1. Introduction
With the aging of the world’s population, the number of neurodegenerative disease cases has increased exponentially. It is predicted that this number will rise to around 135 million in 2050, with a projected economic burden of up to 20 billion US dollars and rising [
1]. The most prominent neurodegenerative diseases, Alzheimer’s disease, Parkinson’s disease (PD), Huntington’s disease and amyotrophic lateral sclerosis, share neuropathological hallmarks such as oxidative stress, abnormal protein glycation, abnormal protein deposition (proteotoxic stress), dysfunction in ubiquitin-proteosome and autophagosomal/lysosomal systems, neuroinflammation, progressive neuronal dysfunction and cell death [
2,
3,
4]. A key central player in the pathophysiology of neurodegenerative diseases is mitochondrial dysfunction. The central nervous system is highly dependent on this organelle to function correctly for energy supply and calcium (Ca
2+) buffering [
5,
6,
7]. Manifestations of mitochondrial dysfunction in neurodegenerative diseases range from altered dynamics (fusion/fission and axonal transport) leading to abnormal organelle morphology and cellular distribution, to oxidative stress, Ca
2+ dyshomeostasis, mitochondrial membrane depolarization, decreased adenosine triphosphate (ATP) production, and mitochondrial DNA (mtDNA) damage with concomitant cell death [
8,
9].
No useful diagnostic tools exist for the early detection of neurodegenerative diseases, with the neuropathological evaluation at autopsy being the actual golden diagnostic standard [
2]. Moreover, when clinical symptoms appear, the cellular and metabolic alterations have already reached a point of no return towards neurodegeneration and neuronal cell death. Due to the massive social and economic impact of neurodegenerative diseases, significant efforts to learn and identify risk factors and mechanisms behind the neuropathology of these diseases have been put in place [
4]. One of the most promising approaches is targeting mitochondria, since the previously mentioned structural and metabolic alterations in mitochondria start at the early stages of neurodegeneration, prior to neuronal cell death [
9,
10]. A certain threshold of mitochondrial dysfunction and decreased ATP production must occur for visible clinical signs [
11,
12,
13].
Differentiated SH-SY5Y cells are useful in vitro models to investigate cellular and mitochondrial morphological and bioenergetic alterations in mitochondrial dysfunction-related pathologies such as neurodegenerative diseases [
14]. Moreover, 6-hydroxydopamine (6-OHDA) and rotenone have been used to develop PD models [
15]. In this work, differentiated SH-SY5Y cells were treated for different time points and with distinct concentrations of 6-OHDA and rotenone, which were previously shown to affect mitochondrial movement in these cells [
16]. Experimental biological analyses of cell and mitochondrial function parameters were then assessed. The combination of classical statistical methods, and unsupervised and supervised machine learning (ML) algorithms were used to assess the crosstalk between the different biological assays used, attempting to understand the differences between the mechanism of action and the degree of toxicity of 6-OHDA and rotenone. Our main objective was to rely on a ML framework to determine which experimental biological features are the most relevant in neurodegeneration induction, prior to cell death, caused by 6-OHDA and rotenone in vitro. Moreover, we wanted to unravel the different mechanisms of action of these compounds on SH-SY5Y toxicity that may help prevent neuronal cell death using different biological approaches.
3. Discussion
The use of computational applied statistical methodologies in biology, i.e., combining classical statistics with ML algorithms, is a continuously evolving field. In this interdisciplinary work, we applied ML methodologies in original biological data to predict which experimental features could inform us about the mechanism and degree of toxicity of 6-OHDA and rotenone related with neurodegeneration before cell death. Concerning the ML approaches, we used unsupervised and supervised methods. Finally, we performed an interdisciplinary analysis of both biological and computational results retrieving the most important conclusions from the combination of both fields.
Different studies revealed similar results regarding the effects of 6-OHDA and rotenone on SH-SY5Y cells. Firstly, both compounds were previously shown to induce a mitochondrial fragmentation phenotype [
21,
22,
23] and prompt a loss of ΔΨm, release of cytochrome c from mitochondria, reduction of mtDNA content, and decreased protein levels of PGC-1α, NRF1, and TFAM [
22,
24]. Additionally, increased oxidative stress and caspase 3 activity were also previously found in SH-SY5Y treated with these compounds [
21,
25,
26,
27]. Using different cell models, both compounds were previously shown to induce mitochondrial bioenergetic deficits. Using rat forebrain mitochondria, 6-OHDA induced a reduction in respiratory control ratio and rate of oxygen consumption at state 3 (state 4 remained unaltered) [
28], while primary rat cortical neurons treated with rotenone presented an evident decrease in OCR-linked ATP production, maximal respiration-linked OCR, and spare respiratory capacity, while non-mitochondrial respiration-linked OCR remained unaltered [
29].
Additionally, both compounds have been reported to induce autophagy. A study by Chu and collaborators, using both primary cortical neurons and undifferentiated SH-SY5Y treated with sublethal concentrations of rotenone and 6-OHDA, showed an evident increase in LC3 levels and LC3 puncta co-localized with mitochondria. A decrease in p62 levels was also present, suggesting an increased autophagic flux [
30].
Nonetheless, 6-OHDA and rotenone are mitochondrial toxicants known to have different mechanisms of action. Firstly, while rotenone is a highly lipophilic and hydrophobic molecule that can easily cross cell membranes without the need for specific membrane transport mechanisms [
31], 6-OHDA cellular uptake needs specific transporters such as the dopamine or norepinephrine transporters [
32,
33]. Regarding mitochondrial-induced toxic effects, these compounds also display different mechanisms of action. Rotenone is a golden-standard high-affinity, time-dependent, irreversible mitochondrial complex I inhibitor [
34] while 6-OHDA inhibits both mitochondrial complexes I and IV [
35]. Complex I inhibition by both compounds leads to superoxide production [
15,
36]. Moreover, 6-OHDA is an easily oxidizable molecule, forming semiquinone radicals, participating in free radical producing reactions and leading to H
2O
2, O
2• − and
•OH production [
37].
These differences could help to explain why, in our work, despite inducing similar effects in caspase activity, mitochondrial-encoded ETC subunits mRNA levels, mitochondrial morphology, Mitosox Red oxidation, mtDNA copy number, and lysosomal protease activity; in the remaining parameters, 6-OHDA and rotenone promoted distinct effects. 6-OHDA induced a decrease in mitochondrial area and ΔΨm, mRNA levels of nuclear-encoded ETC subunits, fusion/fission proteins, and antioxidant enzymes, and ATP production rate. Moreover, 6-OHDA also prompted an acute increase in mitochondrial bioenergetic parameters. Rotenone promoted a decrease in mitochondrial bioenergetic parameters and ATP production rate while inducing an increase in ΔΨm and mitochondrial biogenesis-associated genes.
