
Fentanyl Structure as a Scaffold for Opioid/Non-Opioid Multitarget Analgesics


Abstract
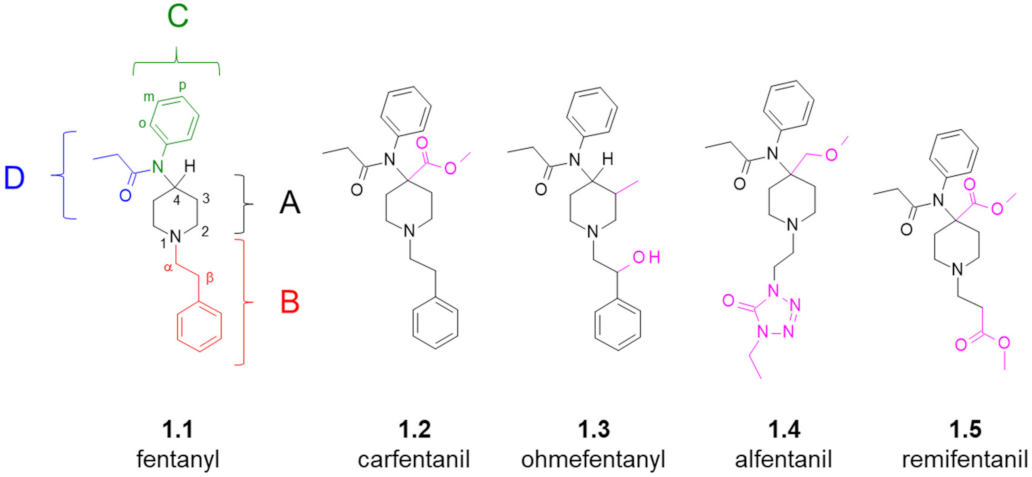
1. Introduction
- (region A): 4-carboxymethyl, 4-methoxymethyl, 3-methyl,
- (region B): α-methyl, β-hydroxyl (if accompanied by 3-methyl in the region A), replacement of the phenyl ring for heterocyclic aromatics,
- (region C): p-fluoro substitution at the ring (some other substitutions and replacements could be tolerated or beneficial, too).
- (region D): alicyclic fragments (e.g., cyclopropyl), linear (elongated) or branched alkyl chains, ether fragments, aromatic fragments (e.g., 2-furanyl).
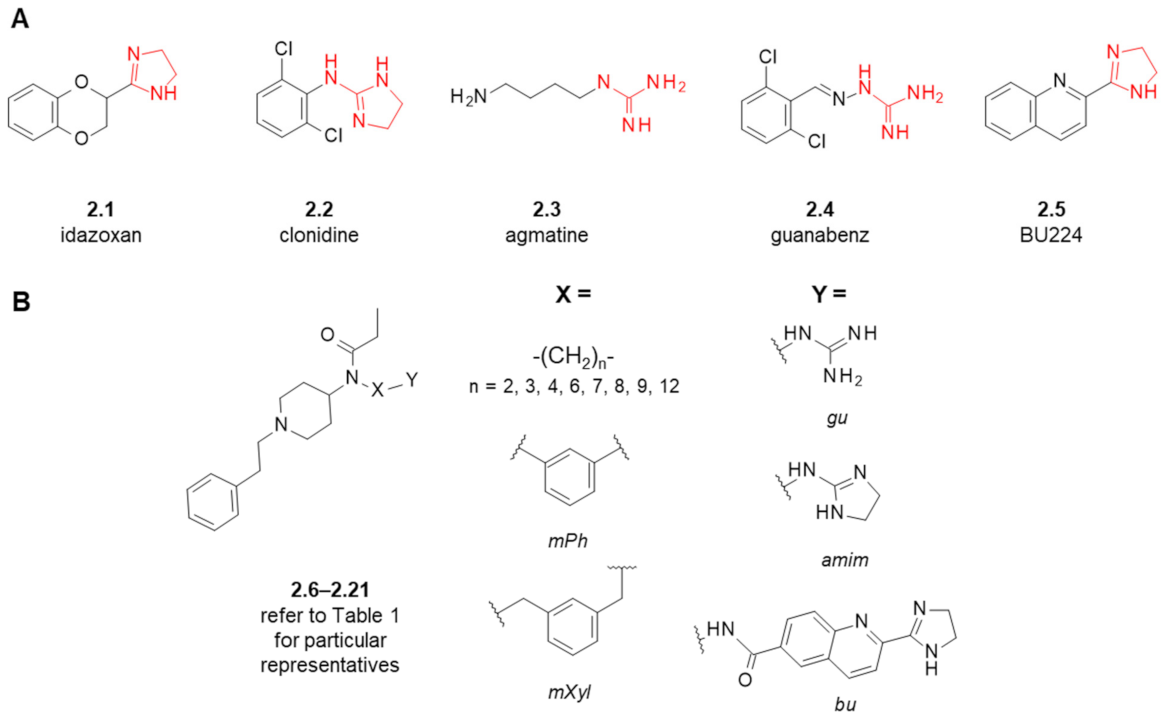
2. Fentanyl-Based MTAs Targeting MOR and I2-Imidazoline Binding Sites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
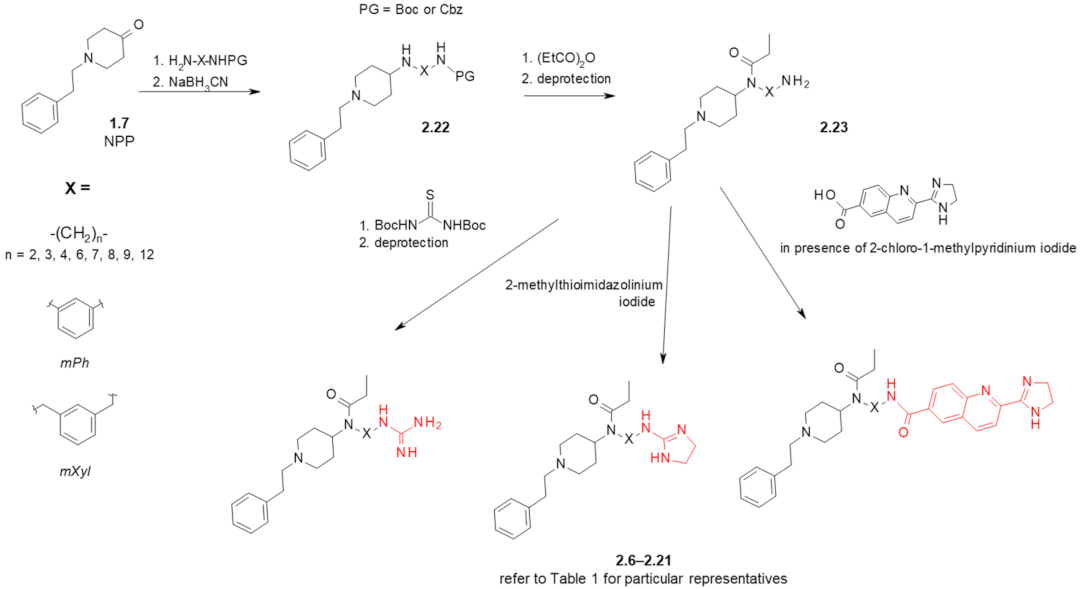
| Structure (Refer to Figure 3B for Structural Explanations) | Affinity [Ki (nM)] 1 | |||||
|---|---|---|---|---|---|---|
| Compound | X | Y | dNC 2 | MOR 3 | I2-IBS 4 | Ref. |
| fentanyl (1.1) | - | - | - | 6 ± 1.5 5 | 5462 ± 1343 6 | [26] |
| 2.9 ± 1.5 | 8593 ± 738 | [28] | ||||
| idazoxan (2.1) | - | - | - | - | 307 ± 183 6 | [26] |
| 28 ± 11 | [27,28] | |||||
| BU224 (2.5) | - | - | - | - | 9.8 ± 0.3 | [48] |
| 2.6 | mPh | gu | 5 | 7.8 ± 2.5 5 | 1890 ± 499 6 | [26] |
| 2.7 | mPh | amim | 5 | 7119 ± 4089 5 | 9630 ± 6731 6 | [26] |
| 2.8 | -(CH2)3- | gu | 5 | 37 ± 12 5 | 2022 ± 949 6 | [26] |
| 23 ± 4.5 | 1920 ± 996 | [27] | ||||
| 2.9 | -(CH2)3- | amim | 5 | 1751 ± 1135 5 | 2327 ± 811 6 | [26] |
| 2.10 | -(CH2)2- | gu | 4 | 433 ± 83 | 437 ± 228 | [27] |
| 2.11 | -(CH2)4- | gu | 6 | 0.59 ± 0.18 | >10,000 | [28] |
| 2.12 | -(CH2)6- | gu | 8 | 1.04 ± 0.28 | 409 ± 238 | [27] |
| 2.13 | -(CH2)7- | gu | 9 | 0.37 ± 0.19 | 6627 ± 3106 | [28] |
| 2.14 | -(CH2)8- | gu | 10 | 37 ± 9.7 | 126 ± 72 | [27] |
| 2.15 | -(CH2)9- | gu | 11 | 26 ± 6 | 58 ± 46 | [28] |
| 2.16 | -(CH2)12- | gu | 14 | 477 ± 75 | 6.5 ± 3.0 | [27] |
| 2.17 | mXyl | gu | 7 | 0.0098 ± 0.0033 | >10,000 | [28] |
| 0.448 ± 0.079 7 | [49] | |||||
| 2.18 | -(CH2)3- | bu | 12 | 6142 ± 2123 | 875 ± 713 | [28] |
| 2.19 | -(CH2)6- | bu | 15 | 2168 ± 66 | 323 ± 270 | [28] |
| 2.20 | -(CH2)8- | bu | 17 | 339 ± 35 | >10,000 | [28] |
| 2.21 | -(CH2)12- | bu | 21 | 545 ± 179 | 547 ± 316 | [28] |
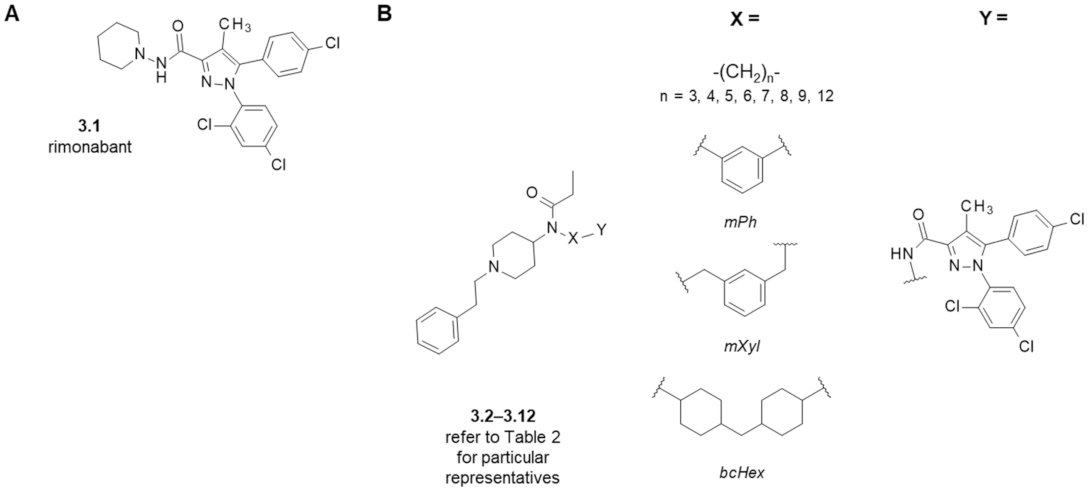
3. Fentanyl-Based MTAs Targeting MOR and CB1R
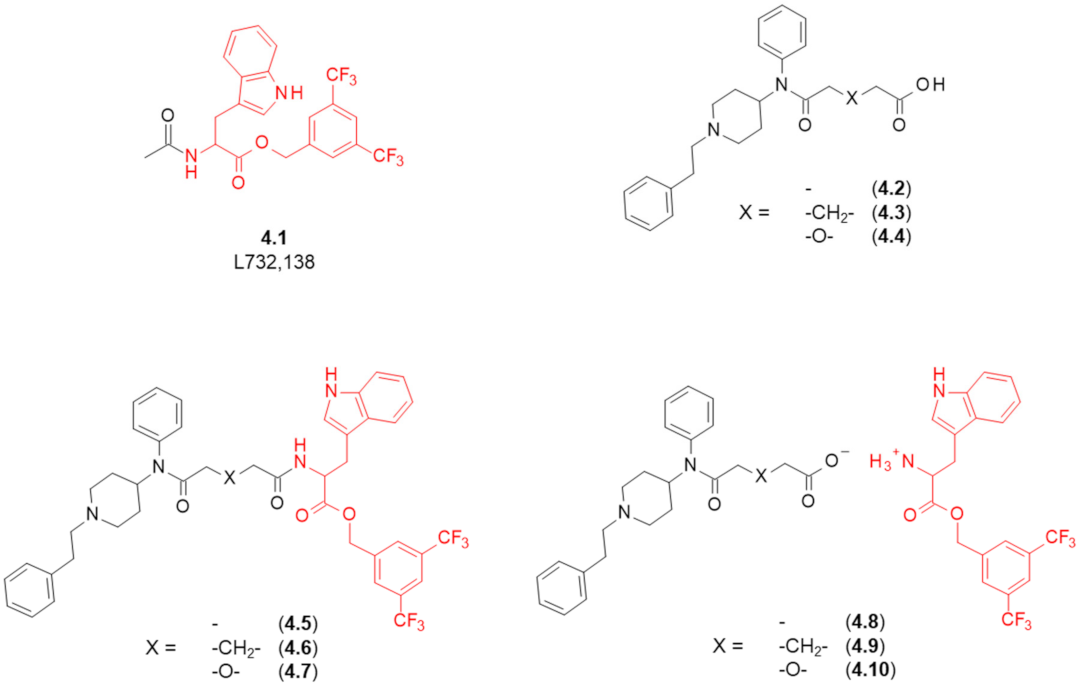
4. Fentanyl-Based MTAs Targeting MOR and NK1R
5. Fentanyl-Based MTAs Targeting MOR and D2-like Dopamine Receptors
6. Fentanyl-Based MTAs Targeting MOR and COX
7. Fentanyl-Based MTAs Targeting MOR and FAAH/MAGL Hydrolases
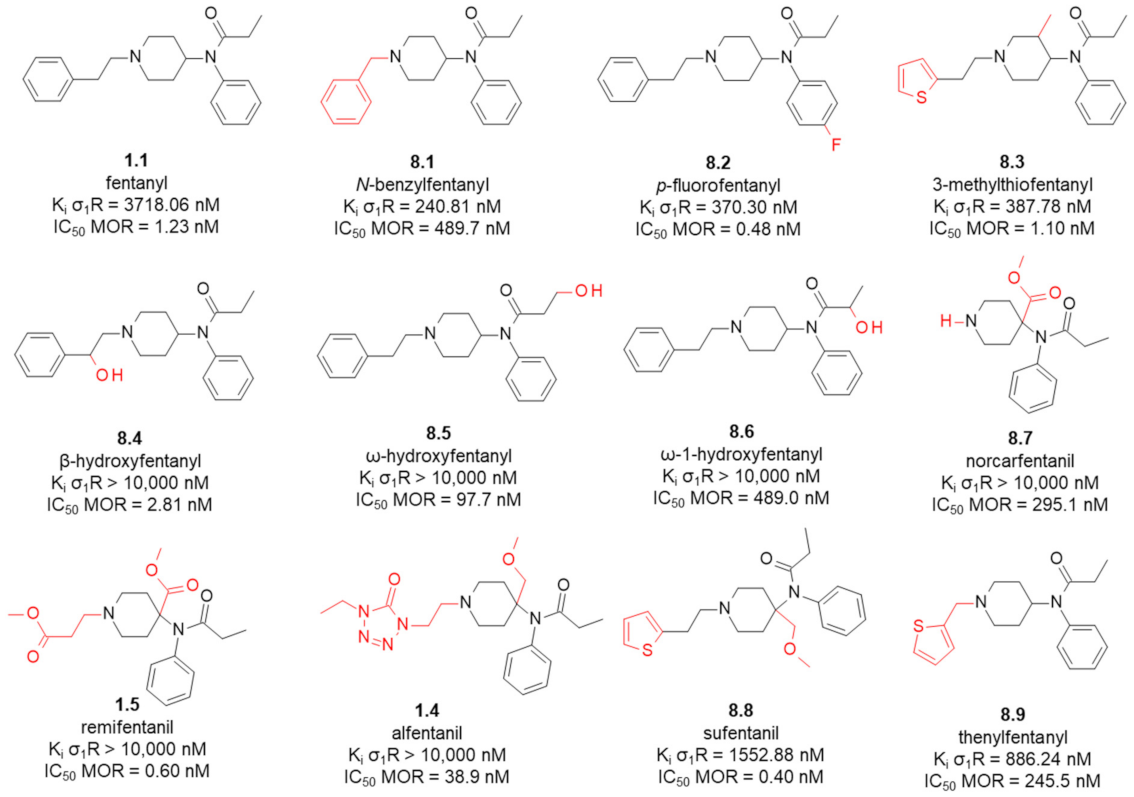
8. Fentanyl-Related MTAs Targeting MOR and σ1R
8.1. Affinity of Fentanyl Analogues for σ1R

- ionic interaction of protonated piperidine’s nitrogen with Glu172,
- direction of the anilide’s ring towards α4 and α5 helices of the receptor,
- positioning of the N-substituent towards the bottom of β-barrel (close to Asp126).
8.2. 1-Oxa-4,9-diazaspiro[5.5]undecane Derivatives
8.3. Amide Derivatives with Piperidine in Their Structures
8.4. Remark on σ2R
9. Outlook
Supplementary Materials
Funding
Conflicts of Interest
References
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef] [PubMed]
- de Vries, F.; Bruin, M.; Lobatto, D.J.; Dekkers, O.M.; Schoones, J.W.; van Furth, W.R.; Pereira, A.M.; Karavitaki, N.; Biermasz, N.R.; Zamanipoor Najafabadi, A.H. Opioids and Their Endocrine Effects: A Systematic Review and Meta-analysis. J. Clin. Endocrinol. Metab. 2020, 105, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, C.J.; Durieux, M.E. Differential Opioid Tolerance and Opioid-induced Hyperalgesia. Anesthesiology 2016, 124, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Machelska, H.; Celik, M.Ö. Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef]
- Dvoracsko, S.; Stefanucci, A.; Novellino, E.; Mollica, A. The design of multitarget ligands for chronic and neuropathic pain. Future Med. Chem. 2015, 7, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Aricò, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.-T.; Hang, L.; Liu, T. Mu Opioid Receptor Heterodimers Emerge as Novel Therapeutic Targets: Recent Progress and Future Perspective. Front. Pharmacol. 2020, 11, 1078. [Google Scholar] [CrossRef] [PubMed]
- Starnowska-Sokół, J.; Przewłocka, B. Multifunctional Opioid-Derived Hybrids in Neuropathic Pain: Preclinical Evidence, Ideas and Challenges Joanna. Molecules 2020, 25, 5520. [Google Scholar] [CrossRef]
- Mollica, A.; Pelliccia, S.; Famiglini, V.; Stefanucci, A.; Macedonio, G.; Chiavaroli, A.; Orlando, G.; Brunetti, L.; Ferrante, C.; Pieretti, S.; et al. Exploring the first Rimonabant analog-opioid peptide hybrid compound, as bivalent ligand for CB1 and opioid receptors. J. Enzyme Inhib. Med. Chem. 2017, 32, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, M.; Akgün, E.; Yekkirala, A.; Lunzer, M.M.; Powers, M.D.; Kalyuzhny, A.E.; Portoghese, P.S. Bivalent Ligands That Target μ Opioid (MOP) and Cannabinoid1 (CB 1) Receptors Are Potent Analgesics Devoid of Tolerance. J. Med. Chem. 2013, 56, 5505–5513. [Google Scholar] [CrossRef]
- Kleczkowska, P.; Nowicka, K.; Bujalska-Zadrozny, M.; Hermans, E. Neurokinin-1 receptor-based bivalent drugs in pain management: The journey to nowhere? Pharmacol. Ther. 2019, 196, 44–58. [Google Scholar] [CrossRef]
- Dyniewicz, J.; Lipiński, P.F.J.; Kosson, P.; Bochyńska-Czyż, M.; Matalińska, J.; Misicka, A. Antinociceptive and Cytotoxic Activity of Opioid Peptides with Hydrazone and Hydrazide Moieties at the C-Terminus. Molecules 2020, 25, 3429. [Google Scholar] [CrossRef]
