
Are the Main Methionine Sources Equivalent? A Focus on DL-Methionine and DL-Methionine Hydroxy Analog Reveals Differences on Rainbow Trout Hepatic Cell Lines Functions

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Only DL-MET Supports Cell Proliferation
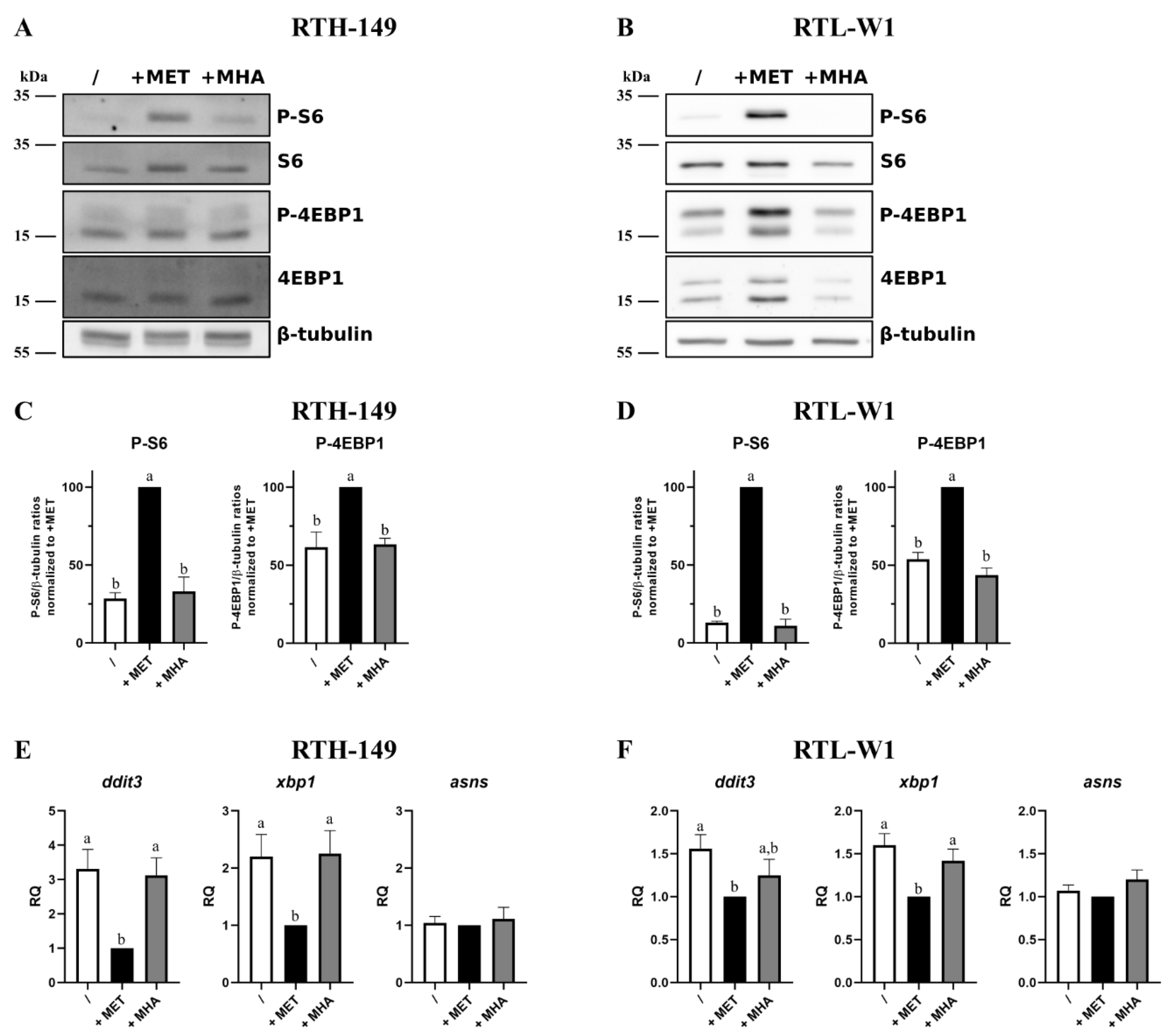
2.2. Responses of Cellular AA Sensing Pathways to Different MET Sources
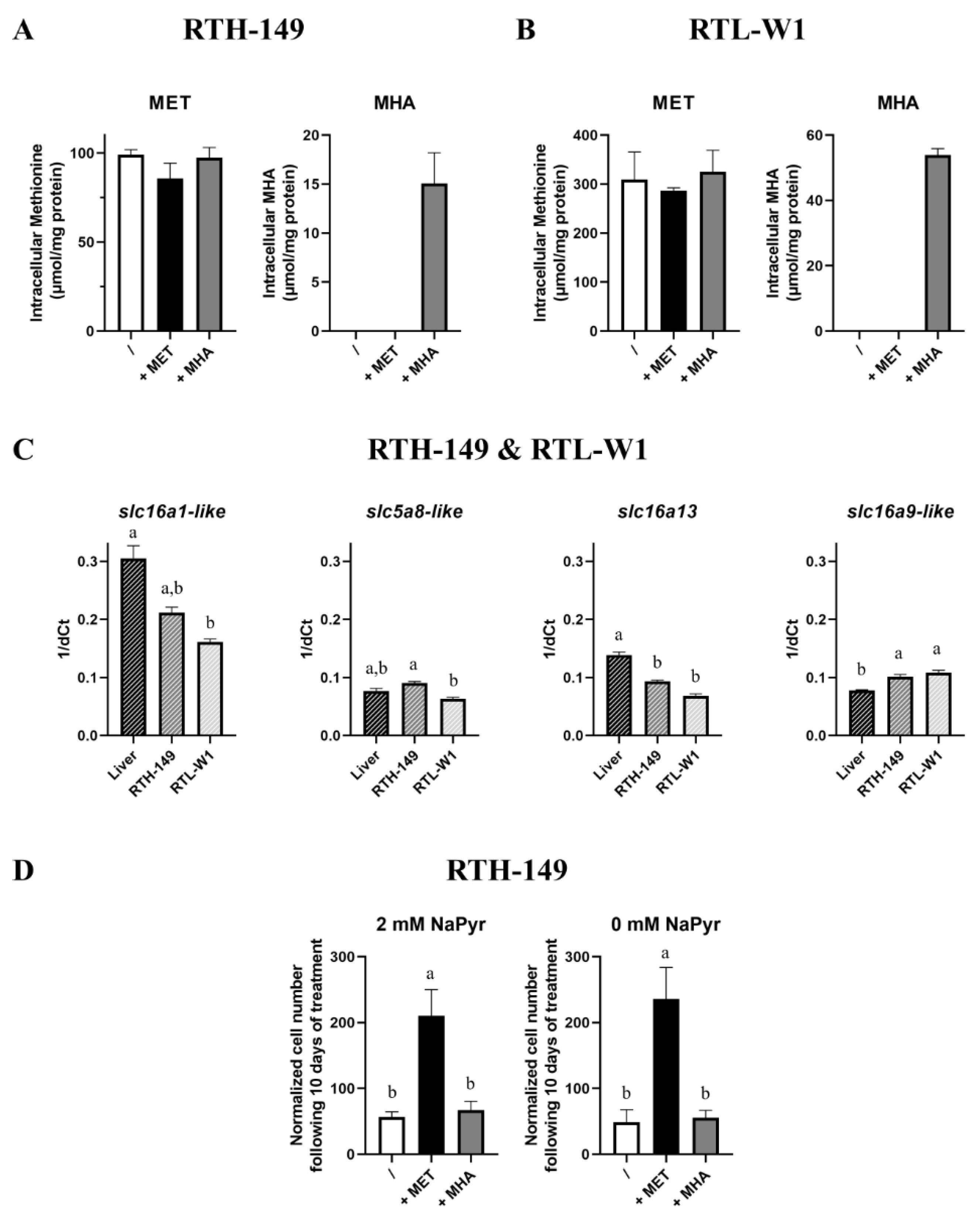
2.3. Assessing the MET-Related Metabolic Pathways When It Is Fueled with DL-MET or MHA
2.4. A Defect in MHA Uptake and Metabolization?
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Proliferation Assay
4.3. Cytotoxicity Assay
4.4. Protein Extraction and Western Blot Analysis
4.5. RNA Extraction and RT-qPCR Analyses
4.6. Liquid Chromatography Procedures
4.6.1. Methanolic Extraction of Polar Metabolites
4.6.2. SAM and GSH Separation and Detection by HPLC-UV
4.6.3. MET and MHA Separation and Detection by UPLC-MS
4.6.4. Biogenic Amino Acid Derivatization, Separation and Detection by HPLC-FL
4.6.5. Metabolites Quantification
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. The State of World Fisheries and Aquaculture 2020: Sustainability in Action; The State of World Fisheries and Aquaculture (SOFIA); FAO: Rome, Italy, 2020; ISBN 978-92-5-132692-3. [Google Scholar]
- Belghit, I.; Skiba-Cassy, S.; Geurden, I.; Dias, K.; Surget, A.; Kaushik, S.; Panserat, S.; Seiliez, I. Dietary Methionine Availability Affects the Main Factors Involved in Muscle Protein Turnover in Rainbow Trout (Oncorhynchus mykiss). Br. J. Nutr. 2014, 112, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonyoung, S.; Haga, Y.; Satoh, S. Preliminary Study on Effects of Methionine Hydroxy Analog and Taurine Supplementation in a Soy Protein Concentrate-Based Diet on the Biological Performance and Amino Acid Composition of Rainbow Trout [Oncorhynchus mykiss (Walbaum)]. Aquac. Res. 2013, 44, 1339–1347. [Google Scholar] [CrossRef]
- Powell, C.D.; Chowdhury, M.A.K.; Bureau, D.P. Assessing the Bioavailability of L-Methionine and a Methionine Hydroxy Analogue (MHA-Ca) Compared to DL-Methionine in Rainbow Trout (Oncorhynchus mykiss). Aquac. Res. 2017, 48, 332–346. [Google Scholar] [CrossRef]
- Keembiyehetty, C.N.; Gatlin, D.M. Performance of Sunshine Bass Fed Soybean-Meal-Based Diets Supplemented with Different Methionine Compounds. Progress. Fish-Cult. 1997, 59, 25–30. [Google Scholar] [CrossRef]
