
Cancer-Related Cachexia: The Vicious Circle between Inflammatory Cytokines, Skeletal Muscle, Lipid Metabolism and the Possible Role of Physical Training

 , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
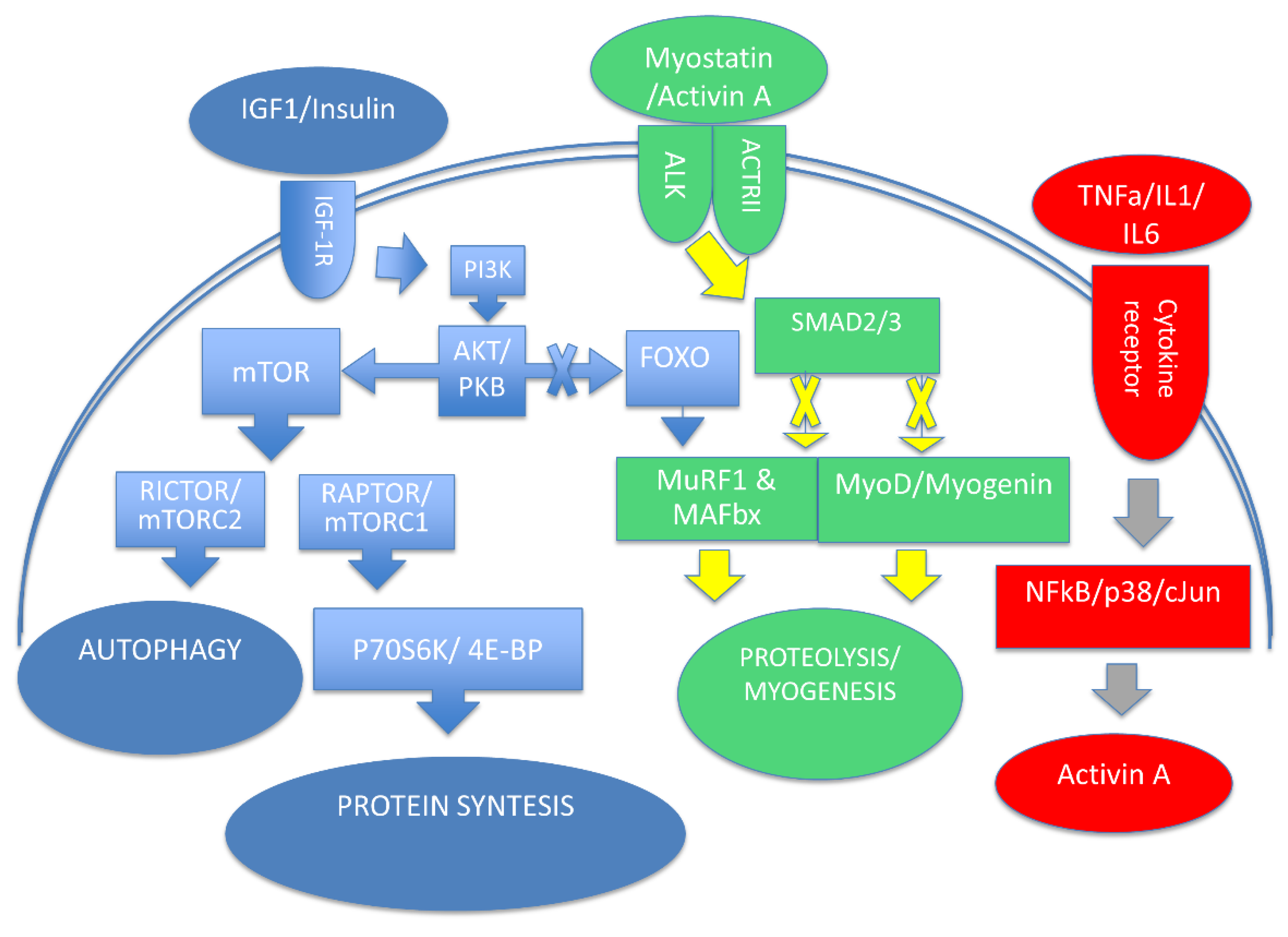
2. Cellular Processes Involved in Muscle Mass Homeostasis
3. Evidence for Inflammation as a Therapeutic Target in Cachexia
4. Inflammation and Lipid Metabolism in Cancer-Related Cachexia
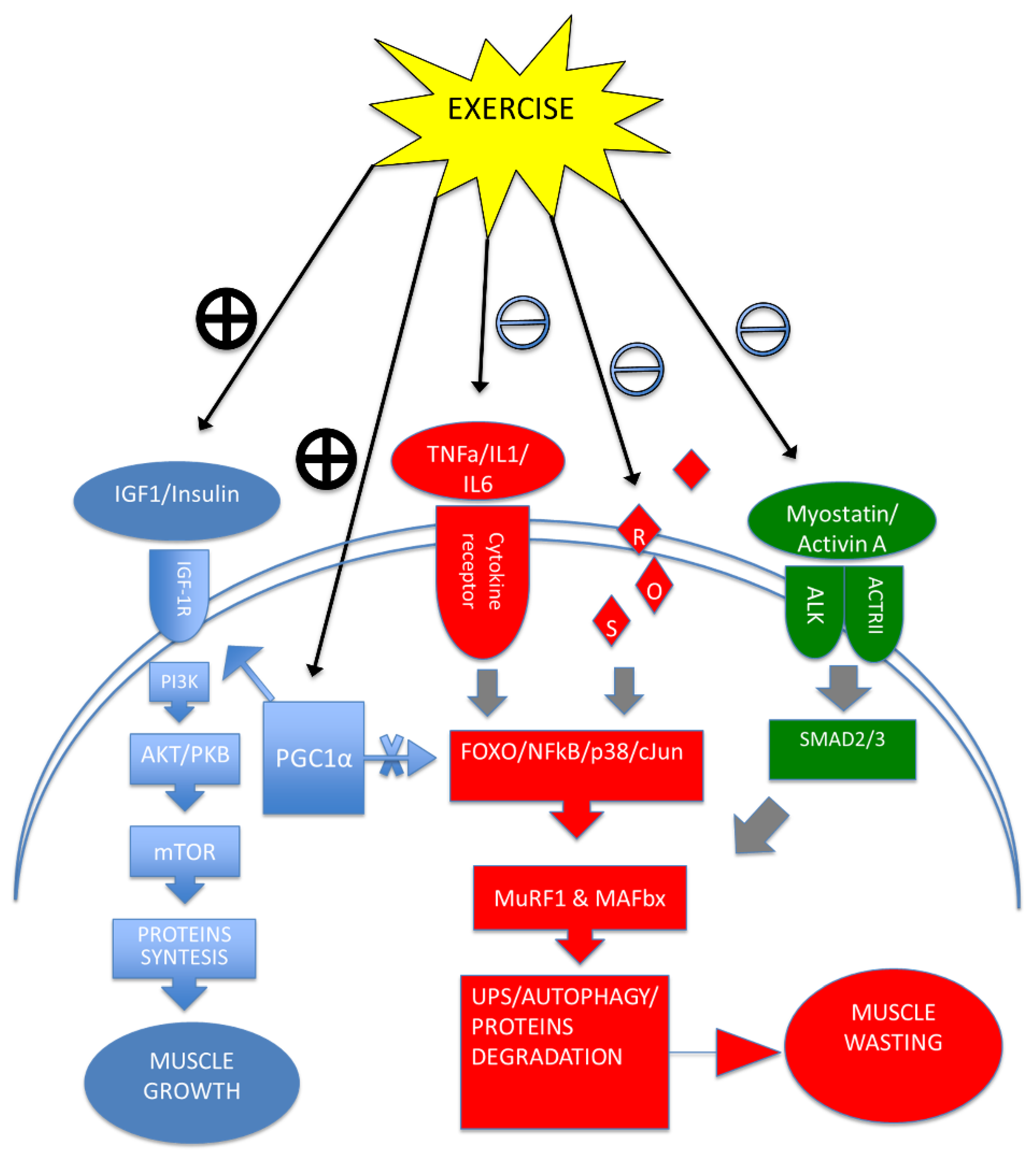
5. Exercise Training Modulates Cytokines, Improving Cachexia Symptoms
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sadeghi, M.; Keshavarz-Fathi, M.; Baracos, V.; Arends, J.; Mahmoudi, M.; Rezaei, N. Cancer cachexia: Diagnosis, assessment, and treatment. Crit. Rev. Oncol. Hematol. 2018, 127, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Cachexia as a major underestimated and unmet medical need: Facts and numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 18, 17105. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Evans, W.J.; Morley, J.E.; Argilés, J.; Bales, C.; Baracos, V.E.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Dev, R.; Hui, D.; Chisholm, G.; Delgado-Guay, M.; Dalal, S.; Del Fabbro, E.; Bruera, E. Hypermetabolism and symptom burden in advanced cancer patients evaluated in a cachexia clinic. J. Cachexia Sarcopenia Muscle 2015, 6, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solheim, T.S.; Laird, B.; Balstad, T.R.; Bye, A.; Stene, G.; Baracos, V.; Strasser, F.; Griffiths, G.; Maddocks, M.; Fallon, M.; et al. Cancer cachexia: Rationale for the MENAC (Multimodal-Exercise, Nutrition and Anti-inflammatory medication for Cachexia) trial. BMJ Supportive Palliat. Care 2018, 8, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/ GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Sacheck, J.M.; Ohtsuka, A.; McLary, S.C.; Goldberg, A.L. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E591–E601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheny, R.W.; Geddis, A.V.; Abdalla, M.N.; Leandry, L.A.; Ford, M.; McClung, H.L.; Pasiakos, S.M. AKT2 is the predominant AKT isoform expressed in human skeletal muscle. Physiol. Rep. 2018, 6, e13652. [Google Scholar] [CrossRef] [PubMed]
- Hodson, N.; McGlory, C.; Oikawa, S.Y.; Jeromson, S.; Song, Z.; Rüegg, M.A.; Hamilton, D.L.; Phillips, S.M.; Philp, A. Differential localization and anabolic responsiveness of mTOR complexes in human skeletal muscle in response to feeding and exercise. Am. J. Physiol. Cell. Physiol. 2017, 313, C604–C611. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Kalista, S.; Bertrand, L.; Lause, P.; Verniers, J.; Ketelslegers, J.M.; Thissen, J.P. Role of Akt/GSK-3beta/beta-catenin transduction pathway in the muscle anti-atrophy action of insulin-like growth factor-I in glucocorticoid-treated rats. Endocrinology 2008, 149, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.D.; Esser, K.A. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2005, 289, C853–C859. