamed, Y.A.M.; Abuelliel, H.A.A.; Rady, A.s.S.M.; Soliman, M.E.S.; Ahmed, M.N.; Mohamed, L.A.; Moussa, N.A.M. σ-Hole interactions of tetrahedral group iv–viii lewis acid centers with lewis bases: A comparative study. ChemistrySelect 2021, 6, 11856–11864. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Telb, E.M.Z. Sigma-hole and lone-pair hole interactions in chalcogen-containing complexes: A comparative study. ACS Omega 2020, 5, 21631–21640. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Telb, E.M.Z. Comparison of ±σ-hole and ±R-hole interactions formed by tetrel-containing complexes: A computational study. RSC Adv. 2021, 11, 4011–4021. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Mahmoud, A.H.M.; Moussa, N.A.M. Comparative investigation of ±σ–hole interactions of carbon-containing molecules with Lewis bases, acids and di-halogens. Chem. Pap. 2020, 74, 3569–3580. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Telb, E.M.Z. A computational investigation of unconventional lone-pair hole interactions of group V–VIII elements. ChemistrySelect 2019, 4, 5489–5495. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Saad, S.M.A.; Al-Fahemi, J.H.; Mekhemer, G.A.H.; Ahmed, S.A.; Shawky, A.M.; Moussa, N.A.M. External electric field effects on the σ-hole and lone-pair hole interactions of group V elements: A comparative investigation. RSC Adv. 2021, 11, 4022–4034. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The nature of halogen...halogen synthons: Crystallographic and theoretical studies. Chemistry 2006, 12, 8952–8960. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Moussa, N.A.M.; Soliman, M.E.S.; Moustafa, M.F.; Al-Fahemi, J.H.; El-Mageed, H.R.A. On the potentiality of X-T-X3 compounds (T = C, Si, and Ge, and X = F, Cl, and Br) as tetrel- and halogen-bond donors. ACS Omega 2021, 6, 19330–19341. [Google Scholar] [CrossRef]
- Cukrowski, I.; de Lange, J.H.; Adeyinka, A.S.; Mangondo, P. Evaluating common QTAIM and NCI interpretations of the electron density concentration through IQA interaction energies and 1D cross-sections of the electron and deformation density distributions. Comput. Theor. Chem. 2015, 1053, 60–76. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting and Cholesky decomposition approximations in symmetry-adapted perturbation theory: Implementation and application to probe the nature of pi-pi interactions in linear acenes. J. Chem. Phys. 2010, 132, 184111–184120. [Google Scholar] [CrossRef] [Green Version]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. PSI4: An open-source ab initio electronic structure program. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Ahmed, O.A.M.; Moussa, N.A.M.; El-Taher, S.; Moustafa, H. Comparative investigation of interactions of hydrogen, halogen and tetrel bond donors with electron-rich and electron-deficient π-systems. RSC Adv. 2019, 9, 32811–32820. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.A.; Ahmed, O.A.M.; El-Taher, S.; Al-Fahemi, J.H.; Moussa, N.A.M.; Moustafa, H. Cospatial sigma-hole and lone pair interactions of square-pyramidal pentavalent halogen compounds with pi-systems: A quantum mechanical study. ACS Omega 2021, 6, 3319–3329. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Hasb, A.A.M.; Mekhemer, G.A.H. Role and nature of halogen bonding in inhibitor … receptor complexes for drug discovery: Casein kinase-2 (CK2) inhibition as a case study. Theor. Chem. Acc. 2018, 137, 38–47. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Moussa, N.A.M.; Safy, M.E.A. Quantum-mechanical investigation of tetrel bond characteristics based on the point-of-charge (PoC) approach. J. Mol. Model. 2018, 24, 219. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Safy, M.E.A. A new insight for chalcogen bonding based on Point-of-Charge approach. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 444–454. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Molecular Stabilization Energy (Estabilization/Edestabilization, kcal/mol) | |||
|---|---|---|---|---|
| PoC= −0.50 au | PoC= +0.50 au | |||
| x-Axis | z-Axis | x-Axis | z-Axis | |
| C6H5Cl⋯PoC | −2.88 | 1.06 | −4.39 | –9.70 |
| C6H5Br⋯PoC | −6.08 | 0.28 | −3.25 | –11.31 |
| C6H5I⋯PoC | −10.99 | −1.68 | −2.14 | –13.54 |
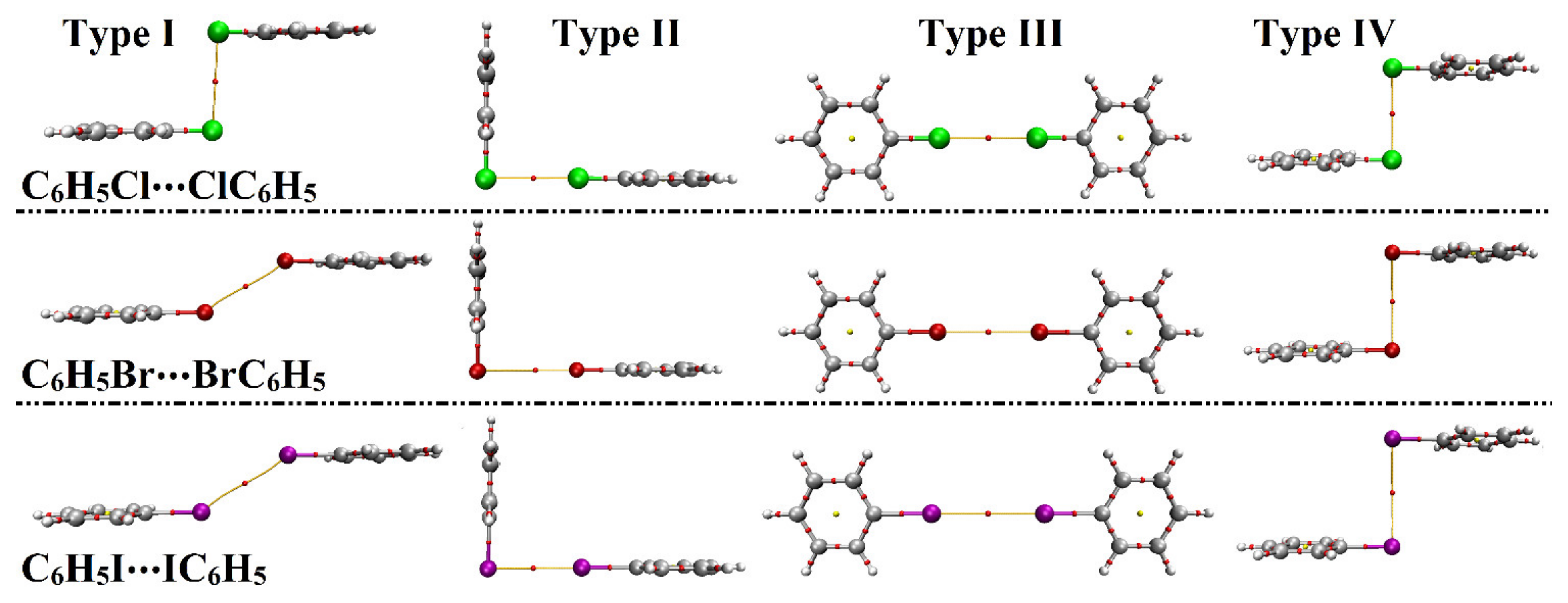
| Homodimer | Complexation Parameters a | Binding Energy | |||
|---|---|---|---|---|---|
| Distance (Å) | θ1b (Degree) | θ2b (Degree) | EMP2/aug−cc−pVDZ (kcal/mol) | ||
| Type I | C6H5Cl⋯ClC6H5 | 3.71 | 92.5° | 92.5° | –1.12 |
| C6H5Br⋯BrC6H5 | 3.71 | 147.5° | 147.5° | –1.15 | |
| C6H5I⋯IC6H5 | 4.02 | 147.5° | 147.5° | –1.49 | |
| Type II | C6H5Cl⋯ClC6H5 | 3.49 | 90° | 180° | –1.32 |
| C6H5Br⋯BrC6H5 | 3.64 | –1.80 | |||
| C6H5I⋯IC6H5 | 3.96 | –2.20 | |||
| Type III | C6H5Cl⋯ClC6H5 | 3.58 | 180° | 180° | –0.60 |
| C6H5Br⋯BrC6H5 | 3.77 | –0.74 | |||
| C6H5I⋯IC6H5 | 4.15 | –0.79 | |||
| Type IV | C6H5Cl⋯ClC6H5 | 3.68 | 90° | 90° | –1.27 |
| C6H5Br⋯BrC6H5 | 3.97 | –1.25 | |||
| C6H5I⋯IC6H5 | 4.43 | –1.16 | |||
| Homodimer | ρb (au) | ∇2ρb (au) | Hb (au) | |
|---|---|---|---|---|
| Type I | C6H5Cl⋯ClC6H5 | 0.00573 | 0.01575 | 0.00050 |
| C6H5Br⋯BrC6H5 | 0.00584 | 0.01839 | 0.00092 | |
| C6H5I⋯IC6H5 | 0.00604 | 0.01499 | 0.00064 | |
| Type II | C6H5Cl⋯ClC6H5 | 0.00660 | 0.02206 | 0.00091 |
| C6H5Br⋯BrC6H5 | 0.00708 | 0.02255 | 0.00107 | |
| C6H5I⋯IC6H5 | 0.00713 | 0.01773 | 0.00072 | |
| Type III | C6H5Cl⋯ClC6H5 | 0.00417 | 0.01567 | 0.00077 |
| C6H5Br⋯BrC6H5 | 0.00406 | 0.01470 | 0.00077 | |
| C6H5I⋯IC6H5 | 0.00374 | 0.01070 | 0.00050 | |
| Type IV | C6H5Cl⋯ClC6H5 | 0.00603 | 0.01676 | 0.00052 |