- Zhou, Y.; He, J.; Su, N.; Masagounder, K.; Xu, M.; Chen, L.; Liu, Q.; Ye, H.; Sun, Z.; Ye, C. Effects of DL-Methionine and a Methionine Hydroxy Analogue (MHA-Ca) on Growth, Amino Acid Profiles and the Expression of Genes Related to Taurine and Protein Synthesis in Common Carp (Cyprinus carpio). Aquaculture 2021, 532, 735962. [Google Scholar] [CrossRef]
- Teodósio, R.; Engrola, S.; Cabano, M.; Colen, R.; Masagounder, K.; Aragão, C. Metabolic and Nutritional Responses of Nile Tilapia Juveniles to Dietary Methionine Sources. Br. J. Nutr. 2021, 127, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.H.; Allen, O.W., Jr.; Poe, W.E.; Wilson, R.P. Utilization of Dietary Sulfur Compounds by Fingerling Channel Catfish: L-Methionine, DL-Methionine, Methionine Hydroxy Analogue, Taurine and Inorganic Sulfate. J. Nutr. 1978, 108, 1932–1936. [Google Scholar] [CrossRef]
- Ma, R.; Hou, H.; Mai, K.; Bharadwaj, A.S.; Cao, H.; Ji, F.; Zhang, W. Comparative Study on the Effects of L-Methionine or 2-Hydroxy-4-(Methylthio) Butanoic Acid as Dietary Methionine Source on Growth Performance and Anti-Oxidative Responses of Turbot (Psetta maxima). Aquaculture 2013, 412, 136–143. [Google Scholar] [CrossRef]
- Zhao, J.-X.; Li, X.-Q.; Leng, X.-J.; Peng, S.; Hu, J.; Zhao, X.-X. Comparative Study on the Utilization of Different Methionine Sources by Channel Catfish, Ictalurus punctatus (Rafinesque, 1818). Aquac. Res. 2017, 48, 3618–3630. [Google Scholar] [CrossRef]
- Dibner, J.J. Review of the Metabolism of 2-Hydroxy-4-(Methylthio) Butanoic Acid. Worlds Poult. Sci. J. 2003, 59, 99–110. [Google Scholar] [CrossRef]
- Pham Thi Ha To, V.; Subramaniam, M.; Masagounder, K.; Loewen, M.E. Characterization of the Segmental Transport Mechanisms of DL-Methionine Hydroxy Analogue along the Intestinal Tract of Rainbow Trout with an Additional Comparison to DL-Methionine. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2020, 249, 110776. [Google Scholar] [CrossRef]
- Morin, G.; Pinel, K.; Dias, K.; Seiliez, I.; Beaumatin, F. RTH-149 Cell Line, a Useful Tool to Decipher Molecular Mechanisms Related to Fish Nutrition. Cells 2020, 9, 1754. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.-L. MTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino Acid Sufficiency and MTOR Regulate P70 S6 Kinase and EIF-4E BP1 through a Common Effector Mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino Acid-Dependent Control of MTORC1 Signaling: A Variety of Regulatory Modes. J. Biomed. Sci. 2020, 27, 87. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR Is an S-Adenosylmethionine Sensor for the MTORC1 Pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousse, C.; Bruhat, A.; Harding, H.P.; Ferrara, M.; Ron, D.; Fafournoux, P. Amino Acid Limitation Regulates CHOP Expression through a Specific Pathway Independent of the Unfolded Protein Response. FEBS Lett. 1999, 448, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Lannan, C.N.; Winton, J.R.; Fryer, J.L. Fish Cell Lines: Establishment and Characterization of Nine Cell Lines from Salmonids. In Vitro 1984, 20, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.E.J.; Clemons, J.H.; Bechtel, D.G.; Caldwell, S.J.; Han, K.-B.; Pasitschniak-Arts, M.; Mosser, D.D.; Bols, N.C. Development and Characterization of a Rainbow Trout Liver Cell Line Expressing Cytochrome P450-Dependent Monooxygenase Activity. Cell Biol. Toxicol. 1993, 9, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Bols, N.C.; Pham, P.H.; Dayeh, V.R.; Lee, L.E.J. Invitromatics, Invitrome, and Invitroomics: Introduction of Three New Terms for in Vitro Biology and Illustration of Their Use with the Cell Lines from Rainbow Trout. In Vitro Cell. Dev. Biol. Anim. 2017, 53, 383–405. [Google Scholar] [CrossRef]
- Rangel-Lugo, M.; Austic, R.E. Transamination of 2-Oxo-4-[Methylthio]Butanoic Acid in Chicken Tissues. Poult. Sci. 1998, 77, 98–104. [Google Scholar] [CrossRef]
- FAO. The State of World Fisheries and Aquaculture 2018: Meeting the Sustainable Development Goals; The State of World Fisheries and Aquaculture (SOFIA); FAO: Rome, Italy, 2018; ISBN 978-92-5-130562-1. [Google Scholar]
- Schuhmacher, A.; Wax, C.; Gropp, J.M. Plasma Amino Acids in Rainbow Trout (Oncorhynchus mykiss) Fed Intact Protein or a Crystalline Amino Acid Diet. Aquaculture 1997, 151, 15–28. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Rinaldi, S.; Scalbert, A.; Ferrari, P.; Achaintre, D.; Gunter, M.J.; Appleby, P.N.; Key, T.J.; Travis, R.C. Plasma Concentrations and Intakes of Amino Acids in Male Meat-Eaters, Fish-Eaters, Vegetarians and Vegans: A Cross-Sectional Analysis in the EPIC-Oxford Cohort. Eur. J. Clin. Nutr. 2016, 70, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62.e8. [Google Scholar] [CrossRef] [PubMed]