- Lee, Y.S.; Agnes, R.S.; Davis, P.; Ma, S.; Badghisi, H.; Lai, J.; Porreca, F.; Hruby, V.J. Partial Retro−Inverso, Retro, and Inverso Modifications of Hydrazide Linked Bifunctional Peptides for Opioid and Cholecystokinin (CCK) Receptors. J. Med. Chem. 2007, 50, 165–168. [Google Scholar] [CrossRef][Green Version]
- Agnes, R.S.; Lee, Y.S.; Davis, P.; Ma, S.; Badghisi, H.; Porreca, F.; Lai, J.; Hruby, V.J. Structure−Activity Relationships of Bifunctional Peptides Based on Overlapping Pharmacophores at Opioid and Cholecystokinin Receptors. J. Med. Chem. 2006, 49, 2868–2875. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowska, P.; Kosson, P.; Ballet, S.; Van den Eynde, I.; Tsuda, Y.; Tourwé, D.; Lipkowski, A.W. PK20, a New Opioid-Neurotensin Hybrid Peptide That Exhibits Central and Peripheral Antinociceptive Effects. Mol. Pain 2010, 6, 86. [Google Scholar] [CrossRef]
- Gonzalez, S.; Dumitrascuta, M.; Eiselt, E.; Louis, S.; Kunze, L.; Blasiol, A.; Vivancos, M.; Previti, S.; Dewolf, E.; Martin, C.; et al. Optimized Opioid-Neurotensin Multitarget Peptides: From Design to Structure-Activity Relationship Studies. J. Med. Chem. 2020, 63, 12929–12941. [Google Scholar] [CrossRef] [PubMed]
- Starnowska-Sokół, J.; Piotrowska, A.; Bogacka, J.; Makuch, W.; Mika, J.; Witkowska, E.; Godlewska, M.; Osiejuk, J.; Gątarz, S.; Misicka, A.; et al. Novel hybrid compounds, opioid agonist+melanocortin 4 receptor antagonist, as efficient analgesics in mouse chronic constriction injury model of neuropathic pain. Neuropharmacology 2020, 178, 108232. [Google Scholar] [CrossRef]
- Witkowska, E.; Godlewska, M.; Osiejuk, J.; Gątarz, S.; Wileńska, B.; Kosińska, K.; Starnowska-Sokół, J.; Piotrowska, A.; Lipiński, P.F.J.; Matalińska, J.; et al. Bifunctional Opioid/Melanocortin Peptidomimetics for Use in Neuropathic Pain: Variation in the Type and Length of the Linker Connecting the Two Pharmacophores. Int. J. Mol. Sci. 2022, 23, 674. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-L.L.; Pan, J.-X.X.; Song, J.-J.J.; Tang, H.-H.H.; Yu, H.-P.P.; Li, X.-H.H.; Li, N.; Zhang, T.; Zhang, R.; Zhang, M.-N.N.; et al. Structure-Based Optimization of Multifunctional Agonists for Opioid and Neuropeptide FF Receptors with Potent Nontolerance Forming Analgesic Activities. J. Med. Chem. 2016, 59, 10198–10208. [Google Scholar] [CrossRef]
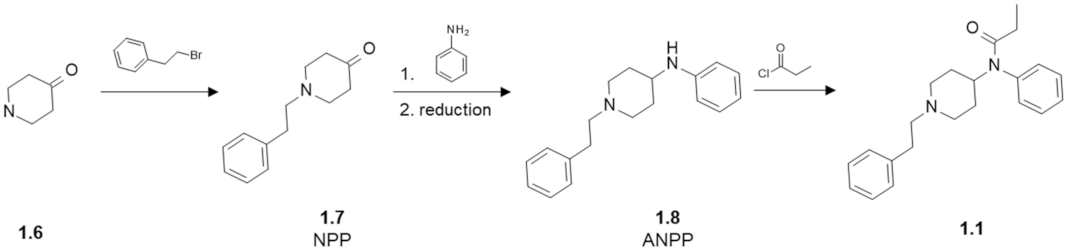
- Vardanyan, R.S.; Hruby, V.J. Substituted 1-Arylalkyl-4-Acylaminopiperidine Compounds and Methods of Producing and Using the Same. WO2017034631A1, 24 August 2015. [Google Scholar]
- Zhuang, T.; Xiong, J.; Hao, S.; Du, W.; Liu, Z.; Liu, B.; Zhang, G.; Chen, Y. Bifunctional μ opioid and σ1 receptor ligands as novel analgesics with reduced side effects. Eur. J. Med. Chem. 2021, 223, 113658. [Google Scholar] [CrossRef]
- Vardanyan, R.; Vijay, G.; Nichol, G.S.; Liu, L.; Kumarasinghe, I.; Davis, P.; Vanderah, T.; Porreca, F.; Lai, J.; Hruby, V.J. Synthesis and investigations of double-pharmacophore ligands for treatment of chronic and neuropathic pain. Bioorg. Med. Chem. 2009, 17, 5044–5053. [Google Scholar] [CrossRef]
- Monti, L.; Stefanucci, A.; Pieretti, S.; Marzoli, F.; Fidanza, L.; Mollica, A.; Mirzaie, S.; Carradori, S.; De Petrocellis, L.; Schiano Moriello, A.; et al. Evaluation of the analgesic effect of 4-anilidopiperidine scaffold containing ureas and carbamates. J. Enzyme Inhib. Med. Chem. 2016, 31, 1638–1647. [Google Scholar] [CrossRef]
- Mollica, A.; Costante, R.; Novellino, E.; Stefanucci, A.; Pieretti, S.; Zador, F.; Samavati, R.; Borsodi, A.; Benyhe, S.; Vetter, I.; et al. Design, Synthesis and Biological Evaluation of Two Opioid Agonist and Ca v 2.2 Blocker Multitarget Ligands. Chem. Biol. Drug Des. 2015, 86, 156–162. [Google Scholar] [CrossRef]
- Montero, A.; Goya, P.; Jagerovic, N.; Callado, L.F.; Meana, J.J.; Girón, R.; Goicoechea, C.; Martín, M.I. Guanidinium and aminoimidazolinium derivatives of N-(4-piperidyl)propanamides as potential ligands for μ opioid and I2-imidazoline receptors: Synthesis and pharmacological screening. Bioorg. Med. Chem. 2002, 10, 1009–1018. [Google Scholar] [CrossRef]
- Dardonville, C.; Jagerovic, N.; Callado, L.F.; Meana, J.J. Fentanyl derivatives bearing aliphatic alkaneguanidinium moieties: A new series of hybrid molecules with significant binding affinity for μ-opioid receptors and I2-imidazoline binding sites. Bioorg. Med. Chem. Lett. 2004, 14, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Dardonville, C.; Fernandez-Fernandez, C.; Gibbons, S.-L.; Ryan, G.J.; Jagerovic, N.; Gabilondo, A.M.; Meana, J.J.; Callado, L.F. Synthesis and pharmacological studies of new hybrid derivatives of fentanyl active at the μ-opioid receptor and I2–imidazoline binding sites. Bioorg. Med. Chem. 2006, 14, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Pawełczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Molecular Consortia—Various Structural and Synthetic Concepts for More Effective Therapeutics Synthesis. Int. J. Mol. Sci. 2018, 19, 1104. [Google Scholar] [CrossRef]
- Burns, S.M.; Cunningham, C.W.; Mercer, S.L. DARK Classics in Chemical Neuroscience: Fentanyl. ACS Chem. Neurosci. 2018, 9, 2428–2437. [Google Scholar] [CrossRef]
- Clotz, M.A.; Nahata, M.C. Clinical uses of fentanyl, sufentanil, and alfentanil. Clin. Pharm. 1991, 10, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Jannetto, P.J.; Helander, A.; Garg, U.; Janis, G.C.; Goldberger, B.; Ketha, H. The Fentanyl Epidemic and Evolution of Fentanyl Analogs in the United States and the European Union. Clin. Chem. 2019, 65, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.A.; Leif, R.N.; Mayer, B.P. An Efficient, Optimized Synthesis of Fentanyl and Related Analogs. PLoS ONE 2014, 9, e108250. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.A.J.; Gardocki, J.F. Method for Producing Analgesia. U.S. Patent 3141823, 21 July 1964. [Google Scholar]