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.E.; Bhasin, S.; Artaza, J.; Byhower, F.; Azam, M.; Willard, D.H.J.; Kull, F.C.J.; Gonzalez-Cadavid, N. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E221–E228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004, 20, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lehar, A.; Liu, Y.; Ly, C.H.; Pham, Q.M.; Michaud, M.; Rydzik, R.; Youngstrom, D.W.; Shen, M.M.; Kaartinen, V.; et al. Functional redundancy of type I and type II receptors in the regulation of skeletal muscle growth by myostatin and activin A. Proc. Natl. Acad. Sci. USA 2020, 117, 30907–30917. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Matsumoto, M.; Katoh, Y.; Liu, L.; Ochiai, K.; Aizawa, Y.; Nagatomi, R.; Okuno, H.; Itoi, E.; Igarashi, K. Bach1 promotes muscle regeneration through repressing Smad-mediated inhibition of myoblast differentiation. PLoS ONE 2020, 15, e0236781. [Google Scholar] [CrossRef]
- Conery, A.R.; Cao, Y.; Thompson, E.A.; Townsend, C.M.J.; Ko, T.C.; Luo, K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat. Cell Biol. 2004, 6, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Khalil, R. Ubiquitin-proteasome pathway and muscle atrophy. Adv. Exp. Med. Biol. 2018, 1088, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Vanderveen, B.N.; Fix, D.K.; Carson, J.A. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: A role for inflammation. Oxid. Med. Cell. Longev. 2017, 2017, 3292087. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.M.Y.; Poljak, A.; Bentley, N.L.; Turner, N.; Tan, T.C.; Polly, P. Proteomic profiling of skeletal and cardiac muscle in cancer cachexia: Alterations in sarcomeric and mitochondrial protein expression. Oncotarget 2018, 9, 22001–22022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, S.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Age decreases mitochondrial motility and increases mitochondrial size in vascular smooth muscle. J. Physiol. 2016, 594, 4283–4295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leduc-Gaudet, J.P.; Picard, M.; Pelletier, F.S.J.; Sgarioto, N.; Auger, M.J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Keep, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Margeta, M. Autophagy defects in skeletal myopathies. Annu. Rev. Pathol. 2020, 15, 261–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef] [PubMed]
- Smuder, A.J.; Kavazis, A.N.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic. Biol. Med. 2010, 49, 1152–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezeoke, C.C.; Morley, J.E. Pathophysiology of anorexia in the cancer cachexia syndrome. J. Cachexia Sarcopenia Muscle 2015, 6, 287–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Cella, P.S.; Marinello, P.C.; Borges, F.H.; Ribeiro, D.F.; Chimin, P.; Testa, M.T.J.; Guirro, P.B.; Duarte, J.A.; Cecchini, R.; Guarnier, F.A.; et al. Creatine supplementation in Walker-256 tumor-bearing rats prevents skeletal muscle atrophy by attenuating