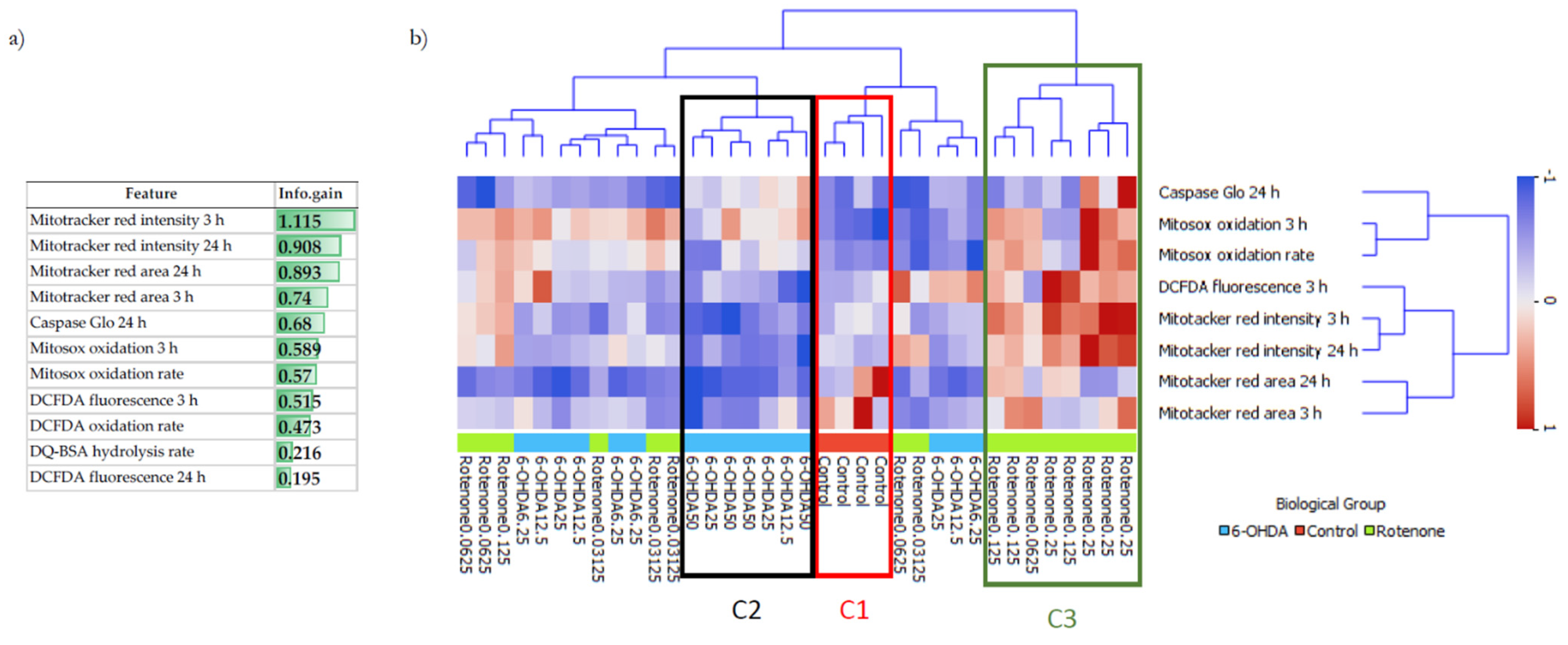
To understand the differences and similarities of non-toxic concentrations of 6-OHDA and rotenone on differentiated SH-SY5Y cells and to complement our biological data analysis, ML methodologies, such as hierarchical clustering and supervised classification algorithms, were used to clarify two main questions. Firstly, was it possible to distinguish between control and treated cells? And if so, is it also possible to distinguish between control cells and cells treated with different 6-OHDA or rotenone concentrations? Hierarchical clustering is an unsupervised approach that estimates the dissimilarities (based on distances) between any pair of observations. Being an unsupervised method, hierarchical clustering aimed to discover hidden patterns in the data without using the respective class labels for each observation. This methodology allowed us to have a broader idea of how the different observations were organized. Information gain revealed that the features MitoTracker area and intensity (3 and 24 h), mitochondrial (Mitosox oxidation 3 h and oxidation rate) and cellular (DCFDA fluorescence 3 h)-derived oxidative stress, and Caspase activity, seem to have an enhanced ability to signal neurodegeneration induction, prior to cell death, caused by 6-OHDA and rotenone. Using this subset of eight features with higher information gain, hierarchical clustering automatically grouped control cells in one cluster and cells treated with higher concentrations of 6-OHDA and higher rotenone concentrations in distinct clusters. This occurred because the higher the concentration of each compound, the more evidently different biologic effects on SH-SY5Y cells were demonstrated. On the other hand, milder rotenone and 6-OHDA concentrations were clustered together since they caused smaller and similar effects. Biological results were also used to train a decision tree predictive model. In this methodology, the output labels are known during model training (contrary to hierarchical clustering). The decision tree algorithm could correctly classify a high percentage (>75%) of cells treated with the highest 6-OHDA and rotenone concentrations (50 µM and 0.25 µM, respectively), but only 50% of control cells. Interestingly, this model was able to predict correctly 75% of cells treated with 6.25 µM 6-OHDA, being an improvement from the results derived from the hierarchical clustering analysis. Regarding cells treated with the lowest rotenone concentration, the hierarchical clustering results already suggest that classification methods may be unable to classify samples in their respective class correctly. Misclassified samples were predicted to be in classes corresponding to the remaining rotenone concentrations or the class of cells treated with 6.25 µM 6-OHDA. Similar to what happened in the hierarchical clustering analysis, the predictive model was unable to distinguish between cells treated with a milder concentration of both compounds (except on cells treated with 6.25 µM 6-OHDA), since the biological effects caused by these conditions only induced smaller and similar effects.
4. Materials and Methods
4.1. Cell Culture
SH-SY5Y human cell line (ECACC, cat. 94030304, Merck KGaA, Darmstadt, Germany) was cultured in a humidified atmosphere with 5% CO2, 95% air at 37 °C. The media used for undifferentiated cells was Dulbecco’s Modified Eagle’s Medium (DMEM D5030, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) supplemented with 4.5 g/L D-(+)-glucose (cat. G7021, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 0.8 g/L L-glutamine (cat. G3126, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 1.2 g/L HEPES (cat. H4024, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 3.7 g/L sodium bicarbonate (cat. S6014, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 0.1 g/L sodium pyruvate (cat. P2256, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 10% fetal bovine serum (FBS, cat. 41F6445K, Gibco, Thermo Fisher Scientific, Hampton, NH, USA) and 1% penicillin/streptomycin (cat 1772652 Thermo Fisher Scientific, Hampton, NH, USA). Cell media was changed every 2 to 3 days and cells were subcultured, until the 23rd passage, when reaching 90–100% of confluence.
We previously described the protocol used for SH-SY5Y cell differentiation in ref. [
14]. Briefly, undifferentiated cells were plated at a density of 30,000 cells/cm
2 in a low D-(+)-glucose (0.9 g/L) media supplemented with 1% FBS and 10 µM retinoic acid (cat. A6947 Panreac AppliChem ITW Reagents, Darmstadt, Germany) for 3 days. Exceptionally, cells were plated at a density of 120,000 cells/cm
2 in the Seahorse experiments (Seahorse XF Cell Mito Stress Test and Real-Time ATP Rate Assay) due to equipment sensitivity.