- Janssen, P.A.J. 1-Aralkyl-4-(N-Aryl-Carbonyl Amino)-Piperidines and Related Compounds. U.S. Patent 3164600, 5 January 1965. [Google Scholar]
- Vardanyan, R.S.; Hruby, V.J. Fentanyl-related compounds and derivatives: Current status and future prospects for pharmaceutical applications. Future Med. Chem. 2014, 6, 385–412. [Google Scholar] [CrossRef] [PubMed]
- Ringuette, A.E.; Spock, M.; Lindsley, C.W.; Bender, A.M. DARK Classics in Chemical Neuroscience: Carfentanil. ACS Chem. Neurosci. 2020, 11, 3955–3967. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Zhu, Y.-C.; Jin, W.-Q.; Chen, X.-J.; Chen, J.; Ji, R.-Y.; Chi, Z.-Q. Stereoisomers of N-[1-(2-Hydroxy-2-phenylethyl)-3-methyl-4-piperidyl]- N-phenylpropanamide: Synthesis, Stereochemistry, Analgesic Activity, and Opioid Receptor Binding Characteristics. J. Med. Chem. 1995, 38, 3652–3659. [Google Scholar] [CrossRef]
- Janssens, F.; Torremans, J.; Janssen, P.A.J. Synthetic 1,4-disubstituted 1,4-dihydro-5H-tetrazol-5-one derivatives of fentanyl: Alfentanil (R 39209), a potent, extremely short-acting narcotic analgesic. J. Med. Chem. 1986, 29, 2290–2297. [Google Scholar] [CrossRef]
- Feldman, P.L.; James, M.K.; Brackeen, M.F.; Bilotta, J.M.; Schuster, S.V.; Lahey, A.P.; Lutz, M.W.; Johnson, M.R.; Leighton, H.J. Design, synthesis, and pharmacological evaluation of ultrashort- to long-acting opioid analgesics. J. Med. Chem. 1991, 34, 2202–2208. [Google Scholar] [CrossRef]
- Regunathan, S.; Reis, D.J. Imidazoline Receptors and Their Endogenous Ligands. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 511–544. [Google Scholar] [CrossRef]
- Li, J.-X. Imidazoline I 2 receptors: An update. Pharmacol. Ther. 2017, 178, 48–56. [Google Scholar] [CrossRef]
- Kimura, A.; Tyacke, R.J.; Robinson, J.J.; Husbands, S.M.; Minchin, M.C.W.; Nutt, D.J.; Hudson, A.L. Identification of an imidazoline binding protein: Creatine kinase and an imidazoline-2 binding site. Brain Res. 2009, 1279, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, P.; Hudson, A.; García-Sevilla, J.A.; Li, J.-X. Imidazoline Receptor System: The Past, the Present, and the Future. Pharmacol. Rev. 2020, 72, 50–79. [Google Scholar] [CrossRef]
- Li, J.-X.; Zhang, Y. Imidazoline I2 receptors: Target for new analgesics? Eur. J. Pharmacol. 2011, 658, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Rovati, L.C.; Brambilla, N.; Blicharski, T.; Connell, J.; Vitalini, C.; Bonazzi, A.; Giacovelli, G.; Girolami, F.; D’Amato, M. Efficacy and safety of the first-in-class imidazoline-2 receptor ligand CR4056 in pain from knee osteoarthritis and disease phenotypes: A randomized, double-blind, placebo-controlled phase 2 trial. Osteoarthr. Cartil. 2020, 28, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gomez, J.I.; Ruiz, J.; Callado, L.F.; Garibi, J.M.; Aguinaco, L.; Barturen, F.; Meana, J.J. Increased density of I2-imidazoline receptors in human glioblastomas. Neuroreport 1996, 7, 1393–1396. [Google Scholar] [CrossRef] [PubMed]
- Weltrowska, G.; Chung, N.N.; Lemieux, C.; Guo, J.; Lu, Y.; Wilkes, B.C.; Schiller, P.W. “Carba”-Analogues of Fentanyl are Opioid Receptor Agonists. J. Med. Chem. 2010, 53, 2875–2881. [Google Scholar] [CrossRef]
- Jagerovic, N.; Fernández-Fernández, C.; Erdozain, A.M.; Girón, R.; Sánchez, E.; López-Moreno, J.A.; Morales, P.; Rodríguez de Fonseca, F.; Goya, P.; Meana, J.J.; et al. Combining rimonabant and fentanyl in a single entity: Preparation and pharmacological results. Drug Des. Devel. Ther. 2014, 8, 263–277. [Google Scholar] [CrossRef]
- Zádor, F.; Wollemann, M. Receptome: Interactions between three pain-related receptors or the “Triumvirate” of cannabinoid, opioid and TRPV1 receptors. Pharmacol. Res. 2015, 102, 254–263. [Google Scholar] [CrossRef]
- Trang, T.; Sutak, M.; Jhamandas, K. Involvement of cannabinoid (CB1)-receptors in the development and maintenance of opioid tolerance. Neuroscience 2007, 146, 1275–1288. [Google Scholar] [CrossRef]
- Dvorácskó, S.; Keresztes, A.; Mollica, A.; Stefanucci, A.; Macedonio, G.; Pieretti, S.; Zádor, F.; Walter, F.R.; Deli, M.A.; Kékesi, G.; et al. Preparation of bivalent agonists for targeting the mu opioid and cannabinoid receptors. Eur. J. Med. Chem. 2019, 178, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Seely, K.A.; Brents, L.K.; Franks, L.N.; Rajasekaran, M.; Zimmerman, S.M.; Fantegrossi, W.E.; Prather, P.L. AM-251 and rimonabant act as direct antagonists at mu-opioid receptors: Implications for opioid/cannabinoid interaction studies. Neuropharmacology 2012, 63, 905–915. [Google Scholar] [CrossRef]
- Zádor, F.; Ötvös, F.; Benyhe, S.; Zimmer, A.; Páldy, E. Inhibition of forebrain μ-opioid receptor signaling by low concentrations of rimonabant does not require cannabinoid receptors and directly involves μ-opioid receptors. Neurochem. Int. 2012, 61, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Zádor, F.; Kocsis, D.; Borsodi, A.; Benyhe, S. Micromolar concentrations of rimonabant directly inhibits delta opioid receptor specific ligand binding and agonist-induced G-protein activity. Neurochem. Int. 2014, 67, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Carai, M.A.; Brunetti, G.; Gomez, R.; Melis, S.; Vacca, G.; Colombo, G.; Gessa, G.L. The cannabinoid receptor antagonist SR 141716 prevents acquisition of drinking behavior in alcohol-preferring rats. Eur. J. Pharmacol. 2001, 430, 369–371. [Google Scholar] [CrossRef]