- Pasquariello, R.; Verdile, N.; Pavlovic, R.; Panseri, S.; Schirmer, K.; Brevini, T.A.L.; Gandolfi, F. New Stable Cell Lines Derived from the Proximal and Distal Intestine of Rainbow Trout (Oncorhynchus mykiss) Retain Several Properties Observed In Vivo. Cells 2021, 10, 1555. [Google Scholar] [CrossRef]
- Kawano, A.; Haiduk, C.; Schirmer, K.; Hanner, R.; Lee, L.E.J.; Dixon, B.; Bols, N.C. Development of a Rainbow Trout Intestinal Epithelial Cell Line and Its Response to Lipopolysaccharide. Aquac. Nutr. 2011, 17, e241–e252. [Google Scholar] [CrossRef]
- Seite, S.; Masagounder, K.; Heraud, C.; Veron, V.; Marandel, L.; Panserat, S.; Seiliez, I. Early Feeding of Rainbow Trout (Oncorhynchus mykiss) with Methionine-Deficient Diet over a 2 Week Period: Consequences for Liver Mitochondria in Juveniles. J. Exp. Biol. 2019, 222, jeb203687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seite, S.; Pioche, T.; Ory, N.; Plagnes-Juan, E.; Panserat, S.; Seiliez, I. The Autophagic Flux Inhibitor Bafilomycine A1 Affects the Expression of Intermediary Metabolism-Related Genes in Trout Hepatocytes. Front. Physiol. 2019, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Seiliez, I.; Belghit, I.; Gao, Y.; Skiba-Cassy, S.; Dias, K.; Cluzeaud, M.; Rémond, D.; Hafnaoui, N.; Salin, B.; Camougrand, N.; et al. Looking at the Metabolic Consequences of the Colchicine-Based in Vivo Autophagic Flux Assay. Autophagy 2016, 12, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Wischhusen, P.; Heraud, C.; Skjærven, K.; Kaushik, S.J.; Fauconneau, B.; Prabhu, P.A.J.; Fontagné-Dicharry, S. Long-Term Effect of Parental Selenium Supplementation on the One-Carbon Metabolism in Rainbow Trout (Oncorhynchus mykiss) Fry Exposed to Hypoxic Stress. Br. J. Nutr. 2021, 127, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, C.T.; Larroquet, L.; Véron, V.; Robaina, L.; Izquierdo, M.S.; Panserat, S.; Kaushik, S.; Fontagné-Dicharry, S. Influence of Dietary Astaxanthin on the Hepatic Oxidative Stress Response Caused by Episodic Hyperoxia in Rainbow Trout. Antioxidants 2019, 8, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontagné-Dicharry, S.; Alami-Durante, H.; Aragão, C.; Kaushik, S.J.; Geurden, I. Parental and Early-Feeding Effects of Dietary Methionine in Rainbow Trout (Oncorhynchus mykiss). Aquaculture 2017, 469, 16–27. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Abbr. | Gene ID/Ref | Primers (5′→3′) | Eff. |
|---|---|---|---|---|