4.2. Cell Treatments
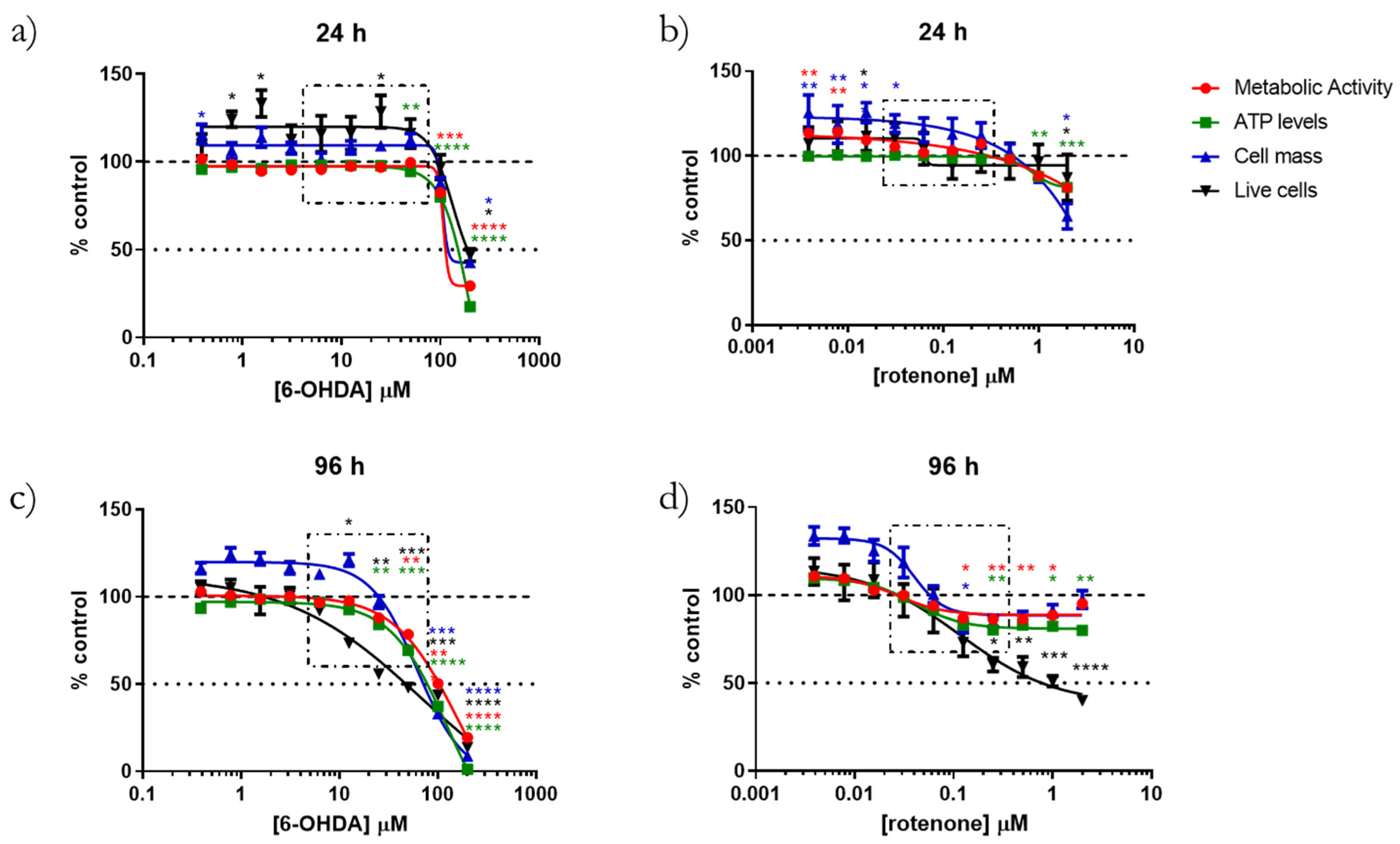
Two mitochondrial poisons were used, 6-OHDA (cat. H4381 Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) and rotenone (Cat. MKBS1062V, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). Rotenone was dissolved in dimethyl sulfoxide (DMSO, Thermo Fisher Scientific, Hampton, NH, USA) while 6-OHDA is water-soluble. The final concentration of DMSO used was never higher than 0.1% (v/v). Cell treatment strategies are specified in the respective results subsection and in the respective figure legend.
4.3. Cell Proliferation Measurements by Sulforhodamine B Method
To evaluate the cytotoxic effects of 6-OHDA and rotenone on cell mass, the sulforhodamine B (SRB) assay [
38] was used to indirectly evaluate the total cell protein [
39]. After cell treatments, the medium was removed, and cells were fixed in 1% acetic acid in ice-cold methanol overnight at −20 °C. Then, cells were incubated with 0.05% (
w/
v) SRB (Cat. S9012, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) for 1 h at 37 °C. Subsequently, SRB was removed, and wells were washed with 1% acetic acid to remove the unbound stain. Dye bound to cell proteins was extracted with 10 mM Tris-base solution, pH 10, and absorbance was read at 510 nm and 620 nm (reference wavelength) in a Cytation 3 reader (BioTek Instruments Inc., Winooski, VT, USA). Then, we subtracted the 620 nm absorbance value from the 510 nm absorbance value for each condition. Data were normalized to the average control (100%) condition.
4.4. Cellular Metabolic Viability Assessment Using the Resazurin Assay
The effect of 6-OHDA and rotenone toxicity was also evaluated by measuring the cellular metabolic activity using the resazurin reduction assay. The resazurin assay is based on the reduction of resazurin (Cat. R7017-5G, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) to resorufin by dehydrogenases present in viable cells [
39]. After cell treatments, the medium was replaced by a fresh medium containing resazurin (10 μg/mL) and kept in a humidified atmosphere, 5% CO
2, at 37 °C for 3 h. The fluorescent signal was monitored using a 540 nm excitation wavelength and 590 nm emission wavelength in a Cytation 3 microplate reader. A blank condition (wells without cells) was used as a fluorescent reference signal and subtracted from the remaining conditions. Data were normalized to the average control (100%) condition.
4.5. ATP Levels Determination
Intracellular ATP levels were assessed using CellTiter-Glo Luminescent Cell Viability Assay (cat. G7570, Promega, Madison, WI, USA). Cell differentiation and treatments were undertaken in 96-well white opaque-bottom plates (cat. 136101, Thermo Fisher Scientific, Hampton, NH, USA) and, at the end of the cell treatments, the medium was removed from the wells, replaced by 50 µL of fresh medium plus 50 µL of the Cell Titer-Glo reagent, and mixed for 2 min on an orbital shaker to promote cell lysis. The plate was incubated for 10 min and the luminescent signal was recorded using a Cytation 3 microplate reader (BioTek Instruments Inc., Winooski, VT, USA). Data were normalized to the average control (100%) condition.