- Bagley, J.R.; Wynn, R.L.; Rudo, F.G.; Doorley, B.M.; Spencer, H.K.; Spaulding, T. New 4-(heteroanilido)piperidines, structurally related to the pure opioidagonist fentanyl, with agonist and/or antagonist properties. J. Med. Chem. 1989, 32, 663–671. [Google Scholar] [CrossRef]
- Qin, Y.; Ni, L.; Shi, J.; Zhu, Z.; Shi, S.; Lam, A.; Magiera, J.; Sekar, S.; Kuo, A.; Smith, M.T.; et al. Synthesis and Biological Evaluation of Fentanyl Analogues Modified at Phenyl Groups with Alkyls. ACS Chem. Neurosci. 2019, 10, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Zubrzycka, M.; Janecka, A. Substance P: Transmitter of nociception (Minireview). Endocr. Regul. 2000, 34, 195–201. [Google Scholar]
- Xiao, J.; Zeng, S.; Wang, X.; Babazada, H.; Li, Z.; Liu, R.; Yu, W. Neurokinin 1 and opioid receptors: Relationships and interactions in nervous system. Transl. Perioper. Pain Med. 2016, 1, 11–21. [Google Scholar] [PubMed]
- Ossipov, M.H.; Lai, J.; King, T.; Vanderah, T.W.; Porreca, F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers 2005, 80, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Powell, K.J.; Quirion, R.; Jhamandas, K. Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur. J. Neurosci. 2003, 18, 1572–1583. [Google Scholar] [CrossRef] [PubMed]
- Kream, R.M.; Kato, T.; Shimonaka, H.; Marchand, J.E.; Wurm, W.H. Substance P markedly potentiates the antinociceptive effects of morphine sulfate administered at the spinal level. Proc. Natl. Acad. Sci. USA 1993, 90, 3564–3568. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R.; Kumirov, V.K.; Nichol, G.S.; Davis, P.; Liktor-Busa, E.; Rankin, D.; Varga, E.; Vanderah, T.; Porreca, F.; Lai, J.; et al. Synthesis and biological evaluation of new opioid agonist and neurokinin-1 antagonist bivalent ligands. Bioorg. Med. Chem. 2011, 19, 6135–6142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- MacLeod, A.M.; Merchant, K.J.; Brookfield, F.; Kelleher, F.; Stevenson, G.; Owens, A.P.; Swain, C.J.; Cascieri, M.A.; Sadowski, S. Identification of L-Tryptophan Derivatives with Potent and Selective Antagonist Activity at the NK1 Receptor. J. Med. Chem. 1994, 37, 1269–1274. [Google Scholar] [CrossRef]
- Ambrose, L.M.; Unterwald, E.M.; Van Bockstaele, E.J. Ultrastructural evidence for co-localization of dopamine D2 and μ-opioid receptors in the rat dorsolateral striatum. Anat. Rec. 2004, 279A, 583–591. [Google Scholar] [CrossRef]
- Qian, M.; Vasudevan, L.; Huysentruyt, J.; Risseeuw, M.D.P.; Stove, C.; Vanderheyden, P.M.L.; Van Craenenbroeck, K.; Van Calenbergh, S. Design, Synthesis, and Biological Evaluation of Bivalent Ligands Targeting Dopamine D 2 -Like Receptors and the μ-Opioid Receptor. ChemMedChem 2018, 13, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.C.; Beck-Schimmer, B.; Kajdi, M.-E.; Müller, D.; Tobler, P.N.; Quednow, B.B. Dopamine D2/3- and μ-opioid receptor antagonists reduce cue-induced responding and reward impulsivity in humans. Transl. Psychiatry 2016, 6, e850. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Bonifazi, A.; Ellenberger, M.P.; Keck, T.M.; Pommier, E.; Rais, R.; Slusher, B.S.; Gardner, E.; You, Z.-B.; Xi, Z.-X.; et al. Highly Selective Dopamine D 3 Receptor (D 3 R) Antagonists and Partial Agonists Based on Eticlopride and the D 3 R Crystal Structure: New Leads for Opioid Dependence Treatment. J. Med. Chem. 2016, 59, 7634–7650. [Google Scholar] [CrossRef]
- Gago, B.; Suárez-Boomgaard, D.; Fuxe, K.; Brené, S.; Reina-Sánchez, M.D.; Rodríguez-Pérez, L.M.; Agnati, L.F.; de la Calle, A.; Rivera, A. Effect of acute and continuous morphine treatment on transcription factor expression in subregions of the rat caudate putamen. Marked modulation by D4 receptor activation. Brain Res. 2011, 1407, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Gago, B.; Fuxe, K.; Agnati, L.; Peñafiel, A.; De La Calle, A.; Rivera, A. Dopamine D4 receptor activation decreases the expression of μ-opioid receptors in the rat striatum. J. Comp. Neurol. 2007, 502, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Hagelberg, N.; Kajander, J.K.; Någren, K.; Hinkka, S.; Hietala, J.; Scheinin, H. μ-Receptor agonism with alfentanil increases striatal dopamine D2 receptor binding in man. Synapse 2002, 45, 25–30. [Google Scholar] [CrossRef]
- Park, Y.; Kang Ho, I.; Fan, L.-W.; Loh, H.H.; Ko, K.H. Region specific increase of dopamine receptor D1/D2 mRNA expression in the brain of μ-opioid receptor knockout mice. Brain Res. 2001, 894, 311–315. [Google Scholar] [CrossRef]
- Bonifazi, A.; Battiti, F.O.; Sanchez, J.; Zaidi, S.A.; Bow, E.; Makarova, M.; Cao, J.; Shaik, A.B.; Sulima, A.; Rice, K.C.; et al. Novel Dual-Target μ-Opioid Receptor and Dopamine D 3 Receptor Ligands as Potential Nonaddictive Pharmacotherapeutics for Pain Management. J. Med. Chem. 2021, 64, 7778–7808. [Google Scholar] [CrossRef] [PubMed]
- Jevtic, I.; Penjisevic, J.; Ivanovic, M.; Kostic-Rajacic, S. Synthetic route towards potential bivalent ligands possessing opioid and D2/D3 pharmacophores. J. Serbian Chem. Soc. 2019, 84, 639–647. [Google Scholar] [CrossRef]
- Jevtic, I.; Penjisevic, J.; Savic-Vujovic, K.; Srebro, D.; Vuckovic, S.; Ivanovic, M.; Kostic-Rajacic, S. μ-opioid/D2 dopamine receptor pharmacophore containing ligands: Synthesis and pharmacological evaluation. J. Serbian Chem. Soc. 2020, 85, 711–720. [Google Scholar] [CrossRef]
- Jevtić, I. Synthesis, Pharmacological Evaluation and Docking Analisys of Novel Anilidopiperidines (in Serbian). Ph.D. Thesis, University of Belgrade, Belgrade, Serbia, 2020. Available online: https://nardus.mpn.gov.rs/handle/123456789/17614 (accessed on 11 December 2020).