| Elongation factor 1α | ef1α | 100136004 [13] | Fwd: TCCTCTTGGTCGTTTCGCTG Rev: ACCCGAGGGACATCCTGTG | 1.91 |
| Asparagine synthetase | asns | 110488144 [31] | Fwd: CTGCACACGGTCTGGAGCTG Rev: GGATCTCGTCTGGGATCAGGTT | 1.94 |
| DNA damage-inducible transcript 3 | ddit3 | 110494779 [32] | Fwd: CGACAATGTCCAACAACCTG Rev: ACGAGGAGAACGAGGTGCTA | 1.97 |
| X-box binding protein 1 | xbp1 | 110526029 [31] | Fwd: TGCAACCAAGCCAATTCTTC Rev: GCGAGAACTTCGTCTTCCAG | 1.95 |
| Monocarboxylate transporter 1-like | slc16a1-like | 110492722 [12] | Fwd: GAAGAAGGCGGAGTCTAATC Rev: TAGCGTAGTTGGAGAGGAA | 2.04 |
| Monocarboxylate transporter 9-like | slc16a9-like | 110502533 [12] | Fwd: GTTGTTGGGTGGTTCTTTG Rev: GTCGATGTCAGCCTTCTT | 1.97 |
| Monocarboxylate transporter 13 | slc16a13 | 110531725 [12] | Fwd: GTAGGCTATGCGTGAGTAAG Rev: GCCTCGAGCTAGTTGAATAA | 1.88 |
| Sodium-coupled monocarboxylate transporter 1-like | slc5a8-like | 110494699 [12] | Fwd: GGCATCAGAACCTGAGATAA Rev: CAGTTGACAGAGTGCATTTAG | 1.82 |
| methionine adenosyltransferase 2b | mat2b | 110533399 * | Fwd: GGCTCCAGGACCCATCAATA Rev: AGCTCAAGACGGGAACACTC | 1.90 |
| methionine adenosyltransferase 2aa | mat2aa | 110509902 * | Fwd: GGCTATGACGACTCCTCCAA Rev: GCATAACCAAACATCAGACCCT | 1.94 |
| methionine adenosyltransferase 2al | mat2al | 110537066 * | Fwd: ATCGGAGTCAGTTGGAGAGG Rev: TGACCTCTCCACACAGCAT | 1.91 |
| DNA methyltransferase | dnmt1 | 110486372 [33] | Fwd: TTGCCAGAAGAGGAGATGCC Rev: CCCAGGTCAGCTTGCCATTA | 1.99 |
| adenosylhomocysteinase | ahcy | 110527644 [33] | Fwd: ATCAAACGGGCCACAGATGT Rev: TCGTACCTTCCATGGCAGC | 1.94 |
| Cystathionine-beta-synthase | cbs1 | 100136726 [33] | Fwd: CCACCTCAGGCAATACAGGT Rev: AACATCCACCTTCTCCATGC | 1.98 |
| Cystathionine-beta-synthase | cbs2 | 110520495 [33] | Fwd: CAAGGCTCTCAGCACATCCA Rev: ACCATCATCGAGCCCACCT | 2.06 |
| Cystathionine gamma-lyase | cth1 | 110524183 [33] | Fwd: CACCAACCCCACCATGAAAG Rev: GCGCTGGAAGTAGGCTGACA | 1.95 |
| Cystathionine gamma-lyase | cth2 | 110498361 * | Fwd: TGGCTTGAGACTCCCACCAA Rev: GCGCTGGAAGTAGGCTGACA | 2.03 |
| glutamate-cysteine ligase catalytic subunit | gclc | 110499382 [34] | Fwd: CAACCAACTGGCAGACAATG Rev: CCTTTGACAAGGGGATGAGA | 1.99 |
| 5-methyltetrahydrofolate-homocysteine methyltransferase | mtr1 | 110519474 [35] | Fwd: AATGCAGGTCTGCCCAATAC Rev: CTGATGTGTGCAGGAGTCGT | 1.99 |
| 5-methyltetrahydrofolate-homocysteine methyltransferase | mtr2 | 110499554 * | Fwd: CCAGGAGTGTGGTGGTGTG Rev: CAGGAAGCGCCTCTCCTTTA | 2.00 |
