
Chemical Synthesis, Pharmacokinetic Properties and Biological Effects of JM-00266, a Putative Non-Brain Penetrant Cannabinoid Receptor 1 Inverse Agonist

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
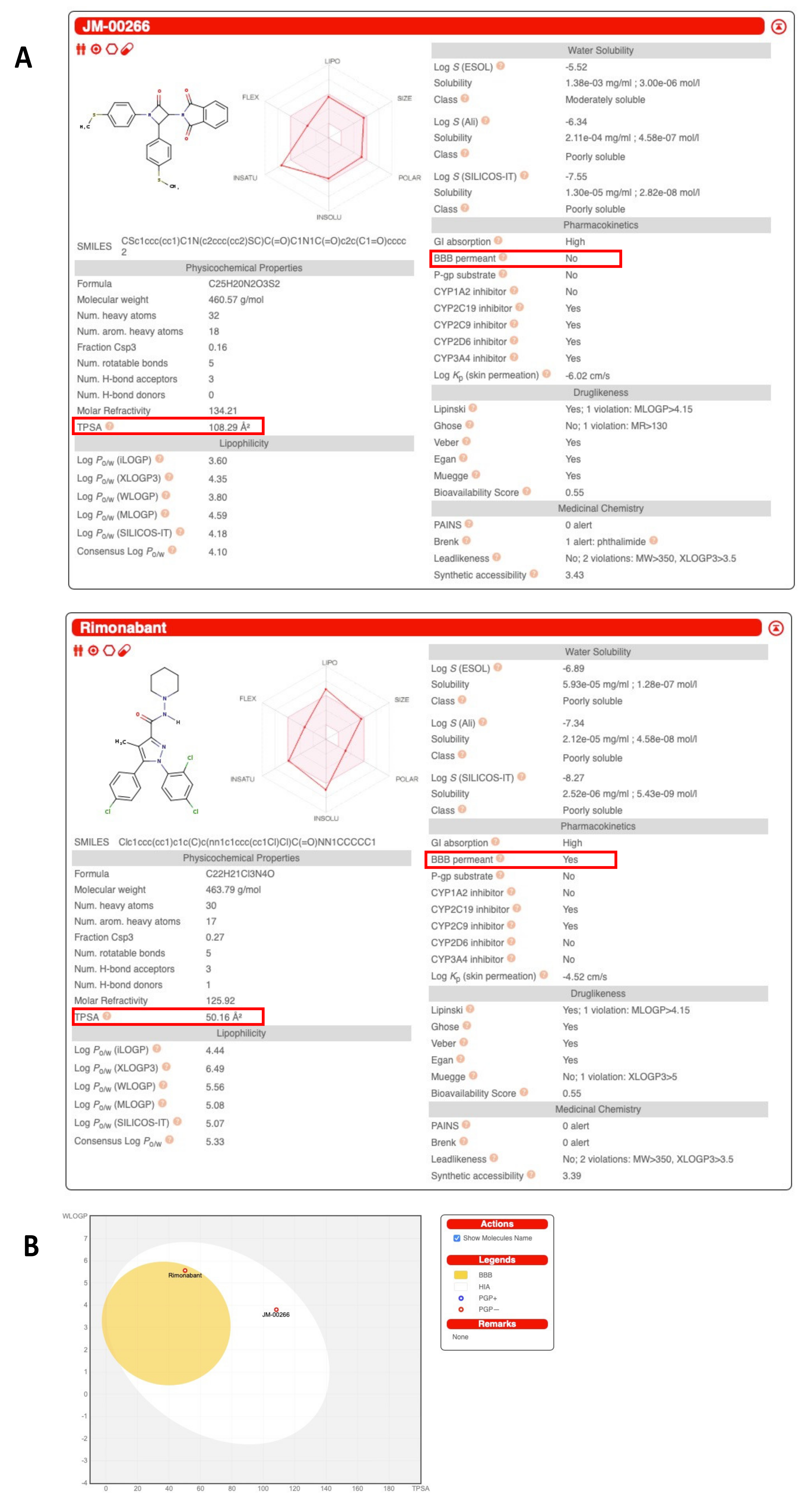
2.1. Structure, Pharmacological Properties and Bioavailability of JM-00266