- Varrassi, G.; Yeam, C.T.; Rekatsina, M.; Pergolizzi, J.V.; Zis, P.; Paladini, A. The Expanding Role of the COX Inhibitor/Opioid Receptor Agonist Combination in the Management of Pain. Drugs 2020, 80, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. FAAH and MAGL inhibitors: Therapeutic opportunities from regulating endocannabinoid levels. Curr. Opin. Investig. Drugs 2010, 11, 51–62. [Google Scholar] [PubMed]
- Pertwee, R.G. Elevating endocannabinoid levels: Pharmacological strategies and potential therapeutic applications. Proc. Nutr. Soc. 2014, 73, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Fichna, J.; Sałaga, M.; Stuart, J.; Saur, D.; Sobczak, M.; Zatorski, H.; Timmermans, J.-P.; Bradshaw, H.B.; Ahn, K.; Storr, M.A. Selective inhibition of FAAH produces antidiarrheal and antinociceptive effect mediated by endocannabinoids and cannabinoid-like fatty acid amides. Neurogastroenterol. Motil. 2014, 26, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Maccarrone, M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin. Drug Discov. 2013, 8, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Morales-Lázaro, S.L.; González-Ramírez, R.; Rosenbaum, T. Molecular Interplay Between the Sigma-1 Receptor, Steroids, and Ion Channels. Front. Pharmacol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Almansa, C.; Vela, J.M. Selective sigma-1 receptor antagonists for the treatment of pain. Future Med. Chem. 2014, 6, 1179–1199. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.J.; Kovalyshyn, I.; Burgman, M.; Neilan, C.; Chien, C.-C.; Pasternak, G.W. σ1 Receptor Modulation of G-Protein-Coupled Receptor Signaling: Potentiation of Opioid Transduction Independent from Receptor Binding. Mol. Pharmacol. 2010, 77, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T. The Sigma Receptor: Evolution of the Concept in Neuropsychopharmacology. Curr. Neuropharmacol. 2005, 3, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, C.G.; Greene, S.F. Sigma receptors [σ Rs]: Biology in normal and diseased states. J. Recept. Signal Transduct. 2015, 36, 1–62. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Qin, W.; Tian, S.; Xu, Q.; Wold, E.A.; Zhou, J.; Zhen, X.-C. Small Molecules Selectively Targeting Sigma-1 Receptor for the Treatment of Neurological Diseases. J. Med. Chem. 2020, 63, 15187–15217. [Google Scholar] [CrossRef] [PubMed]
- Marrazzo, A.; Parenti, C.; Scavo, V.; Ronsisvalle, S.; Maria Scoto, G.; Ronsisvalle, G. In vivo evaluation of (+)-MR200 as a new selective sigma ligand modulating MOP, DOP and KOP supraspinal analgesia. Life Sci. 2006, 78, 2449–2453. [Google Scholar] [CrossRef]
- Sánchez-Fernández, C.; Nieto, F.R.; González-Cano, R.; Artacho-Cordón, A.; Romero, L.; Montilla-García, Á.; Zamanillo, D.; Baeyens, J.M.; Entrena, J.M.; Cobos, E.J. Potentiation of morphine-induced mechanical antinociception by σ1 receptor inhibition: Role of peripheral σ1 receptors. Neuropharmacology 2013, 70, 348–358. [Google Scholar] [CrossRef]
- Puente, B.d.l.; Nadal, X.; Portillo-Salido, E.; Sánchez-Arroyos, R.; Ovalle, S.; Palacios, G.; Muro, A.; Romero, L.; Entrena, J.M.; Baeyens, J.M.; et al. Sigma-1 receptors regulate activity-induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain 2009, 145, 294–303. [Google Scholar] [CrossRef]
- Romero, L.; Zamanillo, D.; Nadal, X.; Sánchez-Arroyos, R.; Rivera-Arconada, I.; Dordal, A.; Montero, A.; Muro, A.; Bura, A.; Segalés, C.; et al. Pharmacological properties of S1RA, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. Br. J. Pharmacol. 2012, 166, 2289–2306. [Google Scholar] [CrossRef] [PubMed]
- Cendán, C.M.; Pujalte, J.M.; Portillo-Salido, E.; Baeyens, J.M. Antinociceptive effects of haloperidol and its metabolites in the formalin test in mice. Psychopharmacology 2005, 182, 485–493. [Google Scholar] [CrossRef]
- Entrena, J.M.; Cobos, E.J.; Nieto, F.R.; Cendán, C.M.; Baeyens, J.M.; Del Pozo, E. Antagonism by haloperidol and its metabolites of mechanical hypersensitivity induced by intraplantar capsaicin in mice: Role of sigma-1 receptors. Psychopharmacology 2009, 205, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, C.; Entrena, J.M.; Baeyens, J.M.; Cobos, E.J. Sigma-1 Receptor Antagonists: A New Class of Neuromodulatory Analgesics. In Sigma Receptors: Their Role in Disease and as Therapeutic Agents; Smith, S., Tsung-Ping, S., Eds.; Springer: Cham, Switzerland, 2017; pp. 109–132. [Google Scholar]
- Chien, C.C.; Pasternak, G.W. Selective antagonism of opioid analgesia by a sigma system. J. Pharmacol. Exp. Ther. 1994, 271, 1583–1590. [Google Scholar] [PubMed]
- Vidal-Torres, A.; de la Puente, B.; Rocasalbas, M.; Touriño, C.; Andreea Bura, S.; Fernández-Pastor, B.; Romero, L.; Codony, X.; Zamanillo, D.; Buschmann, H.; et al. Sigma-1 receptor antagonism as opioid adjuvant strategy: Enhancement of opioid antinociception without increasing adverse effects. Eur. J. Pharmacol. 2013, 711, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Zamanillo, D.; Romero, L.; Merlos, M.; Vela, J.M. Sigma 1 receptor: A new therapeutic target for pain. Eur. J. Pharmacol. 2013, 716, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Cuevas Cordobes, F.; Almansa-Rosales, C.; Garcia-Lopez, M. Piperidine Derivatives Having Multimodal Activity against Pain. WO2015091939A1, 25 June 2015. [Google Scholar]
- Cuevas Cordobes, F.; Almansa-Rosales, C.; Ayet Galcera, E.M.; Serafini Cabanes, M.T.; Garcia-Lopez, M. Piperidine Compounds Having Multimodal Activity Against Pain. WO2015091988A1, 25 June 2015. [Google Scholar]
- Cuevas Cordobes, F.; Almansa-Rosales, C.; Garcia-Lopez, M. Piperazine Derivatives Having Multimodal Activity against Pain. WO2015092009A1, 25 June 2015. [Google Scholar]