systemic inflammation and protein degradation signaling. Eur. J. Nutr. 2020, 59, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, P.; Zhang, J.; Wang, Y.; Zhang, M.; Song, L.; Lu, Z.; Zhang, L.; Zhang, F.; Wang, J.; Zhang, Y.; et al. Reversal of muscle atrophy by Zhimu and Huangbai herb pair via activation of IGF-1/Akt and autophagy signal in cancer cachexia. Support Care Cancer 2016, 24, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grounds, M.D.; Radley, H.G.; Gebski, B.L.; Bogoyevitch, M.A.; Shavlakadze, T. Implications of cross-talk between tumour necrosis factor and insulin-like growth factor-1 signalling in skeletal muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Kempen, L.J.A.P.; Hardy, R.S.; Langen, R.C.J. Inflammation and skeletal muscle wasting during cachexia. Front. Physiol. 2020, 11, 597–675. [Google Scholar] [CrossRef] [PubMed]
- Girgenrath, M.; Weng, S.; Kostek, C.A.; Browning, B.; Wang, M.; Brown, S.A.; Winkles, J.A.; Michaelson, J.S.; Allaire, N.; Schneider, P.; et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 2006, 25, 5826–5839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascoe, A.L.; Johnston, A.J.; Murphy, R.M. Controversies in TWEAK-Fn14 signaling in skeletal muscle atrophy and regeneration. Cell. Mol. Life Sci. 2020, 77, 3369–3381. [Google Scholar] [CrossRef] [PubMed]
- Torelli, G.F.; Meguid, M.M.; Moldawer, L.L.; Edwards, C.K., III; Kim, H.J.; Carter, J.L.; Laviano, A.; Rossi Fanelli, F. Use of recombinant human soluble TNF receptor in anorectic tumor-bearing rats. Am. J. Physiol. 1999, 277, R850–R855. [Google Scholar] [CrossRef] [PubMed]
- Opara, E.I.; Laviano, A.; Meguid, M.M.; Yang, Z.J. Correlation between food intake and CSF IL-1 alpha in anorectic tumor bearing rats. Neuroreport 1995, 6, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Plata-Salamán, C.R.; Ilyin, S.E.; Gayle, D. Brain cytokine mRNAs in anorectic rats bearing prostate adenocarcinoma tumor cells. Am. J. Physiol. 1998, 275, R566–R573. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Gleason, J.R.; Meguid, M.M.; Yang, Z.J.; Cangiano, C.; Rossi Fanelli, F. Effects of intra-VMN mianserin and IL-1ra on meal number in anorectic tumor-bearing rats. J. Investig. Med. 2000, 48, 40–48. [Google Scholar]
- Van Norren, K.; Dwarkasing, J.T.; Witkamp, R.F. The role of hypothalamic inflammation, the hypothalamic-pituitary-adrenal axis and serotonin in the cancer anorexia-cachexia syndrome. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Ohe, Y.; Podack, E.R.; Olsen, K.J.; Miyahara, Y.; Miura, K.; Saito, H.; Koishihara, Y.; Ohsugi, Y.; Ohira, T.; Nishio, K. Interleukin-6 cDNA transfected Lewis lung carcinoma cells show unaltered net tumour growth rate but cause weight loss and shortened survival in syngeneic mice. Br. J. Cancer 1993, 67, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujinaka, T.; Fujita, J.; Ebisui, C.; Yano, M.; Kominami, E.; Suzuki, K.; Tanaka, K.; Katsume, A.; Ohsugi, Y.; Shiozaki, H.; et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J. Clin. Investig. 