2.1.1. Synthesis and Structure of JM-00266 and JM-00252
2.1.2. Tissue Distribution and Brain Penetrance
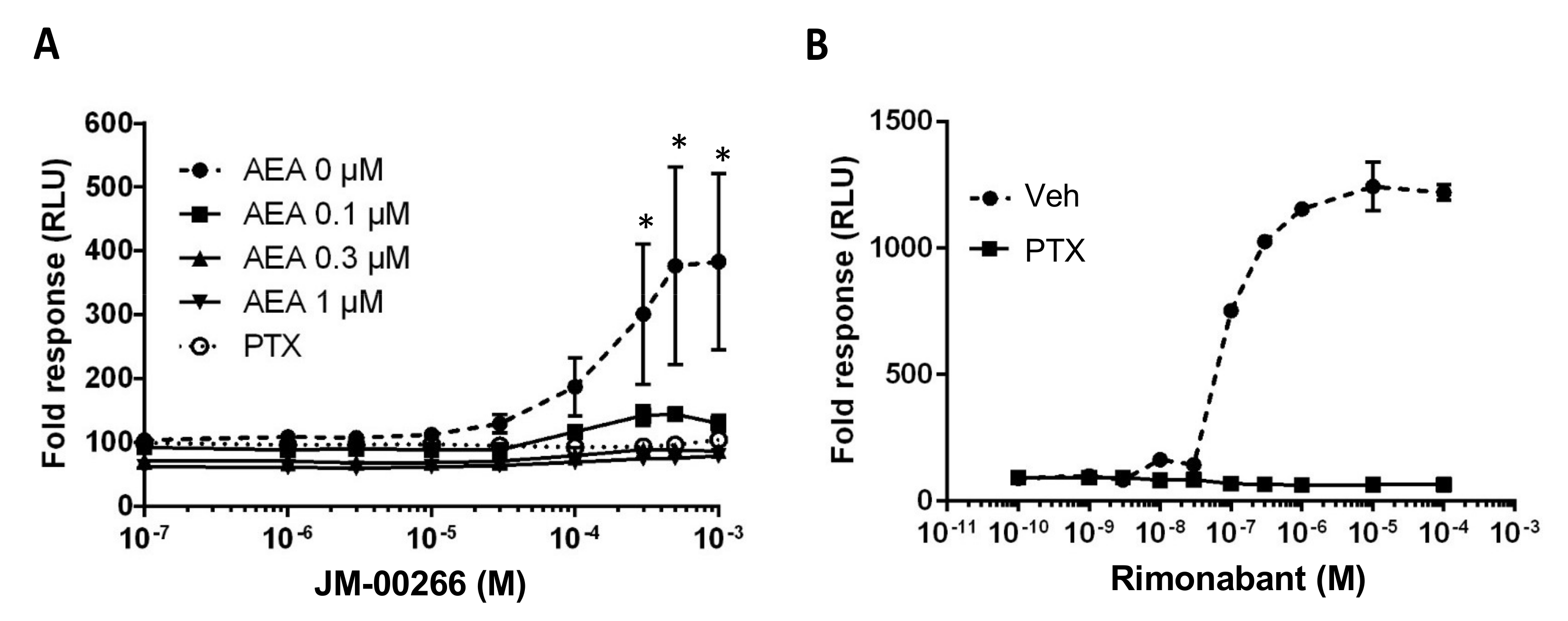
2.2. cAMP Functional Assay
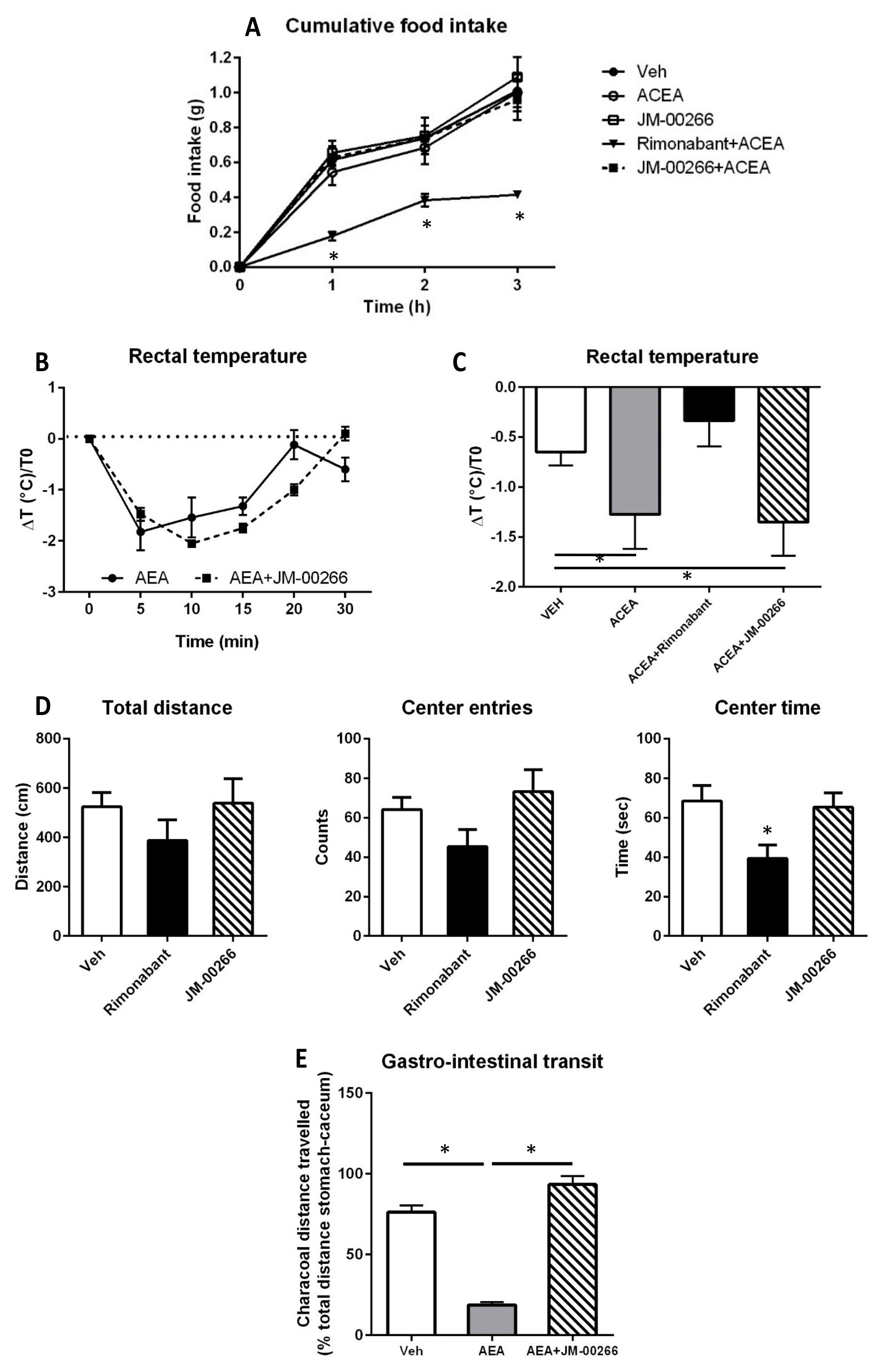
2.3. Effect of JM-00266 on Brain-Mediated Functions and Gastro-Intestinal Transit
2.3.1. Food Intake
2.3.2. Body Temperature