4.6. Live Cell Calculation
Cell membrane integrity and cell death induction were evaluated using propidium iodide (PI) and Hoechst 33,342 staining. After the differentiation protocol and 6-OHDA and rotenone incubations, cell nuclei were stained with 4 µM PI (cat. P21493, Thermo Fisher Scientific, Hampton, NH, USA) and 1 µg/mL of Hoechst 33342 (cat. B2261, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). For being a cell-impermeant stain, only cells without an intact membrane were labeled with PI, while Hoechst 33342 labeled all cell nuclei. Both PI and Hoechst 33,342 were added directly to the media without aspirating it in order to avoid removing dead, PI-positive, cells. Cells were incubated with both stains for 5 min and visualized using an INCell Analyzer 2200 (GE Healthcare, Chicago, IL, USA) cell imaging system with 10× magnification (Nikon 10×/0.45, Plan Apo, CFI/60, Tokyo, Japan). The image analysis was performed using IN Cell Analyzer 1000 analysis Software-Developer Toolbox (GE Healthcare, Chicago, IL, USA). The PI negative cells were divided by the total number of cells to calculate the live cell population. Data were normalized to the average control (100%) condition.
4.7. Caspase 3/7 Activity
Caspase 3/7 activity was measured using the Caspase-Glo 3/7 Assay (Promega, Madison, WI, USA). Cell treatments were performed in 96-well white opaque-bottom plates. Some cells were treated with 1 µM Doxorubicin (Cat. BP2516, Thermo Fisher Scientific, Hampton, NH, USA) for 24 h as a positive control. At the time of the assay, the cell medium was removed from the wells and replaced by 50 µL of fresh medium. Then, 50 µL of the Caspase-Glo 3/7 reagent was added and mixed for 2 min on an orbital shaker to promote cell lysis. The plate was incubated for 30 min and the luminescent signal was recorded using a Cytation 3 microplate reader. A blank condition (wells without cells) was used as a luminescent reference signal and subtracted from the remaining conditions.
4.8. Mitochondrial Polarization and Network Distribution Analysis by Live Cell High Throughput Microscopy
Live cell high-throughput microscopy was used to detect alterations in mitochondrial polarization and network distribution in differentiated SH-SY5Y cells exposed to increasing (non-toxic) concentrations of 6-OHDA and rotenone. Cells were differentiated on Corning 96-well black polystyrene microplates (Cat. 3603, Corning GmbH, Kaiserslautern, Germany) and after the treatment with either compound, mitochondria were stained for 30 min with 50 nM of the mitochondrial fluorescent dye Mitotracker Red CMXRos (Cat. M7512, Thermo Fisher Scientific, Hampton, NH, USA) in a humidified atmosphere, 5% CO2, at 37 °C. For positive control, 10 µM carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP, Cat.024M4003V, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) was used and incubated at the same time as the dye incubation. Cells were also stained with 1 µg/mL Hoechst 33,342 for normalization purposes. An INCell Analyzer 2200 (GE Healthcare, Chicago, IL, USA) cell imaging system was used for visualization and image acquisition