| C6H5Br⋯BrC6H5 | 0.00516 | 0.01368 | 0.00065 | |
| C6H5I⋯IC6H5 | 0.00439 | 0.00905 | 0.00036 |
| Homodimer | Eelst | Eind | Edisp | Eexch | ESAPT2+(3)b | ∆∆E c | |
|---|---|---|---|---|---|---|---|
| Type I | C6H5Cl⋯ClC6H5 | –0.21 | –0.17 | –2.43 | 1.64 | –1.17 | 0.41 |
| C6H5Br⋯BrC6H5 | –0.74 | –0.30 | –2.12 | 1.91 | –1.24 | 0.09 | |
| C6H5I⋯IC6H5 | –1.13 | –0.53 | –2.65 | 2.62 | –1.68 | 0.19 | |
| Type II | C6H5Cl⋯ClC6H5 | –0.08 | –0.14 | –2.19 | 1.26 | –1.15 | –0.17 |
| C6H5Br⋯BrC6H5 | –0.67 | –0.27 | –2.03 | 1.74 | –1.24 | –0.56 | |
| C6H5I⋯IC6H5 | –1.61 | –0.77 | –3.59 | 3.60 | –2.36 | 0.16 | |
| Type III | C6H5Cl⋯ClC6H5 | 0.09 | –0.10 | –1.44 | 0.77 | –0.68 | 0.08 |
| C6H5Br⋯BrC6H5 | 0.17 | –0.15 | –1.75 | 0.91 | –0.82 | 0.08 | |
| C6H5I⋯IC6H5 | 0.32 | –0.24 | –1.99 | 0.98 | –0.93 | 0.14 | |
| Type IV | C6H5Cl⋯ClC6H5 | –0.28 | –0.18 | –2.67 | 1.83 | –1.30 | 0.03 |
| C6H5Br⋯BrC6H5 | –0.27 | –0.18 | –2.65 | 1.80 | –1.30 | 0.05 | |
| C6H5I⋯IC6H5 | –0.33 | –0.19 | –2.54 | 1.77 | –1.29 | 0.13 | |
| Homodimer | trans Configuration | cis Configuration | Trans → cis Interconversion | |
|---|---|---|---|---|
| Etrans (kcal/mol) | Ecis (kcal/mol) | Emax (kcal/mol) | Φa (Degree) | |
| C6H5Cl⋯ClC6H5 | –1.27 | –4.20 | –4.91 | 25° |
| C6H5Br⋯BrC6H5 | –1.25 | –3.96 | –4.25 | 20° |
| C6H5I⋯IC6H5 | –1.16 | –2.93 | –2.93 | 10° |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Saeed, R.R.A.; Shehata, M.N.I.; Ahmed, M.N.; Shawky, A.M.; Khowdiary, M.M.; Elkaeed, E.B.; Soliman, M.E.S.; Moussa, N.A.M. Type I–IV Halogen⋯Halogen Interactions: A Comparative Theoretical Study in Halobenzene⋯Halobenzene Homodimers. Int. J. Mol. Sci. 2022, 23, 3114. https://doi.org/10.3390/ijms23063114
Ibrahim MAA, Saeed RRA, Shehata MNI, Ahmed MN, Shawky AM, Khowdiary MM, Elkaeed EB, Soliman MES, Moussa NAM. Type I–IV Halogen⋯Halogen Interactions: A Comparative Theoretical Study in Halobenzene⋯Halobenzene Homodimers. International Journal of Molecular Sciences. 2022; 23(6):3114. https://doi.org/10.3390/ijms23063114
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Rehab R. A. Saeed, Mohammed N. I. Shehata, Muhammad Naeem Ahmed, Ahmed M. Shawky, Manal M. Khowdiary, Eslam B. Elkaeed, Mahmoud E. S. Soliman, and Nayra A. M. Moussa. 2022. "Type I–IV Halogen⋯Halogen Interactions: A Comparative Theoretical Study in Halobenzene⋯Halobenzene Homodimers" International Journal of Molecular Sciences 23, no. 6: 3114. https://doi.org/10.3390/ijms23063114
APA StyleIbrahim, M. A. A., Saeed, R. R. A., Shehata, M. N. I., Ahmed, M. N., Shawky, A. M., Khowdiary, M. M., Elkaeed, E. B., Soliman, M. E. S., & Moussa, N. A. M. (2022). Type I–IV Halogen⋯Halogen Interactions: A Comparative Theoretical Study in Halobenzene⋯Halobenzene Homodimers. International Journal of Molecular Sciences, 23(6), 3114. https://doi.org/10.3390/ijms23063114