1996, 97, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltgalvis, K.A.; Berger, F.G.; Pena, M.M.; Davis, J.M.; Muga, S.J.; Carson, J.A. Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R393–R401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; McLeod, L.; Alhayyani, S.; Szczepny, A.; Watkins, D.N.; Chen, W.; Enriori, P.; Ferlin, W.; Ruwanpura, S.; Jenkins, B.J. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene 2017, 36, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell. Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, D.M.D.R.; das Neves, R.X.; de Matos-Neto, E.M.; Camargo, R.G.; Lima, J.D.C.C.; Radloff, K.; Alves, M.J.; Costa, R.; Tokeshi, F.; Otoch, J.P.; et al. Plasma lipid profile and systemic inflammation in patients with cancer cachexia. Front. Nutr. 2020, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskiler, G.G.; Bezdegumeli, E.; Ozman, Z.; Ozkan, A.D.; Bilir, C.; Kucukakca, B.N.; Ince, M.N.; Men, A.Y.; Aktas, O.; Horoz, Y.E.; et al. IL-6 mediated JAK/STAT3 signaling pathway in cancer patients with cachexia. Bratisl. Lek. Listy 2019, 66, 819–826. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Matos-Neto, E.M.; Lima, J.D.; de Pereira, W.O.; Figuerêdo, R.G.; Riccardi, D.M.; Radloff, K.; das Neves, R.X.; Camargo, R.G.; Maximiano, L.F.; Tokeshi, F.; et al. Systemic inflammation in cachexia—Is tumor cytokine expression profile the culprit? Front. Immunol. 2015, 6, 629. [Google Scholar] [CrossRef]
- Han, J.; Meng, Q.; Shen, L.; Wu, G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018, 17, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Lu, C.; Meng, Q.; Halim, A.; Yean, T.J.; Wu, G. Plasma concentration of interleukin-6 was upregulated in cancer cachexia patients and was positively correlated with plasma free fatty acid in female patients. Nutr. Metab. 2019, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef] [PubMed]
- De Larichaudy, J.; Zufferli, A.; Serra, F.; Isidori, A.M.; Naro, F.; Dessalle, K.; Desgeorges, M.; Piraud, M.; Cheillan, D.; Vidal, H.; et al. TNF-α- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet. Muscle 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, R.D.S.; Muradás, T.C.; Dagnino, A.P.A.; Rost, F.L.; Costa, K.M.; Venturin, G.T.; Greggio, S.; da Costa, J.C.; Campos, M.M. Targeting FFA1 and FFA4 receptors in cancer-induced cachexia. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E877–E892. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-mediated toxicology and applied pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARalpha: Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 2010, 8, e002. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Jia, Y.; Fu, T.; Viwakarma, N.; Bai, L.; Rao, M.S.; Zhu, Y.; Borensztajn, J.; Reddy, J.K. Sustained activation of PPARα by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J. 2011, 26, 628–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braissant, O.; Wahli, W. Differential expression of peroxisome proliferator-activated receptor-α, -β, and -γ during rat embryonic development. Endocrinology 1998, 139, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-α signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manickam, R.; Wahli, W. Roles of peroxisome proliferator-activated receptorβ/δ in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, R.H.; Mathews, S.T.; Camp, H.S.; Ding, L.; Leff, T. Selective activation of PPAR gamma in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2010, 298, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; He, W.; Barak, Y.; Le, J.; Bandyopadhyay, G.; Olson, P.; Wilkes, J.; Evans, R.M.; Olefsky, J. Muscle-specific Pparg deletion causes insulin resistance. Nat. Med. 2003, 9, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, C.; Dos Santos, B.S.; Rosa, D.D. Impact of cancer cachexia on cardiac and skeletal muscle: Role of exercise training. Cancers 2022, 14, 342. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, N.; Stoner, L.; Farajivafa, V.; Hanson, E.D. Exercise training, circulating cytokine levels and immune function in cancer survivors: A meta-analysis. Brain Behav. Immun. 2019, 81, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Kader, S.M.; Al-Shreef, F.M. Inflammatory cytokines and immune system modulation by aerobic versus resisted exercise training for elderly. Afr. Health Sci. 2018, 18, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilha, C.S.; Borges, F.H.; Costa Mendes da Silva, L.E.; Frajacomo, F.T.T.; Jordao, A.A.; Duarte, J.A.; Cecchini, R.; Guarnier, F.A.; Deminice, R. Resistance exercise attenuates skeletal muscle oxidative stress, systemic pro-inflammatory state, and cachexia in Walker-256 tumor-bearing rats. Appl. Physiol. Nutr. Metab. 2017, 42, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Daou, H.N. Exercise as an anti-inflammatory therapy for cancer cachexia: A focus on interleukin-6 regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R296–R310. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A.; Keller, C.; Starkie, R.L.; Osada, T.; Febbraio, M.A.; Pedersen, B.K. IL-6 and TNF-alpha expression in, and release from, contracting human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E1272–E1278. [Google Scholar] [CrossRef] [Green Version]
- Liang, A.P.; Drazick, A.T.; Gao, H.; Li, Y. Skeletal muscle secretion of IL-6 is muscle type specific: Ex vivo evidence. Biochem. Biophys. Res. Commun. 2018, 505, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, N.H.; Stanya, K.J.; Hyde, A.L.; Chalom, M.M.; Alexander, R.K.; Liou, Y.H.; Starost, K.A.; Gangl, M.R.; Jacobi, D.; Liu, S.; et al. Interleukin-13 drives metabolic conditioning of muscle to endurance exercise. Science 2020, 368, eaat3987. [Google Scholar] [CrossRef]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Marino Gammazza, A.; Macaluso, F.; Di Felice, V.; Cappello, F.; Barone, R. Hsp60 in skeletal muscle