- Prezzavento, O.; Arena, E.; Sánchez-Fernández, C.; Turnaturi, R.; Parenti, C.; Marrazzo, A.; Catalano, R.; Amata, E.; Pasquinucci, L.; Cobos, E.J. (+)-and (−)-Phenazocine enantiomers: Evaluation of their dual opioid agonist/σ1antagonist properties and antinociceptive effects. Eur. J. Med. Chem. 2017, 125, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.F.J.; Szűcs, E.; Jarończyk, M.; Kosson, P.; Benyhe, S.; Misicka, A.; Dobrowolski, J.C.; Sadlej, J. Affinity of fentanyl and its derivatives for the σ1-receptor. Medchemcomm 2019, 10, 1187–1191. [Google Scholar] [CrossRef]
- Largent, B.L.; Wikström, H.; Gundlach, A.L.; Snyder, S.H. Structural determinants of sigma receptor affinity. Mol. Pharmacol. 1987, 32, 772–784. [Google Scholar]
- Chen, J.C.; Smith, E.R.; Cahill, M.; Cohen, R.; Fishman, J.B. The opioid receptor binding of dezocine, morphine, fentanyl, butorphanol and nalbuphine. Life Sci. 1993, 52, 389–396. [Google Scholar] [CrossRef]
- Lipiński, P.; Kosson, P.; Matalińska, J.; Roszkowski, P.; Czarnocki, Z.; Jarończyk, M.; Misicka, A.; Dobrowolski, J.; Sadlej, J. Fentanyl Family at the Mu-Opioid Receptor: Uniform Assessment of Binding and Computational Analysis. Molecules 2019, 24, 740. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- García, M.; Virgili, M.; Alonso, M.; Alegret, C.; Fernández, B.; Port, A.; Pascual, R.; Monroy, X.; Vidal-Torres, A.; Serafini, M.-T.; et al. 4-Aryl-1-oxa-4,9-diazaspiro[5.5]undecane Derivatives as Dual μ-Opioid Receptor Agonists and σ1 Receptor Antagonists for the Treatment of Pain. J. Med. Chem. 2020, 63, 2434–2454. [Google Scholar] [CrossRef] [PubMed]
- García, M.; Virgili, M.; Alonso, M.; Alegret, C.; Farran, J.; Fernández, B.; Bordas, M.; Pascual, R.; Burgueño, J.; Vidal-Torres, A.; et al. Discovery of EST73502, a Dual μ-Opioid Receptor Agonist and σ1 Receptor Antagonist Clinical Candidate for the Treatment of Pain. J. Med. Chem. 2020, 63, 15508–15526. [Google Scholar] [CrossRef] [PubMed]
- Laggner, C.; Schieferer, C.; Fiechtner, B.; Poles, G.; Hoffmann, R.D.; Glossmann, H.; Langer, T.; Moebius, F.F. Discovery of high-affinity ligands of sigma1 receptor, ERG2, and emopamil binding protein by pharmacophore modeling and virtual screening. J. Med. Chem. 2005, 48, 4754–4764. [Google Scholar] [CrossRef] [PubMed]
- Pascual, R.; Almansa, C.; Plata-Salamán, C.; Vela, J.M. A New Pharmacophore Model for the Design of Sigma-1 Ligands Validated on a Large Experimental Dataset. Front. Pharmacol. 2019, 10, 519. [Google Scholar] [CrossRef] [PubMed]
- Almansa-Rosales, C.; Garcia-Lopez, M.; Caamano-Moure, A.-M. Spiro-isoquinoline-1,4′-Piperidine Compounds Having Multimodal Activity Against Pain. WO2016078770A1, 26 May 2016. [Google Scholar]
- Almansa-Rosales, C. Spiro-Isoquinoline-4,4′-Piperidine Compounds Having Multimodal Activity against Pain. WO2016177472A1, 10 November 2016. [Google Scholar]
- Garcia-Lopez, M.; Almansa-Rosales, C. Amide Derivatives Having Multimodal Activity against Pain. WO2017016668A1, 2 February 2017. [Google Scholar]
- Torrens-Jover, A.; Jagerovic, N.; Almansa-Rosales, C. Piperidinylalkylamide Derivatives Having Multimodal Activity Against Pain. WO2017178515A1, 19 October 2017. [Google Scholar]
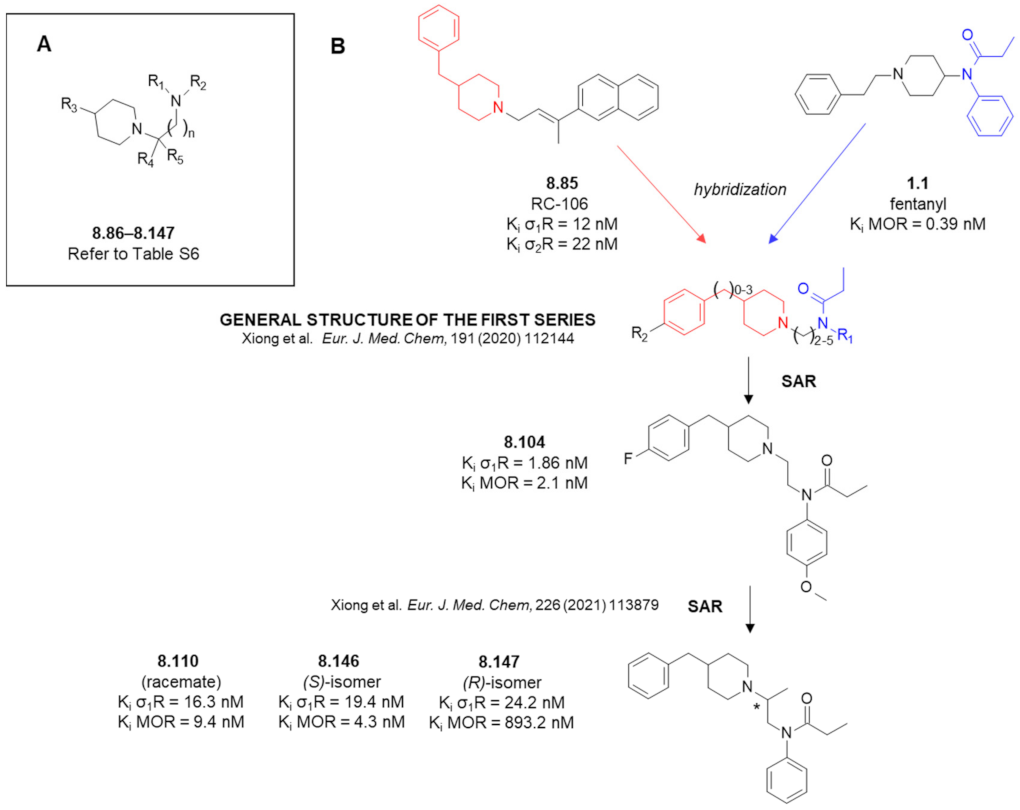
- Xiong, J.; Jin, J.; Gao, L.; Hao, C.; Liu, X.; Liu, B.-F.; Chen, Y.; Zhang, G. Piperidine propionamide as a scaffold for potent sigma-1 receptor antagonists and mu opioid receptor agonists for treating neuropathic pain. Eur. J. Med. Chem. 2020, 191, 112144. [Google Scholar] [CrossRef]
- Xiong, J.; Zhuang, T.; Ma, Y.; Xu, J.; Ye, J.; Ma, R.; Zhang, S.; Liu, X.; Liu, B.-F.; Hao, C.; et al. Optimization of bifunctional piperidinamide derivatives as σ1R Antagonists/MOR agonists for treating neuropathic pain. Eur. J. Med. Chem. 2021, 226, 113879. [Google Scholar] [CrossRef]
- Abate, C.; Niso, M.; Berardi, F. Sigma-2 receptor: Past, present and perspectives on multiple therapeutic exploitations. Future Med. Chem. 2018, 10, 1997–2018. [Google Scholar] [CrossRef]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Development of a Sigma-2 Receptor affinity filter through a Monte Carlo based QSAR analysis. Eur. J. Pharm. Sci. 2017, 106, 94–101. [Google Scholar] [CrossRef]
- Zeng, C.; Mach, R.H. The Evolution of the Sigma-2 (σ2) Receptor from Obscure Binding Site to Bona Fide Therapeutic Target. In Sigma Receptors: Their Role in Disease and as Therapeutic Agents; Smith, S., Tsung-Ping, S., Eds.; Springer: Cham, Switzerland, 2017; pp. 49–61. [Google Scholar]
- Mathia, F.; Marchalín, Š.; Végh, D.; Bobošíková, M.; Halinkovičová, M. Laboratory and industrial synthesis of remifentanil. Acta Chim. Slovaca 2012, 5, 145–152. [Google Scholar] [CrossRef][Green Version]
- Vellemäe, E.; Mastitski, A.; Järv, J.; Veli Hiltunen, J. A Convenient Methanolysis in the Synthesis of Carfentanyl. Org. Prep. Proced. Int. 2018, 50, 522–526. [Google Scholar] [CrossRef]
- Marton, J.; Glaenzel, B.; Roessler, J.; Golaszewski, D.; Henriksen, G. A Convenient Route to 4-Carboxy-4-Anilidopiperidine Esters and Acids. Molecules 2012, 17, 2823–2832. [Google Scholar] [CrossRef] [PubMed]
- Walz, A.J.; Hsu, F.-L. Synthesis of 4-anilinopiperidine methyl esters, intermediates in the production of carfentanil, sufentanil, and remifentanil. Tetrahedron Lett. 2014, 55, 501–502. [Google Scholar] [CrossRef]
- Li, J.; Zhang, T.; Sun, J.; Ren, F.; Jia, H.; Yu, Z.; Cheng, J.; Shi, W. Synthesis and evaluation of peptide–fentanyl analogue conjugates as dual μ/δ-opioid receptor agonists for the treatment of pain. Chin. Chem. Lett. 2021, in press. [Google Scholar] [CrossRef]