2.3.3. Anxiety-like Behavior
2.3.4. Gastro-Intestinal Transit
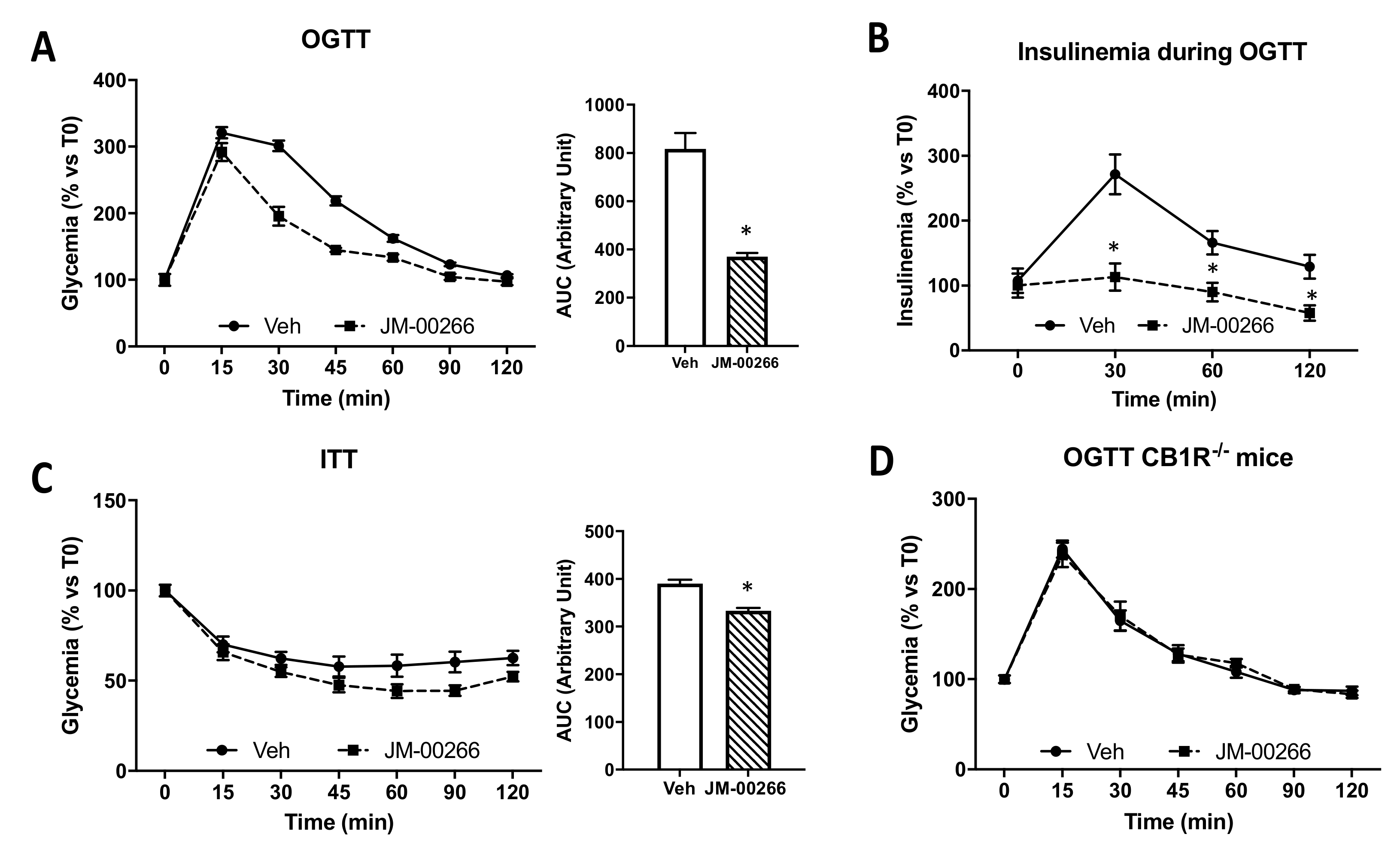
2.4. Metabolic Effects of JM-00266
2.4.1. Glucose Tolerance
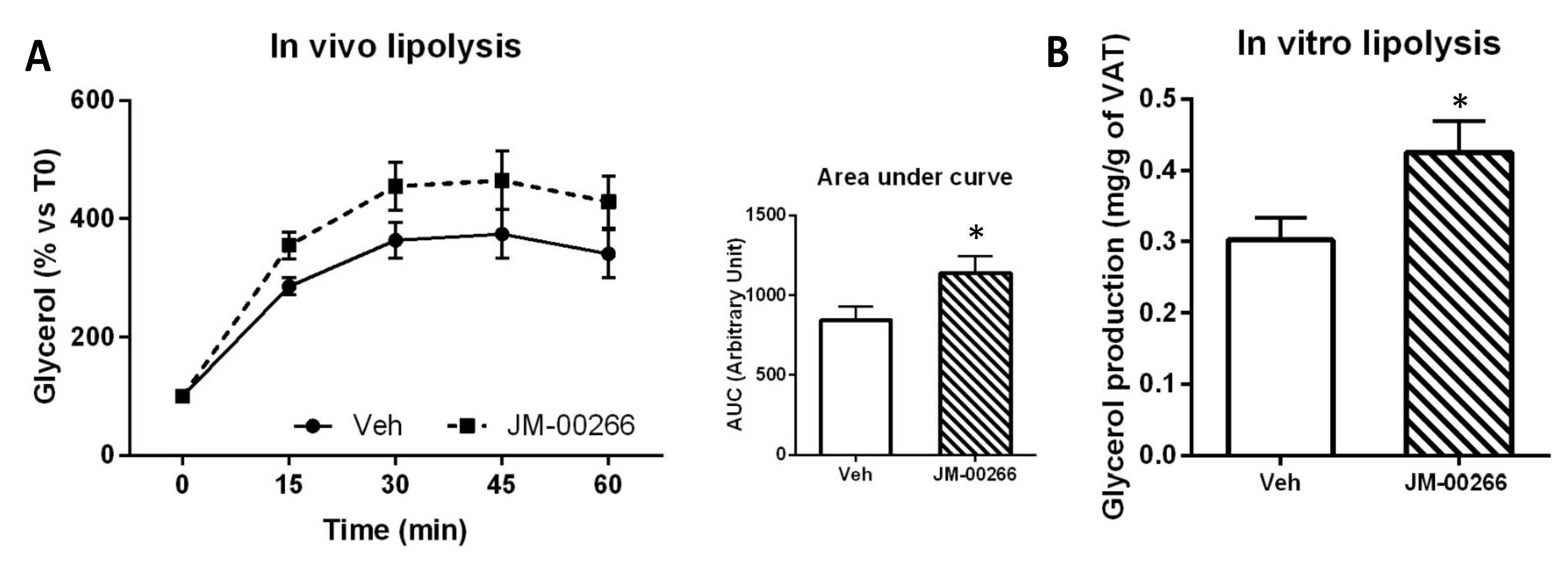
2.4.2. Adipose Tissue Lipolysis
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Synthesis of 4-Méthylthiobenzyl-4-méthylthiobenzaldimine
- MP °C = 143–144 (Diisopropyle oxide).
- 1H-RMN (CDCl3): δ 2.51, s, 3H, 4-SCH3; 2.54, s, 3H, 4′-SCH3; 7.18, d, 2H, H3H5, J = 6.7 Hz; 7.29, d, 2H, H2H6; 7.30, d, 2H, H3′H5′, J = 8.4 Hz; 7.80, d, 2H, H2′H6′; 8.41, s, 1H, HC=N.
- SM (ESI) m/z (%): 274 [M + H]+
- IR (KBr, cm−1): 1552.53 (ν C=N).