fiber biogenesis and homeostasis: From physical exercise to skeletal muscle pathology. Cells 2018, 7, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, R.; Macaluso, F.; Sangiorgi, C.; Campanella, C.; Marino Gammazza, A.; Moresi, V.; Coletti, D.; Conway de Macario, E.; Macario, A.J.; Cappello, F.; et al. Skeletal muscle heat shock protein 60 increases after endurance training and induces peroxisome proliferator-activated receptor gamma coactivator 1 α1 expression. Sci. Rep. 2016, 6, 19781. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, D.; Fiore, R.; Caporossi, D.; Di Felice, V.D.; Cappello, F.; Dimauro, I.; Barone, R. Function and fiber-type specific distribution of Hsp60 and αB-crystallin in skeletal muscles: Role of physical exercise. Biology 2021, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Sangiorgi, C.; Marino Gammazza, A.; D’Amico, D.; Salerno, M.; Cappello, F.; Pomara, C.; Zummo, G.; Farina, F.; Di Felice, V.; et al. Effects of conjugated linoleic acid associated with endurance exercise on muscle fibres and peroxisome proliferator-activated receptor γ coactivator 1 α isoforms. J. Cell. Physiol. 2017, 232, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, D.; Marino Gammazza, A.; Macaluso, F.; Paladino, L.; Scalia, F.; Spinoso, G.; Dimauro, I.; Caporossi, D.; Cappello, F.; Di Felice, V.; et al. Sex-based differences after a single bout of exercise on PGC1α isoforms in skeletal muscle: A pilot study. FASEB J. 2021, 35, e21328. [Google Scholar] [CrossRef] [PubMed]

 ; Negative modulation =
; Negative modulation =  .
; Negative modulation = .
.
; Negative modulation = .
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangano, G.D.; Fouani, M.; D’Amico, D.; Di Felice, V.; Barone, R. Cancer-Related Cachexia: The Vicious Circle between Inflammatory Cytokines, Skeletal Muscle, Lipid Metabolism and the Possible Role of Physical Training. Int. J. Mol. Sci. 2022, 23, 3004. https://doi.org/10.3390/ijms23063004
Mangano GD, Fouani M, D’Amico D, Di Felice V, Barone R. Cancer-Related Cachexia: The Vicious Circle between Inflammatory Cytokines, Skeletal Muscle, Lipid Metabolism and the Possible Role of Physical Training. International Journal of Molecular Sciences. 2022; 23(6):3004. https://doi.org/10.3390/ijms23063004
Chicago/Turabian StyleMangano, Giuseppe Donato, Malak Fouani, Daniela D’Amico, Valentina Di Felice, and Rosario Barone. 2022. "Cancer-Related Cachexia: The Vicious Circle between Inflammatory Cytokines, Skeletal Muscle, Lipid Metabolism and the Possible Role of Physical Training" International Journal of Molecular Sciences 23, no. 6: 3004. https://doi.org/10.3390/ijms23063004
APA StyleMangano, G. D., Fouani, M., D’Amico, D., Di Felice, V., & Barone, R. (2022). Cancer-Related Cachexia: The Vicious Circle between Inflammatory Cytokines, Skeletal Muscle, Lipid Metabolism and the Possible Role of Physical Training. International Journal of Molecular Sciences, 23(6), 3004. https://doi.org/10.3390/ijms23063004