- Ivanovic, M.; Micovic, I.V.; Vuckovic, S.; Prostran, M.; Todorovic, Z.; Kricojevic, V.D.; Djordjevic, J.B.; Dosen-Micovic, L. The synthesis and preliminary pharmacological evaluation of racemic cis and trans 3-alkylfentanyl analogues. J. Serbian Chem. Soc. 2004, 69, 511–526. [Google Scholar] [CrossRef]
- Jevtić, I.; Došen-Mićović, L.; Ivanović, E.; Todorović, N.; Ivanović, M. Synthesis of Orthogonally Protected (±)-3-Amino-4-anilidopiperidines and (±)-3-N-Carbomethoxyfentanyl. Synthesis (Stuttg) 2017, 49, 3126–3136. [Google Scholar]
- Mićović, I.V.; Roglić, G.M.; Ivanović, M.D.; Došen-Mićović, L.; Kiricojević, V.D.; Popović, J.B. The synthesis of lactam analogues of fentanyl. J. Chem. Soc. Perkin Trans. 1996, 1, 2041–2050. [Google Scholar] [CrossRef]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- Lee, Y.; Basith, S.; Choi, S. Recent Advances in Structure-Based Drug Design Targeting Class A G Protein-Coupled Receptors Utilizing Crystal Structures and Computational Simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the µ-opioid receptor–Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.F.J.; Jarończyk, M.; Dobrowolski, J.C.; Sadlej, J. Molecular dynamics of fentanyl bound to μ-opioid receptor. J. Mol. Model. 2019, 25, 144. [Google Scholar] [CrossRef]
- Vo, Q.N.; Mahinthichaichan, P.; Shen, J.; Ellis, C.R. How μ-opioid receptor recognizes fentanyl. Nat. Commun. 2021, 12, 984. [Google Scholar] [CrossRef]
- Mahinthichaichan, P.; Vo, Q.N.; Ellis, C.R.; Shen, J. Kinetics and Mechanism of Fentanyl Dissociation from the μ-Opioid Receptor. JACS Au 2021, 1, 2208–2215. [Google Scholar] [CrossRef]
- Ricarte, A.; Dalton, J.A.R.; Giraldo, J. Structural Assessment of Agonist Efficacy in the μ-Opioid Receptor: Morphine and Fentanyl Elicit Different Activation Patterns. J. Chem. Inf. Model. 2021, 61, 1251–1274. [Google Scholar] [CrossRef]
- Lešnik, S.; Hodošček, M.; Bren, U.; Stein, C.; Bondar, A.-N. Potential Energy Function for Fentanyl-Based Opioid Pain Killers. J. Chem. Inf. Model. 2020, 60, 3566–3576. [Google Scholar] [CrossRef] [PubMed]
- Jarończyk, M.; Lipiński, P.F.J.; Dobrowolski, J.C.; Sadlej, J. The FMO analysis of the molecular interaction of fentanyl derivatives with the μ-opioid receptor. Chem. Pap. 2017, 71, 1429–1443. [Google Scholar] [CrossRef]
- Eshleman, A.J.; Nagarajan, S.; Wolfrum, K.M.; Reed, J.F.; Nilsen, A.; Torralva, R.; Janowsky, A. Affinity, potency, efficacy, selectivity, and molecular modeling of substituted fentanyls at opioid receptors. Biochem. Pharmacol. 2020, 182, 114293. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hruby, V.J. Toward a Universal μ-Agonist Template for Template-Based Alignment Modeling of Opioid Ligands. ACS Omega 2019, 4, 17457–17476. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Betz, R.M.; Dror, R.O.; Kruse, A.C. Structural basis for σ1 receptor ligand recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Alon, A.; Lyu, J.; Braz, J.M.; Tummino, T.A.; Craik, V.; O’Meara, M.J.; Webb, C.M.; Radchenko, D.S.; Moroz, Y.S.; Huang, X.-P.; et al. Structures of the σ2 receptor enable docking for bioactive ligand discovery. Nature 2021, 600, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chapman, K.; Clark, L.D.; Shao, Z.; Borek, D.; Xu, Q.; Wang, J.; Rosenbaum, D.M. Crystal structure of the human NK 1 tachykinin receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 13264–13269. [Google Scholar] [CrossRef]
- Schöppe, J.; Ehrenmann, J.; Klenk, C.; Rucktooa, P.; Schütz, M.; Doré, A.S.; Plückthun, A. Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun. 2019, 10, 17. [Google Scholar] [CrossRef]
- Chen, S.; Lu, M.; Liu, D.; Yang, L.; Yi, C.; Ma, L.; Zhang, H.; Liu, Q.; Frimurer, T.M.; Wang, M.-W.; et al. Human substance P receptor binding mode of the antagonist drug aprepitant by NMR and crystallography. Nat. Commun. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Thom, C.; Ehrenmann, J.; Vacca, S.; Waltenspühl, Y.; Schöppe, J.; Medalia, O.; Plückthun, A. Structures of neurokinin 1 receptor in complex with G q and G s proteins reveal substance P binding mode and unique activation features. Sci. Adv. 2021, 7, eabk2872. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.A.; Faust, B.; Gondin, A.B.; Dämgen, M.A.; Suomivuori, C.-M.; Veldhuis, N.A.; Cheng, Y.; Dror, R.O.; Thal, D.M.; Manglik, A. Selective G protein signaling driven by substance P–neurokinin receptor dynamics. Nat. Chem. Biol. 2022, 18, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; McCorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.-P.; Dror, R.O.; Shoichet, B.K.; et al. D 4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cao, C.; He, L.; Wang, X.; Zhang, X.C. Crystal structure of dopamine receptor D4 bound to the subtype selective ligand, L745870. Elife 2019, 8, e48822. [Google Scholar] [CrossRef] [PubMed]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Won Han, G.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Huang, S.; Mao, C.; Krumm, B.E.; Zhou, X.E.; Tan, Y.; Huang, X.-P.; Liu, Y.; Shen, D.-D.; Jiang, Y.; et al. Structures of the human dopamine D3 receptor-Gi complexes. Mol. Cell 2021, 81, 1147–1159.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Tan, L.; Chen, Z.; Qi, J.; Nie, F.; Luo, Z.; Cheng, J.; Wang, S. Haloperidol bound D2 dopamine receptor structure inspired the discovery of subtype selective ligands. Nat. Commun. 2020, 11, 1074. [Google Scholar] [CrossRef]