4.1.2. Synthesis of 1,4-Di-(4-Méthylthiophenyl)-3-Phtaloylazetidin-2-one (JM-00266)
- C25H20N2S2O3; MP °C = 118–120 (Diethyl ether)
- 1H-RMN (CDCl3): δ 2.44, s, 3H, CH3; 2.49, s, 3H, CH3; 5.26, d, 1H, Ha; 5.33, d, 1H, Hb (JHaHb = 2.4 Hz); 7.18, d, 2 aromH, (J = 8.4 Hz); 7.25–7.30, m, 4 aromH; 7.78, m, 2H, H4″-H5″; 7.88, m, 2H, H3″-H6″.
- 13C-RMN (100.6 MHz, CDCl) 16.45 (CH3); 16.50 (CH3); 60.99 (Cb); 62.77 (Ca); 118.15 (2C); 123.85 (C3″–C6″); 126.63 (2C); 127.00 (2C); 127.91 (2C); 131.65 (C2″–C7″); 132.10 (C1); 134.14 (C4–C1′); 134.61 (C4″–C5″); 140.11 (C4′); 161.75 (C1″–C8″); 166.65 (Cc).
- SM (ESI) m/z (%): 461.6 [M + H]+
- IR (KBr, cm−1): 3064 (ν aromCH); 2974, 2922, 2835 (ν aliphCH); 1759, 1714 (ν C=O).
4.1.3. Synthesis of 1-(Trifluoromethyphenyl)-4-(4-Méthylthiophenyl)-3-Phtaloylazetidin-2-One (JM-00252)
- C25H17F3N2SO3; Mr = 482,48; MP °C = 140 (Diethyl ether).
- 1H-RMN (CDCl3): δ: 2.49, s, 3H; 5.38, d, 1H, J = 2.6 Hz; 5.31, d, 1H, J = 2.6 Hz; 7.28–7.33, m, 4 aromH; 7.43, d, 2 aromH, J = 8.6 Hz; 7.54, d, 2 aromH, J = 8.6 Hz; 7.76–7.79, m, 4 aromH;
- SM (ESI) m/z (%): 483.5 [M + H]+
- IR (KBr, cm−1): 1730, 1778 (ν CO); 2924–2974 (ν aliphC-H); 3070 (ν aromC-H).
4.2. Drugs
4.3. cAMP Assay
4.4. Animals and Diets
4.5. Pharmacokinetic Experiments
4.6. Quantitation of JM-00266 and Rimonabant by LC-MS/MS
4.7. Brain Penetrance
4.8. Body Temperature Measurement
4.9. Behavioral Tests
4.10. Gastro-Intestinal Transit
4.11. Oral Glucose and Insulin Tolerance Tests
4.12. Lipolysis Experiments
4.13. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mechoulam, R.; Parker, L.A. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazier, W.; Saucisse, N.; Gatta-Cherifi, B.; Cota, D. The endocannabinoid system: Pivotal orchestrator of obesity and metabolic disease. Trends Endocrinol. Metab. 2015, 26, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Biro, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluher, M.; Engeli, S.; Kloting, N.; Berndt, J.; Fasshauer, M.; Batkai, S.; Pacher, P.; Schon, M.R.; Jordan, J.; Stumvoll, M. Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes 2006, 55, 3053–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Batkai, S.; Jarai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Engeli, S.; Bohnke, J.; Feldpausch, M.; Gorzelniak, K.; Janke, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Luft, F.C.; Sharma, A.M.; et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes 2005, 54, 2838–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi-Carmona, M.; Barth, F.; Heaulme, M.; Alonso, R.; Shire, D.; Congy, C.; Soubrie, P.; Breliere, J.C.; Le Fur, G. Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci. 1995, 56, 1941–1947. [Google Scholar] [CrossRef]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Despres, J.P.; Golay, A.; Sjostrom, L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med. 2005, 353, 2121–2134. [Google Scholar] [CrossRef] [Green Version]
- Van Gaal, L.F.; Rissanen, A.M.; Scheen, A.J.; Ziegler, O.; Rossner, S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 2005, 365, 1389–1397. [Google Scholar] [CrossRef]
- Jones, D. End of the line for cannabinoid receptor 1 as an anti-obesity target? Nat. Rev. Drug Discov. 2008, 7, 961–962. [Google Scholar] [CrossRef] [PubMed]
- Richey, J.M.; Woolcott, O. Re-visiting the endocannabinoid system and its therapeutic potential in obesity and associated diseases. Curr. Diabetes Rep. 2017, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Ravinet Trillou, C.; Arnone, M.; Delgorge, C.; Gonalons, N.; Keane, P.; Maffrand, J.P.; Soubrie, P. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R345–R353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinar, R.; Iyer, M.R.; Kunos, G. The therapeutic potential of second and third generation CB1R antagonists. Pharmacol. Ther. 2020, 208, 107477. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.; Vemuri, V.K.; Liu, J.; Bátkai, S.n.; Mukhopadhyay, B.; Godlewski, G.; Osei-Hyiaman, D.; Ohnuma, S.; Ambudkar, S.V.; Pickel, J.; et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Investig. 2010, 120, 2953–2966. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Iyer, M.R.; Godlewski, G.; Jourdan, T.; Liu, J.; Coffey, N.J.; Zawatsky, C.N.; Puhl, H.L.; Wess, J.; Meister, J.; et al. Functional selectivity of a biased cannabinoid-1 receptor (CB1R) antagonist. ACS Pharmacol. Transl. Sci. 2021, 4, 1175–1187. [Google Scholar] [CrossRef]