using a 20× objective (INCA ASAC 20×/0.45, ELWD Plan Fluor). The image analysis was performed using the IN Cell Analyzer 1000 analysis software-Developer Toolbox. A protocol was created to quantify a specific target’s fluorescence in IN Cell Developer Toolbox for image analysis. The image stacks were uploaded to the program to identify our target set and establish the respective parameters: area, intensity, and number. The representative images were visualized using ImageJ 1.52a (Wayne Rasband, National Institute of Health, Bethesda, MD, USA).
4.9. Mitochondrial Network Evaluation and Lysosomal Analysis by Confocal Microscopy
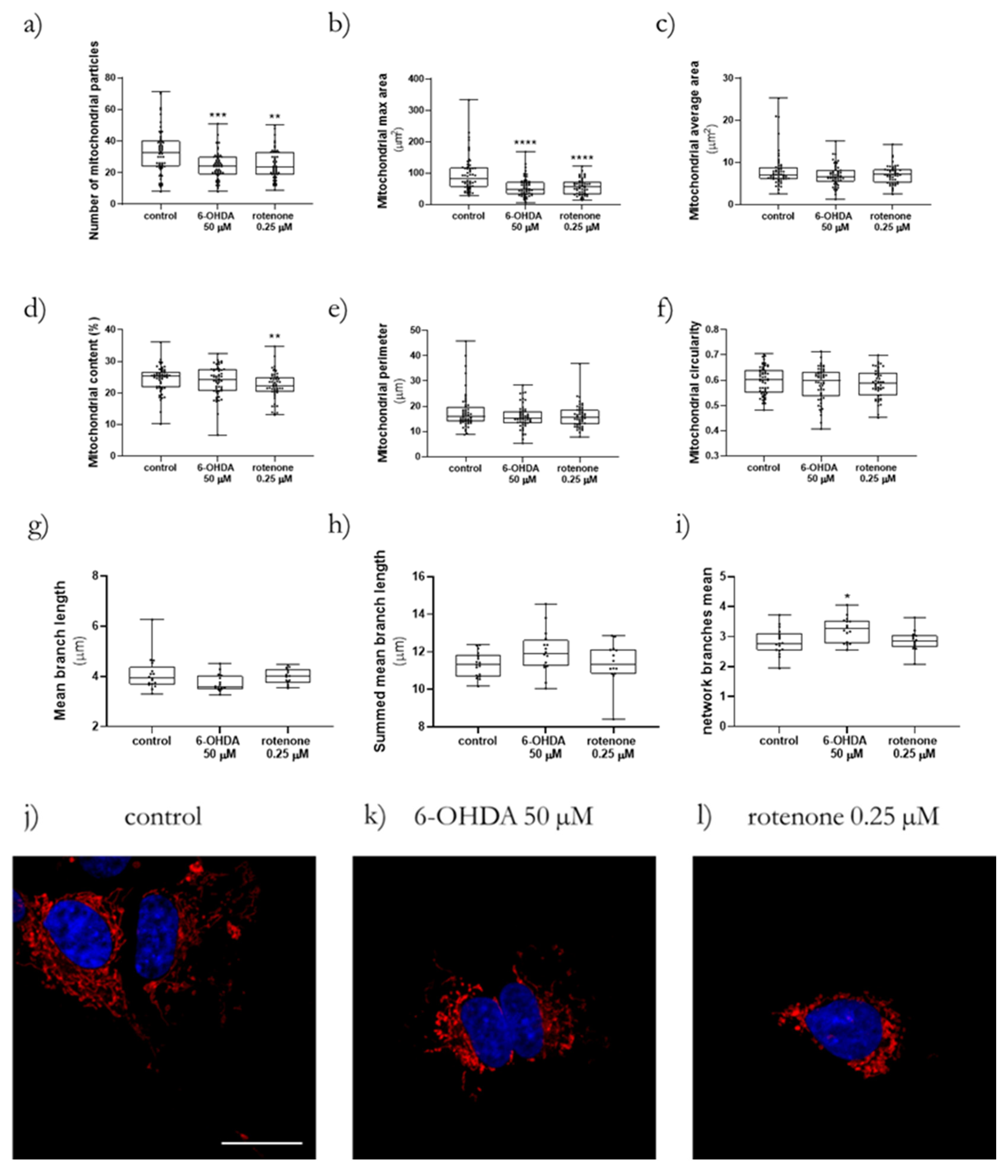
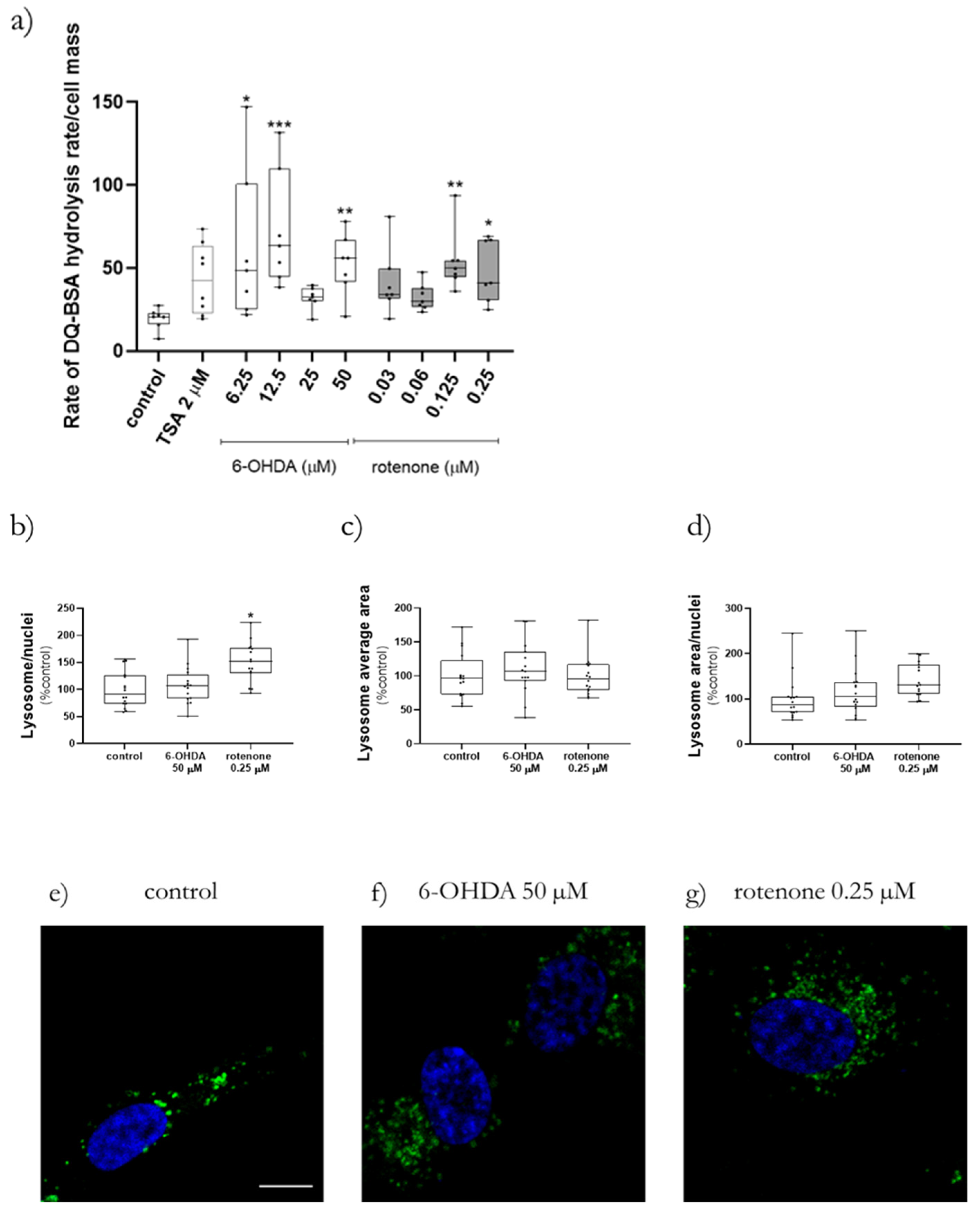
Confocal microscopy was used to accomplish a more detailed definition of the mitochondrial network, to be able to apply the ImageJ macros Mitochondrial Network Analysis (MiNA) toolset [
18] and Mito-Morphology Macro [
17], and to study lysosomal parameters. For mitochondrial network analysis, cells were differentiated in µ-slide 8 well chambers (Cat. 80826, Ibidi GmbH, Gräfelfing, Germany) and subsequently treated with either 50 µM 6-OHDA or 0.25 µM rotenone for 24 h. After these treatments, cells were incubated for 30 min with 50 nM of the mitochondrial fluorescent dye Mitotracker Red CMXRos in a humidified atmosphere, 5% CO
2, at 37 °C. Subsequently, cells were washed 3 times with phosphate buffer saline (PBS) 1× and fixed with 4% paraformaldehyde in PBS 1× overnight at 4 °C. Afterwards, cells were washed 3 times with PBS 1x and incubated with 1 μg/mL Hoechst 33342 in PBS 1× to label cell nuclei. Regarding the study of lysosomal parameters, after cell differentiation and treatment, cells were incubated for 30 min with 75 nM LysoTracker Green DND-26 and 1 μg/μL Hoechst 33,342 in a humidified atmosphere, 5% CO
2, at 37 °C. Finally, cells were imaged under a Carl Zeiss Laser scanning confocal microscope (LSM 710, Carl Zeiss, Jena, Germany) using an oil Plan-Apochromat 63× DIC M27 (Carl Zeiss, Jena, Germany) objective with a numerical aperture of 1.4. A BP 545/25, FT 570, BP 605/70 filter set was used to measure the red fluorescence from Mitotracker Red CMXRos, a BP 470/40, FT 495, BP 525/50 for the measurement of the green fluorescence from LysoTracker Green DND-26 and a G 365, FT 395, BP 445/50 filter set was used to assess the blue fluorescence from Hoechst 33342. Excitation was achieved with an HXP 120V light source. For fixed cells mitochondrial analysis, Z-stack series were made and consisted of 0.2–0.4 µm slice intervals and rendered into a single 2D images using the “maximum intensity projection” processing tool using Zeiss Zen 2 software (Carl Zeiss, Jena, Germany).
By using ImageJ 1.52a, it was possible to apply the MiNA toolset that provided us with data related to the mean of mitochondrial branch length, summed mitochondrial branch length and mitochondrial network branches [
18]. Moreover, with the Mito-Morphology Macro, we obtained parameters such as the number of mitochondrial particles, mitochondria maximal area, average size, content, perimeter, and circularity [
17].