| Betaine-homocysteine methyltransferase | bhmt1 | 110509664 [35] | Fwd: CAGAGAAGCACGGTAACTGG Rev: TTCTTTGTGCTGCATCAGGT | 1.98 |
| Betaine-homocysteine methyltransferase | bhmt2 | 110523446 [35] | Fwd: GCTGAGGAGCTAGCCACAGA Rev: GGCTTCAGCTTCTCCCAGTA | 1.92 |
| glutathione synthetase | gss | 110532297 * | Fwd: TCAATACCATTGCTGCCAGTT Rev: ACTGCCCTTTCTGAGCCATA | 1.93 |
| hydroxyacid oxidase 1 | hao1 | 110505655 * | Fwd: AGTTAGTGTGTGTGGCTGACT Rev: GACACATCCCTCAGTACCCT | 1.94 |
| lactate dehydrogenase D | ldhd | 110494022 * | Fwd: GGCCGACATTCTGATCTGTG Rev: GACTCGTCTCTGCCATGTTG | 2.00 |
| D-amino acid oxidase | dao | 110536118 * | Fwd: CGTTTGACTACCTGCTGAGC Rev: TCCACCATGAGAGCAGTGTT | 1.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinel, K.; Heraud, C.; Morin, G.; Dias, K.; Marcé, A.; Beauclair, L.; Fontagné-Dicharry, S.; Masagounder, K.; Klünemann, M.; Seiliez, I.; et al. Are the Main Methionine Sources Equivalent? A Focus on DL-Methionine and DL-Methionine Hydroxy Analog Reveals Differences on Rainbow Trout Hepatic Cell Lines Functions. Int. J. Mol. Sci. 2022, 23, 2935. https://doi.org/10.3390/ijms23062935
Pinel K, Heraud C, Morin G, Dias K, Marcé A, Beauclair L, Fontagné-Dicharry S, Masagounder K, Klünemann M, Seiliez I, et al. Are the Main Methionine Sources Equivalent? A Focus on DL-Methionine and DL-Methionine Hydroxy Analog Reveals Differences on Rainbow Trout Hepatic Cell Lines Functions. International Journal of Molecular Sciences. 2022; 23(6):2935. https://doi.org/10.3390/ijms23062935
Chicago/Turabian StylePinel, Karine, Cécile Heraud, Guillaume Morin, Karine Dias, Annaëlle Marcé, Linda Beauclair, Stéphanie Fontagné-Dicharry, Karthik Masagounder, Martina Klünemann, Iban Seiliez, and et al. 2022. "Are the Main Methionine Sources Equivalent? A Focus on DL-Methionine and DL-Methionine Hydroxy Analog Reveals Differences on Rainbow Trout Hepatic Cell Lines Functions" International Journal of Molecular Sciences 23, no. 6: 2935. https://doi.org/10.3390/ijms23062935
APA StylePinel, K., Heraud, C., Morin, G., Dias, K., Marcé, A., Beauclair, L., Fontagné-Dicharry, S., Masagounder, K., Klünemann, M., Seiliez, I., & Beaumatin, F. (2022). Are the Main Methionine Sources Equivalent? A Focus on DL-Methionine and DL-Methionine Hydroxy Analog Reveals Differences on Rainbow Trout Hepatic Cell Lines Functions. International Journal of Molecular Sciences, 23(6), 2935. https://doi.org/10.3390/ijms23062935