- Micale, V.; Drago, F.; Noerregaard, P.K.; Elling, C.E.; Wotjak, C.T. The cannabinoid CB1 antagonist TM38837 with limited penetrance to the brain shows reduced fear-promoting effects in mice. Front. Pharmacol. 2019, 10, 207. [Google Scholar] [CrossRef]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef]
- Klumpers, L.E.; Fridberg, M.; de Kam, M.L.; Little, P.B.; Jensen, N.O.; Kleinloog, H.D.; Elling, C.E.; van Gerven, J.M. Peripheral selectivity of the novel cannabinoid receptor antagonist TM38837 in healthy subjects. Br. J. Clin. Pharmacol. 2013, 76, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Howlett, A.C.; Fleming, R.M. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol. Pharmacol. 1984, 26, 532–538. [Google Scholar] [PubMed]
- Janero, D.R.; Makriyannis, A. Cannabinoid receptor antagonists: Pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin. Emerg. Drugs 2009, 14, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Rogers, P.J.; Kirkham, T.C. Hyperphagia in pre-fed rats following oral delta9-THC. Physiol. Behav. 1998, 65, 343–346. [Google Scholar] [CrossRef]
- Garami, A.; Pakai, E.; Oliveira, D.L.; Steiner, A.A.; Wanner, S.P.; Almeida, M.C.; Lesnikov, V.A.; Gavva, N.R.; Romanovsky, A.A. Thermoregulatory phenotype of the Trpv1 knockout mouse: Thermoeffector dysbalance with hyperkinesis. J. Neurosci. 2011, 31, 1721–1733. [Google Scholar] [CrossRef] [Green Version]
- Steiner, A.A.; Molchanova, A.Y.; Dogan, M.D.; Patel, S.; Petervari, E.; Balasko, M.; Wanner, S.P.; Eales, J.; Oliveira, D.L.; Gavva, N.R.; et al. The hypothermic response to bacterial lipopolysaccharide critically depends on brain CB1, but not CB2 or TRPV1, receptors. J. Physiol. 2011, 589, 2415–2431. [Google Scholar] [CrossRef]
- Carai, M.A.; Colombo, G.; Gessa, G.L.; Yalamanchili, R.; Basavarajappa, B.S.; Hungund, B.L. Investigation on the relationship between cannabinoid CB1 and opioid receptors in gastrointestinal motility in mice. Br. J. Pharmacol. 2006, 148, 1043–1050. [Google Scholar] [CrossRef]
- Troy-Fioramonti, S.; Demizieux, L.; Gresti, J.; Muller, T.; Verges, B.; Degrace, P. Acute activation of cannabinoid receptors by anandamide reduces gastrointestinal motility and improves postprandial glycemia in mice. Diabetes 2015, 64, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.; Bayada, D.M.; Delbressine, L.P.; Ploemen, J.P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Mensch, J.; Oyarzabal, J.; Mackie, C.; Augustijns, P. In vivo, in vitro and in silico methods for small molecule transfer across the BBB. J. Pharm. Sci. 2009, 98, 4429–4468. [Google Scholar] [CrossRef]
- Gary-Bobo, M.; Elachouri, G.; Gallas, J.F.; Janiak, P.; Marini, P.; Ravinet-Trillou, C.; Chabbert, M.; Cruccioli, N.; Pfersdorff, C.; Roque, C.; et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology 2007, 46, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Jbilo, O.; Ravinet-Trillou, C.; Arnone, M.; Buisson, I.; Bribes, E.; Peleraux, A.; Penarier, G.; Soubrie, P.; Le Fur, G.; Galiegue, S.; et al. The CB1 receptor antagonist rimonabant reverses the diet-induced obesity phenotype through the regulation of lipolysis and energy balance. FASEB J. 2005, 19, 1567–1569. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Djaouti, L.; Demizieux, L.; Gresti, J.; Verges, B.; Degrace, P. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese mice. Diabetes 2010, 59, 926–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueiras, R.; Veyrat-Durebex, C.; Suchanek, P.M.; Klein, M.; Tschop, J.; Caldwell, C.; Woods, S.C.; Wittmann, G.; Watanabe, M.; Liposits, Z.; et al. Peripheral, but not central, CB1 antagonism provides food intake-independent metabolic benefits in diet-induced obese rats. Diabetes 2008, 57, 2977–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.H.; Kim, H.D.; Chae, Y.N.; Kim, M.K.; Shin, C.Y.; Ahn, G.J.; Choi, S.H.; Yang, E.K.; Park, K.J.; Chae, H.W.; et al. Peripherally acting CB1-receptor antagonist: The relative importance of central and peripheral CB1 receptors in adiposity control. Int. J. Obes. 2010, 34, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Hillard, C.J. Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: Further evidence for an anxiolytic role for endogenous cannabinoid signaling. J. Pharmacol. Exp. Ther. 2006, 318, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Wiley, J.L.; Burston, J.J.; Leggett, D.C.; Alekseeva, O.O.; Razdan, R.K.; Mahadevan, A.; Martin, B.R. CB1 cannabinoid receptor-mediated modulation of food intake in mice. Br. J. Pharmacol. 2005, 145, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Burns, H.D.; Van Laere, K.; Sanabria-Bohorquez, S.; Hamill, T.G.; Bormans, G.; Eng, W.S.; Gibson, R.; Ryan, C.; Connolly, B.; Patel, S.; et al. [18F]MK-9470, a positron emission tomography (PET) tracer for in vivo human PET brain imaging of the cannabinoid-1 receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 9800–9805. [Google Scholar] [CrossRef] [Green Version]
- Fong, T.M.; Guan, X.M.; Marsh, D.J.; Shen, C.P.; Stribling, D.S.; Rosko, K.M.; Lao, J.; Yu, H.; Feng, Y.; Xiao, J.C.; et al. Antiobesity efficacy of a novel cannabinoid-1 receptor inverse agonist, N-[(1S,2S)-3-(4-chlorophenyl)-2-(3-cyanophenyl)-1-methylpropyl]-2-methyl-2 -[[5-(trifluoromethyl)pyridin-2-yl]oxy]propanamide (MK-0364), in rodents. J. Pharmacol. Exp. Ther. 2007, 321, 1013–1022. [Google Scholar] [CrossRef]
- Freeman, A.S.; Martin, B.R. Interactions between phencyclidine and delta 9-tetrahydrocannabinol in mice following smoke exposure. Life Sci. 1983, 32, 1081–1089. [Google Scholar] [CrossRef]
- Wiley, J.L.; Martin, B.R. Cannabinoid pharmacology: Implications for additional cannabinoid receptor subtypes. Chem. Phys. Lipids 2002, 121, 57–63. [Google Scholar] [CrossRef]
- Navarro, M.; Hernandez, E.; Munoz, R.M.; del Arco, I.; Villanua, M.A.; Carrera, M.R.; Rodriguez de Fonseca, F. Acute administration of the CB1 cannabinoid receptor antagonist SR 141716A induces anxiety-like responses in the rat. Neuroreport 1997, 8, 491–496. [Google Scholar] [CrossRef]
- Gamble-George, J.C.; Conger, J.R.; Hartley, N.D.; Gupta, P.; Sumislawski, J.J.; Patel, S. Dissociable effects of CB1 receptor blockade on anxiety-like and consummatory behaviors in the novelty-induced hypophagia test in mice. Psychopharmacology 2013, 228, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinho, E.A.; Oliveira-Lima, A.J.; Santos, R.; Hollais, A.W.; Baldaia, M.A.; Wuo-Silva, R.; Yokoyama, T.S.; Takatsu-Coleman, A.L.; Patti, C.L.; Longo, B.M.; et al. Effects of rimonabant on the development of single dose-induced behavioral sensitization to ethanol, morphine and cocaine in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 58, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, J.; Hinden, L.; Drori, A.; Udi, S.; Azar, S.; Baraghithy, S. The therapeutic potential of targeting the peripheral endocannabinoid/CB1 receptor system. Eur. J. Intern. Med. 2018, 49, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Demizieux, L.; Gresti, J.; Djaouti, L.; Gaba, L.; Verges, B.; Degrace, P. Antagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: Evidence from cultured explants. Hepatology 2012, 55, 790–799. [Google Scholar] [CrossRef]
- Osei-Hyiaman, D.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.I.; Batkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, L.; Xiong, K.; Godlewski, G.; Mukhopadhyay, B.; Tam, J.; Yin, S.; Gao, P.; Shan, X.; Pickel, J.; et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 2012, 142, 1218–1228.e1. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Matias, I.; Gonthier, M.P.; Orlando, P.; Martiadis, V.; De Petrocellis, L.; Cervino, C.; Petrosino, S.; Hoareau, L.; Festy, F.; Pasquali, R.; et al. Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J. Clin. Endocrinol. Metab. 2006, 91, 3171–3180. [Google Scholar] [CrossRef]
- Ruiz de Azua, I.; Mancini, G.; Srivastava, R.K.; Rey, A.A.; Cardinal, P.; Tedesco, L.; Zingaretti, C.M.; Sassmann, A.; Quarta, C.; Schwitter, C.; et al. Adipocyte cannabinoid receptor CB1 regulates energy homeostasis and alternatively activated macrophages. J. Clin. Investig. 2017, 127, 4148–4162. [Google Scholar] [CrossRef] [PubMed]