4.10. Cellular Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) Measurements
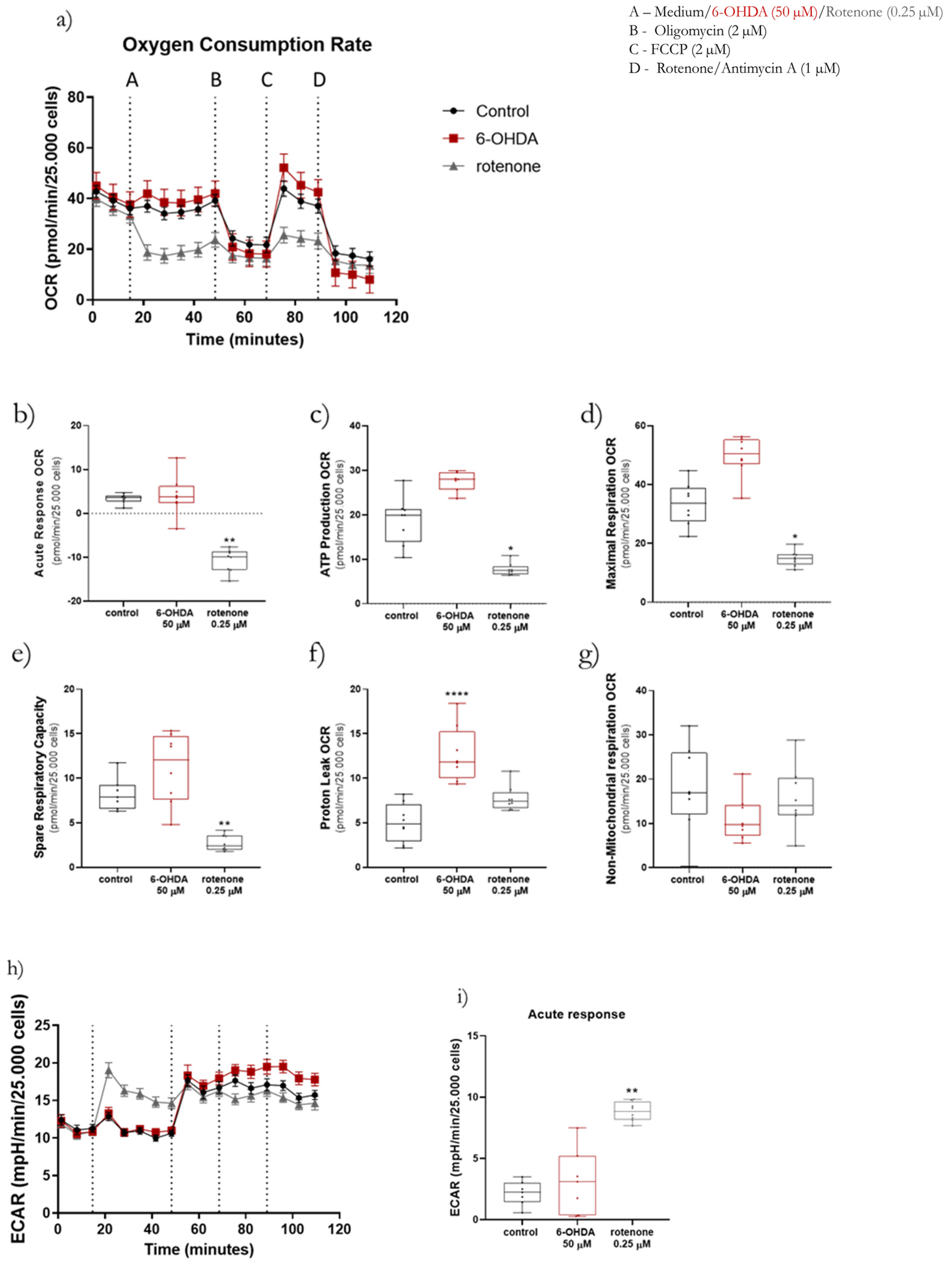
Oxygen consumption and extracellular acidification rates were measured at 37 °C using a Seahorse XFe96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA) following the Seahorse XF Cell Mito Stress Test (cat. 103015-100, Agilent Technologies, Santa Clara, CA, USA) and Real-Time ATP Rate (cat. 103592-100, Agilent Technologies, Santa Clara, CA, USA) Assay protocols. To be in the range of the equipment sensitivity, cells were differentiated at a density of 120,000 cells/cm2. Firstly, a XFe96 sensor cartridge was placed in a 96-well calibration plate containing 200 µL/well of calibration buffer and left to hydrate overnight at 37 °C. Next, the medium in the 96-well plate containing the cells was replaced with 175 µL/well of prewarmed low-buffered serum-free minimal DMEM (D5030, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) medium (pH 7.4) and incubated at 37 °C for 1 h to allow the temperature and pH of the medium to reach equilibrium before the first measurement. For the Seahorse XF Cell Mito Stress Test assay measurement of OCR and ECAR, 2 µM oligomycin (Cat. SLBS6501, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) were injected through port B, 2 µM FCCP were injected through port C and port D was loaded with 1 µM rotenone (Cat. MKBS1062V, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) plus 1 µM antimycin A (Cat. 036M4041V, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). Both the calibration plate and the sensor cartridge were loaded into the XFe96 Extracellular Flux Analyzer for calibration. The calibration plate was then replaced with the study plate. Three baseline rate measurements of SH-SY5Y cell line OCR were made using a 3 min mix, 3 min measure for 3 cycles. Rotenone (0.25 µM), 6-OHDA (50 µM) or medium (control) were then pneumatically injected through port A into the corresponding wells and mixed, with OCR measurements made using a 3 min mix, 3 min measuring time for 5 cycles. Lastly, oligomycin, FCCP, rotenone and antimycin A were sequentially pneumatically injected and mixed, with OCR measurements made using a 3 min mix, 3 min measuring time for 3 cycles. Several parameters were evaluated, such as the acute response, ATP production, proton leak, maximal respiration, spare respiratory capacity and non-mitochondrial respiration-linked OCR. Acute responses were measured as the last OCR measurement before oligomycin injection minus the last measurement before port A injection. After adding 2 µM oligomycin, it was possible to calculate ATP production-linked OCR by subtracting the last measurement before oligomycin injection and the minimum rate measurement after the oligomycin injection. Moreover, it was also possible to calculate the proton leak-linked OCR by subtracting the non-mitochondrial respiration-linked OCR from the minimum rate measurement after oligomycin injection. When cells were treated with 2 µM FCCP it was possible to evaluate the maximal respiration-linked OCR and spare respiratory capacity. Maximal respiration-linked OCR corresponds to the maximum rate measurement after FCCP injection minus the non-mitochondrial respiration-linked OCR, while the spare respiratory capacity is calculated by subtracting the basal from the maximal respiration. Lastly, non-mitochondrial respiration-linked OCR is the minimum rate measurement after rotenone and antimycin A injection.