- Yin, J.; Chen, K.-Y.M.; Clark, M.J.; Hijazi, M.; Kumari, P.; Bai, X.; Sunahara, R.K.; Barth, P.; Rosenbaum, D.M. Structure of a D2 dopamine receptor–G-protein complex in a lipid membrane. Nature 2020, 584, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Im, D.; Inoue, A.; Fujiwara, T.; Nakane, T.; Yamanaka, Y.; Uemura, T.; Mori, C.; Shiimura, Y.; Kimura, K.T.; Asada, H.; et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat. Commun. 2020, 11, 6442. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.-P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942.e18. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Krishna Kumar, K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.D.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.E.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, D.; Shen, L.; Li, F.; Li, Y.; Yang, L.; Xu, T.; Tao, H.; Yao, D.; Wu, L.; et al. A Genetically Encoded F-19 NMR Probe Reveals the Allosteric Modulation Mechanism of Cannabinoid Receptor 1. J. Am. Chem. Soc. 2021, 143, 16320–16325. [Google Scholar] [CrossRef]
















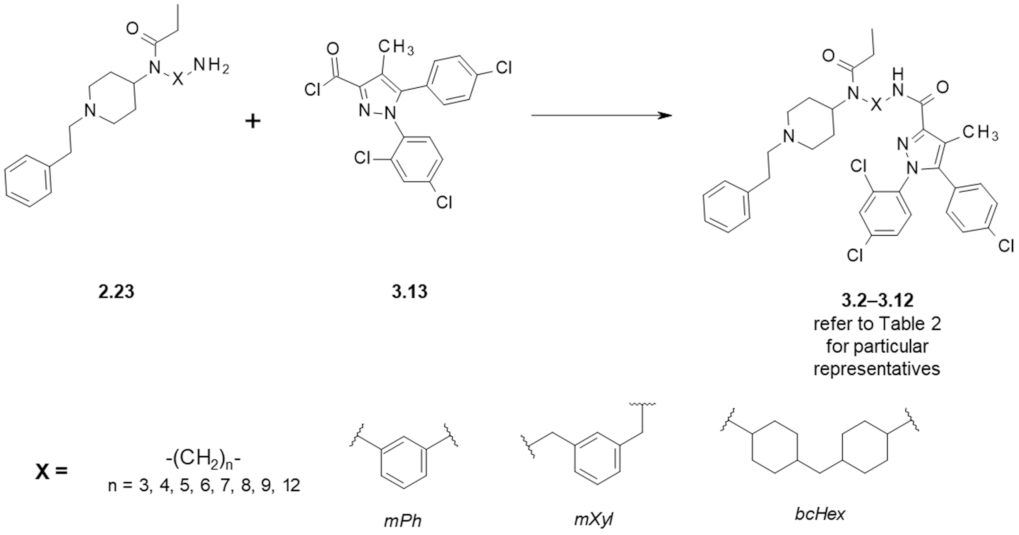
| Structure (Refer to Figure 4) | Affinity [Ki (μM)] 1 | Antagonistic Properties (an Agonist’s EC50 in μM, [35S]GTPγS Binding Assay 2, in Presence of 10 μM of a Tested Compound) | |||
|---|---|---|---|---|---|
| Compound | X | MOR 3 | CB1R 4 | Fentanyl (Alone) EC50 = 0.28 ± 0.04 μM | WIN55,212-2 (Alone) EC50 = 1.1 ± 0.22 μM |
| Fentanyl (1.1) | - | 0.003 ± 0.001 | - | - | - |
| Rimonabant (3.1) | - | 0.20 ± 0.12 5 | 0.004 ± 0.002 | - | 21 ± 3 |
| Naloxone | - | - | - | 456 ± 60 | 1.3 ± 0.17 |
| 3.2 | -(CH2)3- | 3.81 ± 0.39 | 0.19 ± 0.07 | - | - |
| 3.3 | -(CH2)4- | 1.23 ± 0.43 | 0.57 ± 0.20 | 24 ± 5 | 33 ± 8 |
| 3.4 | -(CH2)5- | 0.17 ± 0.10 | >10 | - | - |
| 3.5 | -(CH2)6- | 0.30 ± 0.06 | 2.29 ± 1.86 | 33 ± 2 | 21 ± 2 |
| 3.6 | -(CH2)7- | 6.54 ± 0.95 | 0.70 ± 0.57 | 3 ± 1 | 16 ± 2 |
| 3.7 | -(CH2)8- | 1.24 ± 0.79 | 3.99 ± 1.37 | - | - |
| 3.8 | -(CH2)9- | 0.11 ± 0.06 | >10 | - | - |
| 3.9 | -(CH2)12- | 6.90 ± 1.58 | >10 | - | - |
| 3.10 | mPh | 1.25 ± 0.67 | >10 | - | - |
| 3.11 | mXyl | 1.02 ± 0.25 | >10 | - | - |
| 3.12 | bcHex | 0.66 ± 0.37 | 2.06 ± 0.60 | - | - |
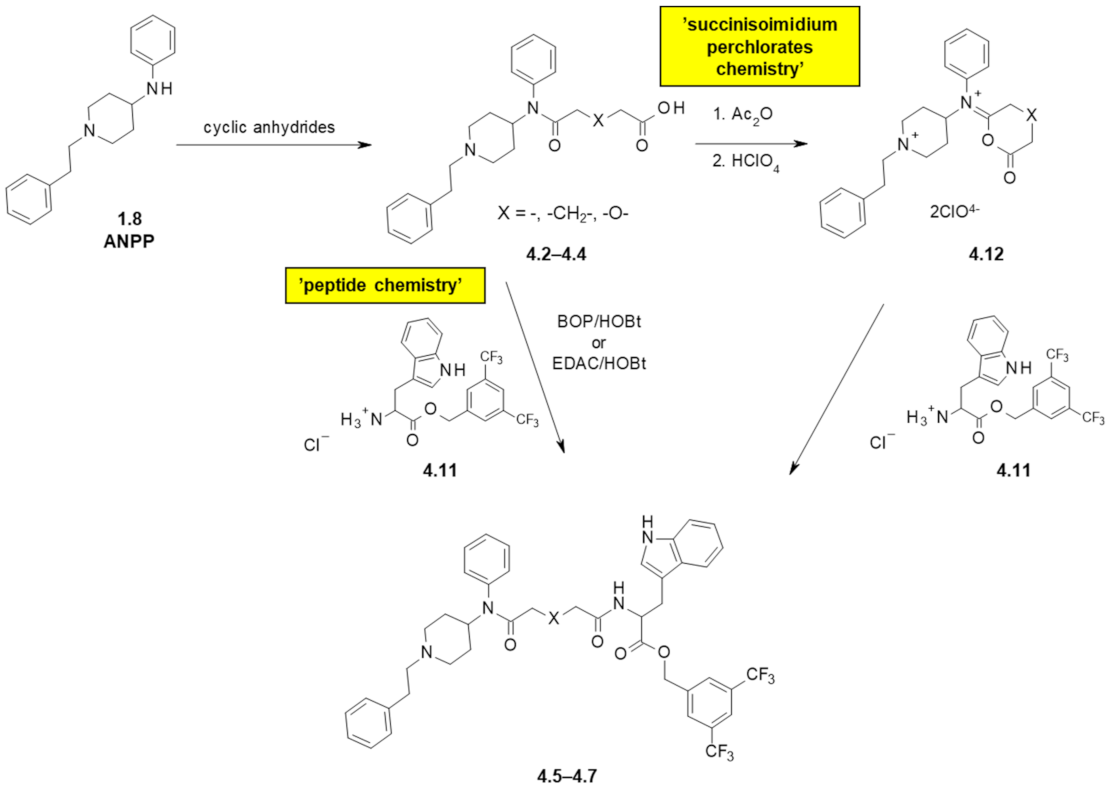
| Structure | Affinity [Ki (nM)] 1 | Functional Tests (Inhibition of the Contractile Response Generated by Electrical Stimulation) | ||||
|---|---|---|---|---|---|---|
| X (Refer to Figure 5) | MOR 2 | NK1R 3 | MOR (GPI/LM/MP 4) | DOR (MVD 5) | NK1R (GPI/LM/MP 4) | |
| Opioid Agonism, Inhibition of Contraction Height (% at 1 μM or IC50 in nM) | Antagonism of SP Action 6 [Ke ± SEM (nM)] (at 1 μM) | |||||
| Covalently linked compounds | ||||||
| 4.5 | - | 130 | 13 | 410 ± 42 nM | 14% | 240 ± 39 |
| 4.6 | -CH2- | 120 | 6.8 | 55 ± 12 nM | 13% | 21 ± 4.3 (at 100 nM) |
| 4.7 | -O- | 400 | 31 | 30% | 30% | 480 ± 12 |
| Ionic pairs | ||||||
| 4.8 | - | >10,000 | 21 | 1.8% | 4.3% | 210 ± 42 |
| 4.9 | -CH2- | 3900 | 44 | 19.5% | 19.8% | 500 ± 130 |
| 4.10 | -O- | 1300 | 23 | 11% | 17.1% | 490 ± 68 |
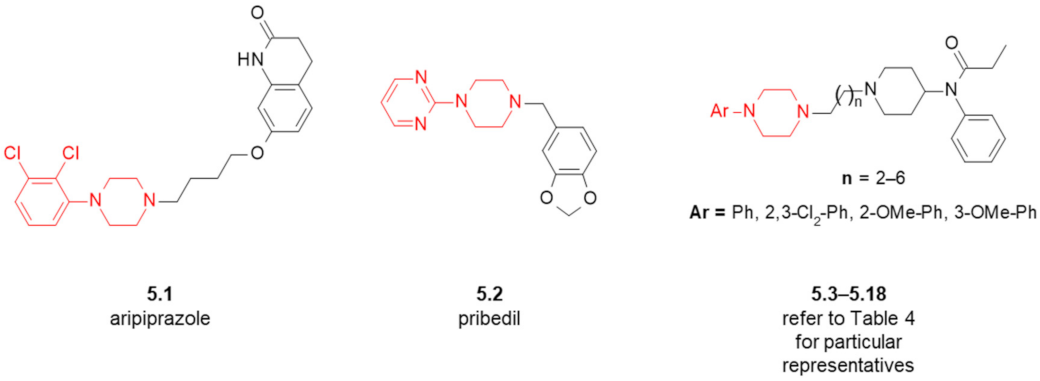
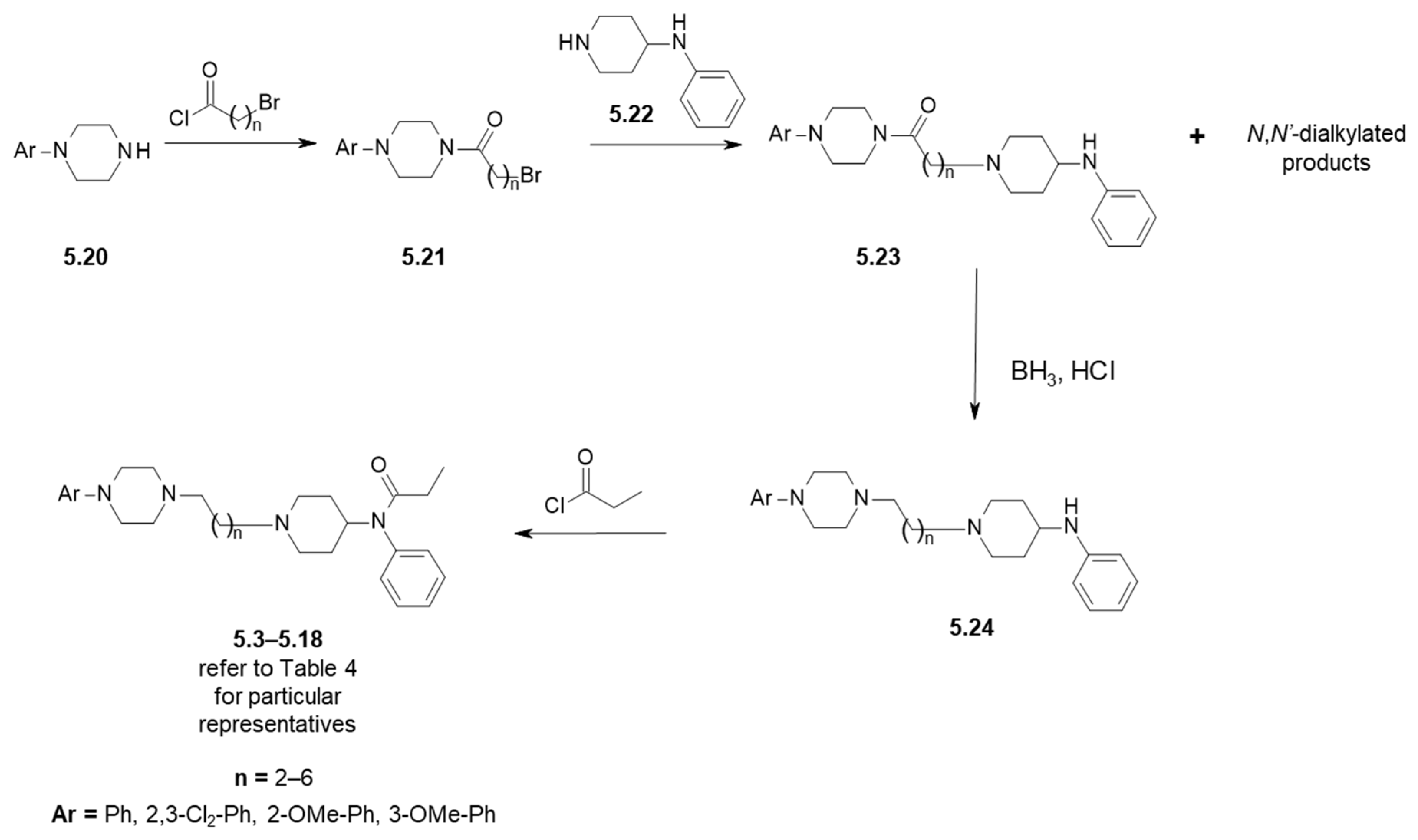
| Compound | Ar (Refer to Figure 6) | n | D2R Receptor Binding, [Ki (nM)] 1 |
|---|---|---|---|
| 5.3 | Ph | 2 | 869 |
| 5.4 | 2-OMe-Ph | 2 | 800 |
| 5.5 | 2,3-Cl2-Ph | 2 | 594 |
| 5.6 | Ph | 3 | 1357 |
| 5.7 | 2-OMe-Ph | 3 | 7992 |
| 5.8 | 3-OMe-Ph | 3 | n/d 2 |
| 5.9 | 2,3-Cl2-Ph | 3 | 6956 |
| 5.10 | Ph | 4 | 7083 |
| 5.11 | 2-OMe-Ph | 4 | 4436 |
| 5.12 | 2,3-Cl2-Ph | 4 | 2376 |
| 5.13 | Ph | 5 | 8105 |
| 5.14 | 2-OMe-Ph | 5 | 3778 |
| 5.15 | 2,3-Cl2-Ph | 5 | 1500 |
| 5.16 | Ph | 6 | 1853 |
| 5.17 | 2-OMe-Ph | 6 | 5454 |
| 5.18 | 2,3-Cl2-Ph | 6 | 5326 |
| Compound | Structure (Refer to Figure 7) | Functional Tests (Inhibition of the Contractile Response Generated by Electrical Stimulation) | ||
|---|---|---|---|---|
| DOR (MVD 1) | MOR (GPI/LM/MP 2) | |||
| R1 | R2 | Opioid Agonism, Inhibition of Contraction Height (% at 1 μM or IC50 in nM) | ||
| 6.4 | Me | H | 17.9% | 0.7% |
| 6.5 | PhCH2 | H | IC50 = 1266 ± 355 nM | IC50 = 5164 ± 2043 nM |
| 6.6 | PhCH2CH2 | H | 19.5% | 3.1% |
| 6.7 | Me | OMe | 2.8% | 0% |
| 6.8 | PhCH2 | OMe | 8.3% | 3% |
| 6.9 | PhCH2CH2 | OMe | 0% | 6% |
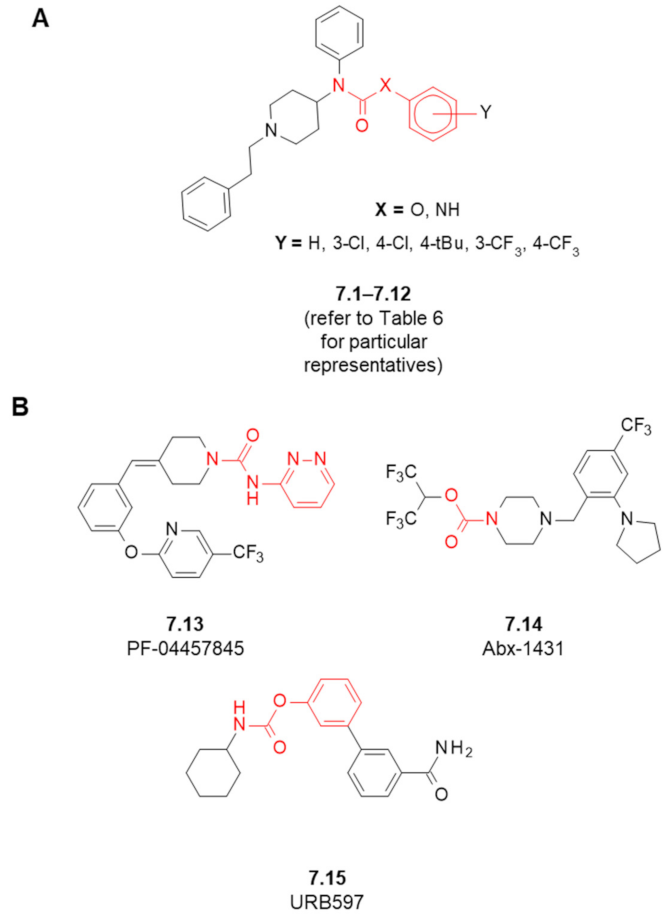
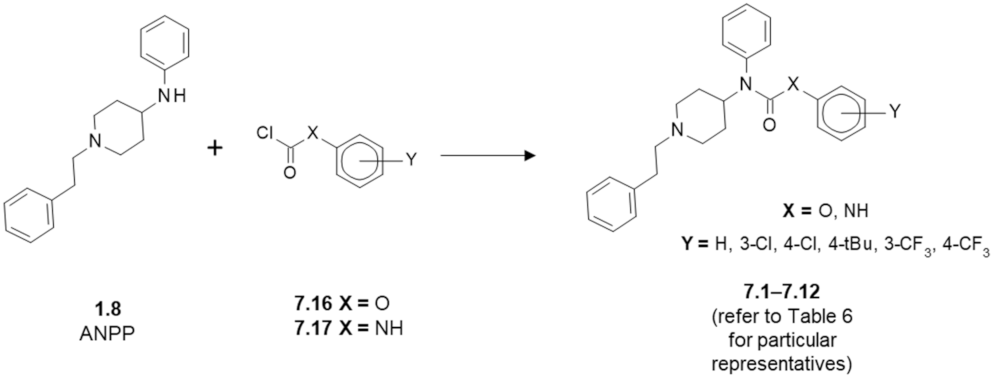
| Structure (Refer to Figure 8) | Maximal Inhibition at 10 μM [%] | ||||
|---|---|---|---|---|---|
| Compound | X | Y | MOR Affinity [IC50 (nM)] 1 | FAAH 2 | MAGL 3 |
| fentanyl (1.1) | - | - | 5.99 | n/d 4 | n/d |
| 7.1 | O | - | 1442 | 0 | 0 |
| 7.2 | O | 4-Cl | 2180 | 0 | 0 |
| 7.3 | O | 3-Cl | 654.2 | 1.90 | 0 |
| 7.4 | O | 4-tBu | 1830 | 10.2 | 0.74 |
| 7.5 | O | 4-CF3 | 2657 | 7.40 | 0 |
| 7.6 | O | 3-CF3 | 4093 | 7.40 | 0.41 |
| 7.7 | NH | - | 516.8 | 7.25 | 0 |
| 7.8 | NH | 4-Cl | 665.1 | 8.26 | 3.50 |
| 7.9 | NH | 3-Cl | 658.9 | 9.76 | 7.32 |
| 7.10 | NH | 4-tBu | 23,050 | 5.82 | 1.28 |
| 7.11 | NH | 4-CF3 | 4031 | 7.49 | 0 |
| 7.12 | NH | 3-CF3 | 1204 | 9.16 | 2.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipiński, P.F.J.; Matalińska, J. Fentanyl Structure as a Scaffold for Opioid/Non-Opioid Multitarget Analgesics. Int. J. Mol. Sci. 2022, 23, 2766. https://doi.org/10.3390/ijms23052766
Lipiński PFJ, Matalińska J. Fentanyl Structure as a Scaffold for Opioid/Non-Opioid Multitarget Analgesics. International Journal of Molecular Sciences. 2022; 23(5):2766. https://doi.org/10.3390/ijms23052766
Chicago/Turabian StyleLipiński, Piotr F. J., and Joanna Matalińska. 2022. "Fentanyl Structure as a Scaffold for Opioid/Non-Opioid Multitarget Analgesics" International Journal of Molecular Sciences 23, no. 5: 2766. https://doi.org/10.3390/ijms23052766
APA StyleLipiński, P. F. J., & Matalińska, J. (2022). Fentanyl Structure as a Scaffold for Opioid/Non-Opioid Multitarget Analgesics. International Journal of Molecular Sciences, 23(5), 2766. https://doi.org/10.3390/ijms23052766