- Cota, D.; Genghini, S.; Pasquali, R.; Pagotto, U. Antagonizing the cannabinoid receptor type 1: A dual way to fight obesity. J. Endocrinol. Investig. 2003, 26, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, V.; Fezza, F.; Pasquariello, N.; Bari, M.; Oddi, S.; Agro, A.F.; Maccarrone, M. Endocannabinoids in adipocytes during differentiation and their role in glucose uptake. Cell Mol. Life Sci. 2007, 64, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Batkai, S.; Harvey-White, J.; Mackie, K.; Offertaler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, C.; Pilon, C.; Calcagno, A.; Urbanet, R.; Rossato, M.; Milan, G.; Bianchi, K.; Rizzuto, R.; Bernante, P.; Federspil, G.; et al. The endogenous cannabinoid system stimulates glucose uptake in human fat cells via phosphatidylinositol 3-kinase and calcium-dependent mechanisms. J. Clin. Endocrinol. Metab. 2007, 92, 4810–4819. [Google Scholar] [CrossRef] [Green Version]
- Vettor, R.; Pagano, C. The role of the endocannabinoid system in lipogenesis and fatty acid metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 51–63. [Google Scholar] [CrossRef]
- Buch, C.; Muller, T.; Leemput, J.; Passilly-Degrace, P.; Ortega-Deballon, P.; Pais de Barros, J.P.; Verges, B.; Jourdan, T.; Demizieux, L.; Degrace, P. Endocannabinoids produced by white adipose tissue modulate lipolysis in lean but not in obese rodent and human. Front. Endocrinol. 2021, 12, 716431. [Google Scholar] [CrossRef]
- Muller, T.; Demizieux, L.; Troy-Fioramonti, S.; Gresti, J.; Pais de Barros, J.P.; Berger, H.; Verges, B.; Degrace, P. Overactivation of the endocannabinoid system alters the antilipolytic action of insulin in mouse adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E26–E36. [Google Scholar] [CrossRef] [Green Version]
- Robert, J.; Troy-Fioramonti, S.; Demizieux, L.; Degrace, P. Preparation of 1,4-bis(4-Methylphenyl)-3-Phtaloylazetidin-2-Ones and Derivatives Thereof Useful for the Treatment of Diseases Associated with Hyperactivity of the Endocannabinoid System. U.S. Patent US20180265498, 2018. [Google Scholar]
- Palomo, C.; Aizpurua, J.M.; Ganboa, I.; Oiarbide, M. From beta-lactams to alpha- and beta-amino acid derived peptides. Amino Acids 1999, 16, 321–343. [Google Scholar] [CrossRef]
- Jiao, L.; Liang, Y.; Xu, J. Origin of the relative stereoselectivity of the beta-lactam formation in the Staudinger reaction. J. Am. Chem. Soc. 2006, 128, 6060–6069. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muller, T.; Demizieux, L.; Troy-Fioramonti, S.; Buch, C.; Leemput, J.; Belloir, C.; Pais de Barros, J.-P.; Jourdan, T.; Passilly-Degrace, P.; Fioramonti, X.; et al. Chemical Synthesis, Pharmacokinetic Properties and Biological Effects of JM-00266, a Putative Non-Brain Penetrant Cannabinoid Receptor 1 Inverse Agonist. Int. J. Mol. Sci. 2022, 23, 2923. https://doi.org/10.3390/ijms23062923
Muller T, Demizieux L, Troy-Fioramonti S, Buch C, Leemput J, Belloir C, Pais de Barros J-P, Jourdan T, Passilly-Degrace P, Fioramonti X, et al. Chemical Synthesis, Pharmacokinetic Properties and Biological Effects of JM-00266, a Putative Non-Brain Penetrant Cannabinoid Receptor 1 Inverse Agonist. International Journal of Molecular Sciences. 2022; 23(6):2923. https://doi.org/10.3390/ijms23062923
Chicago/Turabian StyleMuller, Tania, Laurent Demizieux, Stéphanie Troy-Fioramonti, Chloé Buch, Julia Leemput, Christine Belloir, Jean-Paul Pais de Barros, Tony Jourdan, Patricia Passilly-Degrace, Xavier Fioramonti, and et al. 2022. "Chemical Synthesis, Pharmacokinetic Properties and Biological Effects of JM-00266, a Putative Non-Brain Penetrant Cannabinoid Receptor 1 Inverse Agonist" International Journal of Molecular Sciences 23, no. 6: 2923. https://doi.org/10.3390/ijms23062923
APA StyleMuller, T., Demizieux, L., Troy-Fioramonti, S., Buch, C., Leemput, J., Belloir, C., Pais de Barros, J.-P., Jourdan, T., Passilly-Degrace, P., Fioramonti, X., Le Bon, A.-M., Vergès, B., Robert, J.-M., & Degrace, P. (2022). Chemical Synthesis, Pharmacokinetic Properties and Biological Effects of JM-00266, a Putative Non-Brain Penetrant Cannabinoid Receptor 1 Inverse Agonist. International Journal of Molecular Sciences, 23(6), 2923. https://doi.org/10.3390/ijms23062923