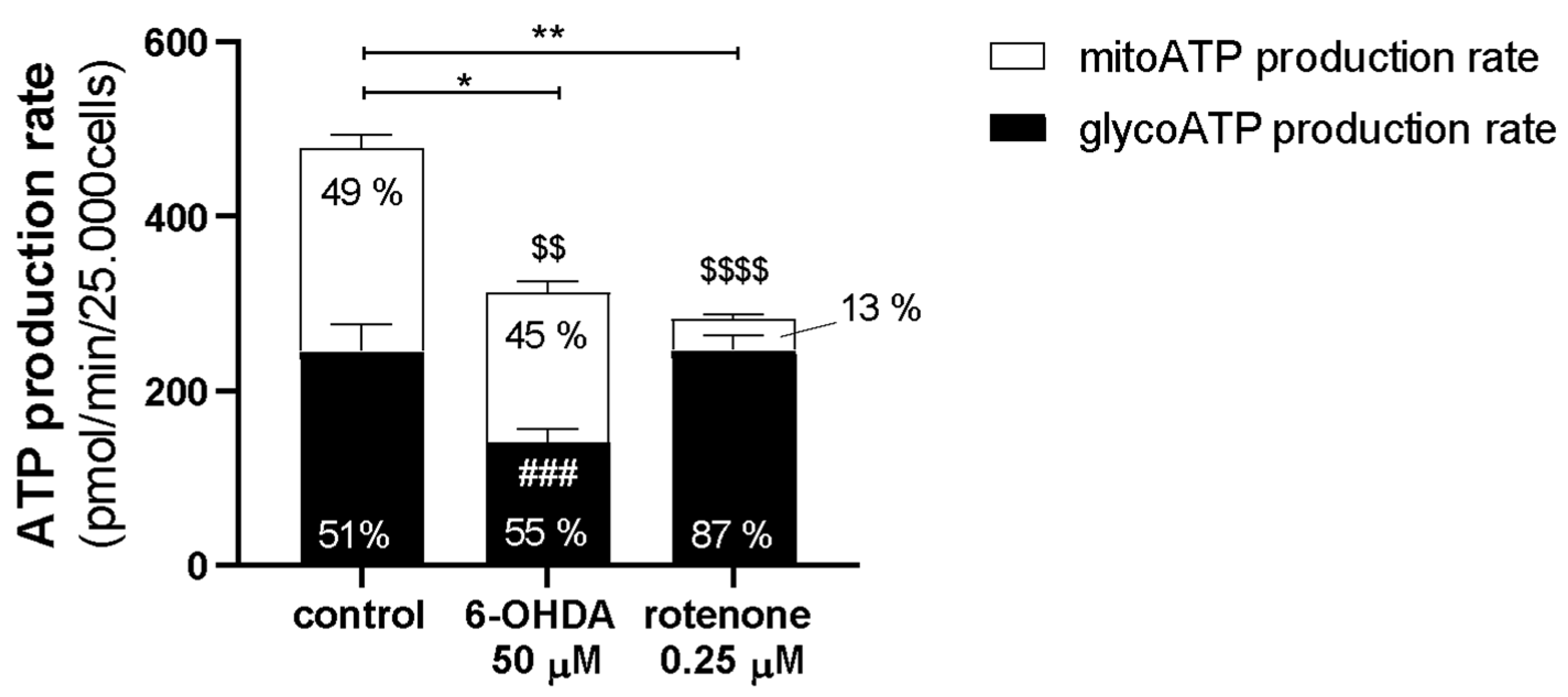
Regarding the Real-Time ATP Rate Assay, the procedure was very similar. However, in this assay, the cells were not injected with FCCP. The cells were firstly acutely treated with rotenone (0.25 µM), 6-OHDA (50 µM) or medium (control) through port A. Subsequently, cells were treated with 2 µM oligomycin through port B followed by 1 µM rotenone plus 1 µM antimycin A through port C. The cycles of mix and measuring were the same used in the Mito Stress Test assays. Oligomycin, FCCP, rotenone, and antimycin A were prepared in DMSO and diluted to the desired final concentration in serum-free minimal DMEM.
For both assays, results were normalized by cell nuclei number in each well and analyzed using the Software Version Wave Desktop 2.6 (Agilent Technologies, Santa Clara, CA, USA).
4.11. Evaluation of Intracellular Oxidative Stress
For studying the effect of 6-OHDA and rotenone on intracellular oxidative stress, the cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, cat. C6827, Molecular Probes, Invitrogen, Eugene, OR, USA) dye was used. Two types of assays were carried out, endpoint and kinetic assays. For the endpoint assay, cells were differentiated in Corning 96-well black polystyrene microplates and subsequently treated with increasing concentrations of 6-OHDA, rotenone or 500 µM of hydrogen peroxide (H2O2, cat. BCBT5298 Panreac AppliChem ITW Reagents, Darmstadt, Germany) as positive control for 3 h or 24 h. At the end of these incubation periods, cells were incubated with 5 µM of H2DCFDA for 30 min at 37 °C, 5% CO2, and the fluorescent signal was monitored using a 485 nm excitation wavelength and 528 nm emission wavelength in a Cytation 3 reader. Regarding the kinetic assay, the differentiation process was the same. Then, cells were incubated with 5 µM H2DCFDA for 30 min at 37 °C, 5% CO2. After this, the medium containing the dye was removed, and cells were treated with increasing concentrations of 6-OHDA, rotenone or 1 mM H2O2 as a positive control. Lastly, the fluorescent signal was monitored using a 485 nm excitation wavelength and 528 nm emission wavelength in a Cytation 3 reader. A kinetic assay was made measuring the fluorescent signal every minute for 5 h in order to determine where dye oxidation was linear. This read always occurs in the first 50 min. Then, a linear regression analysis was calculated, and the slope was used to estimate the H2DCFDA oxidation rate. In these experiments, low D-(+)-glucose medium supplemented with 1% FBS and 10 µM RA but without sodium bicarbonate was used.
4.12. Determination of Mitochondrial Superoxide Anion-Dependent Oxidative Stress
In order to determine if oxidative stress had a mitochondrial contribution, the fluorogenic dye Mitosox Red Mitochondrial Superoxide indicator (cat. 1626551, Thermo Scientific, Waltham, MA, USA) was utilized. This dye is specifically targeted to mitochondria in live cells [
40]. After cell differentiation in Corning 96-well black polystyrene microplates, cells were incubated with 5 µM Mitosox for 30 min at 37 °C, 5% CO
2. Then, and without removing the medium containing the dye, cells were treated with increasing concentrations of 6-OHDA, rotenone or 2 µM antimycin A as positive control. After this, the fluorescent signal was monitored using a 510 nm excitation wavelength and 580 nm emission wavelength in a Cytation 3 reader. A kinetic assay was made by measuring the fluorescent signal every 2 min for 4.5 h in order to determine where ROS production was linear. This occurred between 120 and 200 min. Then, a linear regression analysis was calculated, and the slope was used to estimate Mitosox oxidation rate. Moreover, the value of the fluorescent signal at the final time point was also used as an endpoint assay. In these experiments, low D-(+)-glucose medium supplemented with 1% FBS and 10 µM RA but without sodium bicarbonate was used.
4.13. Calculation of mtDNA Copy Number
After cell differentiation and treatments, trypsinized-collected cells were centrifuged at 2000×
g for 5 min. The pellets were washed in 5 mL of PBS 1× and the suspension was again centrifuged at 2000 g for 5 min. The resulting pellets were stored at −80 °C until total DNA extraction. Total DNA was extracted from cell pellets using the QIAamp DNA mini kit (Qiagen, Dusseldorf, Germany), following the manufacturer’s protocol. The samples were sonicated for 10 min to avoid dilution bias [
41] and then DNA concentration was quantified using a Nanodrop 2000 (Thermo Scientific, Waltham, MA, USA). Real time-PCR was performed using the SsoFast Eva Green Supermix, in a CFX96 real-time PCR system (Bio-Rad, Hercules, CA, USA), with the primers described in
Table 2, at 500 nM. Amplification of 25 ng of total DNA was performed with an initial cycle of 2 min at 98 °C, followed by 40 cycles of 5 s at 98 °C plus 5 s at 60 °C, and 5 s at 65 °C. At the end of each cycle, Eva Green fluorescence was recorded to enable determination of Cq. For each set of primers, amplification efficiency was assessed. To evaluate the reaction efficiency, a standard curve using serial dilutions of a representative sample was generated and then the efficiency for a subsequent gene expression analysis was recorded. The efficiency of the PCR reactions was between 95–105%. mtDNA copy number was determined in each sample by the ratio between the amount of a fragment of the mitochondrial cytochrome B (mito CyB) and the amount of the beta-2-microglobulin (B2m) nuclear gene, using the CFX96 Manager software (v. 3.0; Bio-Rad, Hercules, CA, USA).
4.14. Gene Expression Assessment
PureZOL Reagent (Cat.7326880, Bio-Rad, Hercules, CA, USA) was used to isolate RNA fractions from whole-cell samples. In order to do this, cells were differentiated in 6-well plates and then treated either with 50 µM 6-OHDA or 0.25 µM rotenone for 24 h. After this, the medium was removed and 1 mL of PureZOL was added to each well. The reagent was mixed by pipetting up and down and incubated at room temperature for 5 min to allow nucleoprotein complexes disruption. Total RNA was extracted with RNeasy mini kit (Qiagen, Dusseldorf, Germany), following the manufacturer’s protocol. Total RNA concentration and purity were evaluated using a Nanodrop 2000 (ThermoScientific, Waltham, MA, USA). Subsequently, RNA was converted into cDNA using the iScript cDNA synthesis kit (Cat. 170-8890, Bio-Rad, Hercules, CA, USA), following the manufacturer’s instructions. Quantitative reverse-transcriptase PCR (qRT-PCR) was performed using the SsoFast Eva Green Supermix, in a CFX96 real-time PCR system (Bio-Rad, Hercules, CA, USA), with the primers defined in
Table 2, at 500 nM. Amplification of 25 ng of DNA was performed with an initial cycle of 30 s at 95 °C, followed by 40 cycles of 5 s at 95 °C plus 5 s at 60 °C. At the end of each cycle, Eva Green fluorescence was recorded to enable determination of Cq. For each set of primers, amplification efficiency was assessed. For evaluating the reaction efficiency, a standard curve using serial dilutions of a representative sample was generated and then the efficiency for subsequent gene expression analysis was recorded. The efficiency of the PCR reactions was between 95–105%. Relative normalized expression was determined by the CFX96 Manager software (v. 3.0; Bio-Rad, Hercules, CA, USA), using B2M and TATA-Box Binding Protein (TBP) as reference genes (M value of 0.901 and Stability of 0.101).
4.15. Measurement of Lysosomal Protease Activity
Lysosomal protease activity in cells was measured using DQ Green BSA (cat. D12050, ThermoScientific, Waltham, MA, USA). When this quenched fluorogenic substrate suffers hydrolysis by cellular proteases the quenching disappears, and a bright product is produced. After cell differentiation in Corning 96-well black polystyrene microplates, the medium was replaced with new medium containing 10 µg/mL DQ Green BSA and incubated for 1 h at 37 °C. After dye incubation, cells were treated with increasing concentration of 6-OHDA, rotenone, or 2 µM trichostatin A (TSA, cat. T8552 Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) as positive control in Earle’s Balanced Salt Solution (EBSS) medium. Lysosomal protease activity was measured as fluorescence in a Cytation 3 reader at 495 nm excitation, 525 nm emission each min for 4.5 h in order to find out where DQ Green BSA hydrolysis was linear. This linearity occurred always in the last 170 min. Then, a linear regression analysis was calculated, and the slope was used to estimate the DQ Green BSA hydrolysis rate.
4.16. Applied Statistical Analysis
The complete dataset used in the analysis comprises 36 samples, each described by 11 numeric features and 1 target. The attributes considered were caspase 3/7 activity, Mitotracker Red CMXRos area and intensity (3 h and 24 h incubations with both compounds), Mitosox oxidation (3 h incubation with the referred compounds) and oxidation rate, DCFDA fluorescence (3 h and 24 h incubations with either compound) and oxidation rate, and DQ BSA hydrolysis. The target of each instance corresponds to one of the 9 possible classes (4 samples per class): Control, 6.25, 12.5, 25 and 50 µM for 6-OHDA and 0.03, 0.06, 0.125 and 0.25 µM for rotenone. The dataset is balanced, it does not contain any missing values and data were standardized across features. The small number of samples prevented a full and strong statistical analysis of the results. Nevertheless, it allowed the identification of relevant hidden patterns and trends.
Exploratory data analysis, information gain, hierarchical clustering, and supervised predictive modeling were performed using Orange Data Mining version 3.25.1 [
42]. Hierarchical clustering was performed using the Euclidean distance metric and weighted linkage. Cluster maps were plotted to relate the features with higher mutual information (in rows) with instances (in columns), with the color of each cell representing the normalized level of a particular feature in a specific instance. The information is grouped both in rows and in columns by a two-way hierarchical clustering method using the Euclidean distances and average linkage. Stratified cross-validation was used to train the supervised decision tree. A set of preliminary empirical experiments were performed to choose the best parameters for each algorithm, and we verified that, within moderate variations, there were no significant changes in the outcome. The following settings were adopted for the decision tree algorithm: minimum number of samples in leaves: 2; minimum number of samples required to split an internal node: 5; stop splitting when majority reaches: 95%; criterion: gain ratio. The performance of the supervised model was assessed using accuracy, precision, recall, F-measure and area under the ROC curve (AUC) metrics.
Regarding classical statistics, the data were analyzed using the Version 8.0.1/2 of GraphPad Software, Inc. (San Diego, CA, USA). The cytotoxicity analysis data are presented as points with a connecting line, Mean ± SEM and statistical relevance was evaluated using Kruskal–Wallis test (one-way ANOVA on ranks) pairwise (control vs. 6-OHDA or control vs. rotenone), (*) p < 0.05, (**) p < 0.01, (***) p < 0.001 and (****) p < 0.0001. Seahorse experiment Real-Time ATP Rate Assay data are present in columns and statistical relevance was evaluated using Kruskal–Wallis test (one-way ANOVA on ranks) pairwise (control vs. 6-OHDA or control vs. rotenone) to assess statistical significance of total ATP levels, (*) p < 0.05, (**) p < 0.01. Additionally, two-way ANOVA with Sidak’s multiple comparisons test was used to assess statistical significance of mitoATP production rate (control vs. 6-OHDA or control vs. rotenone) ($$) p < 0.01, ($$$$) p < 0.0001) and glycoATP production rate (control vs. 6-OHDA or control vs. rotenone), (###) p < 0.001. The remaining results are presented in box-and-whisker plots where the middle line represents the median. The applied method—Min to max—shows all points, plots the whiskers down to the minimal value and up to the maximum value and represents each individual value as a point. Kruskal–Wallis test followed by Dunn’s post hoc analysis was used for multiple conditions comparison. The statistical significance was set at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001). The number of experiments carried out is represented in the legend of the figures, as well as the number of replicates (at least 2) and the statistical tests applied.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}