
Low-Dose Delta-9-Tetrahydrocannabinol as Beneficial Treatment for Aged APP/PS1 Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
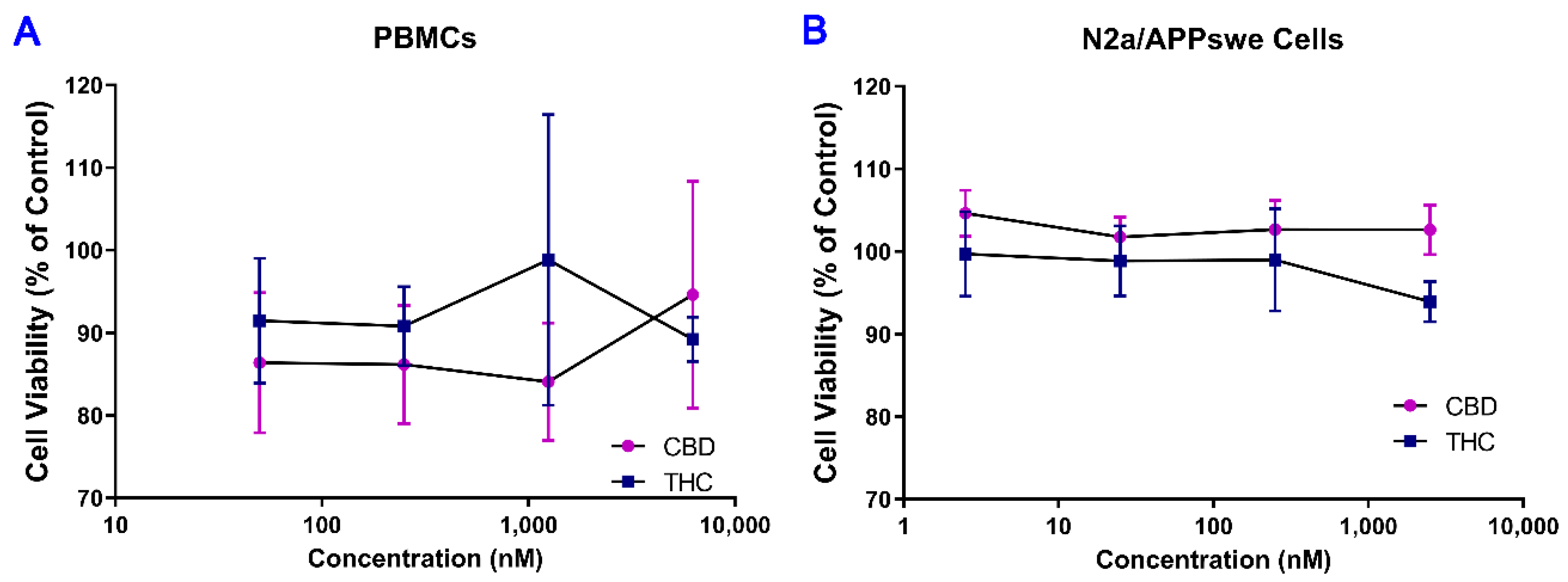
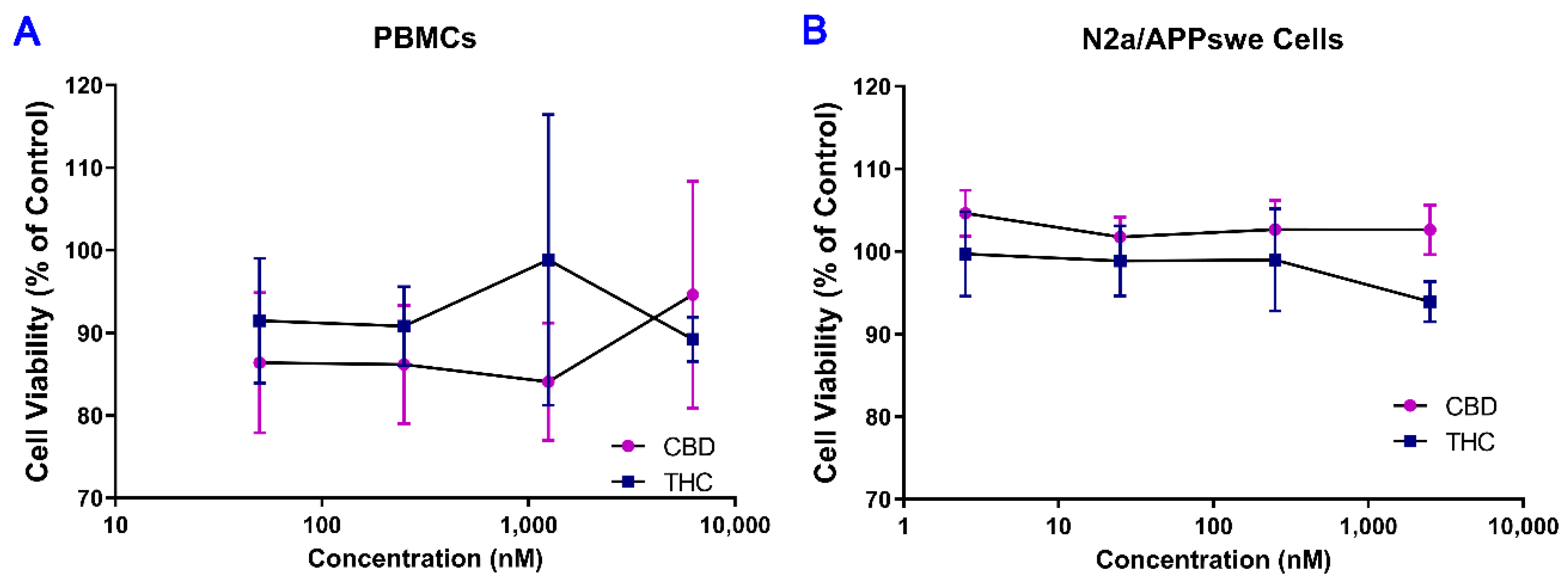
2.1. No Cytotoxic Effect of THC and CBD Observed in Human PBMCs and Mouse N2a/APPswe
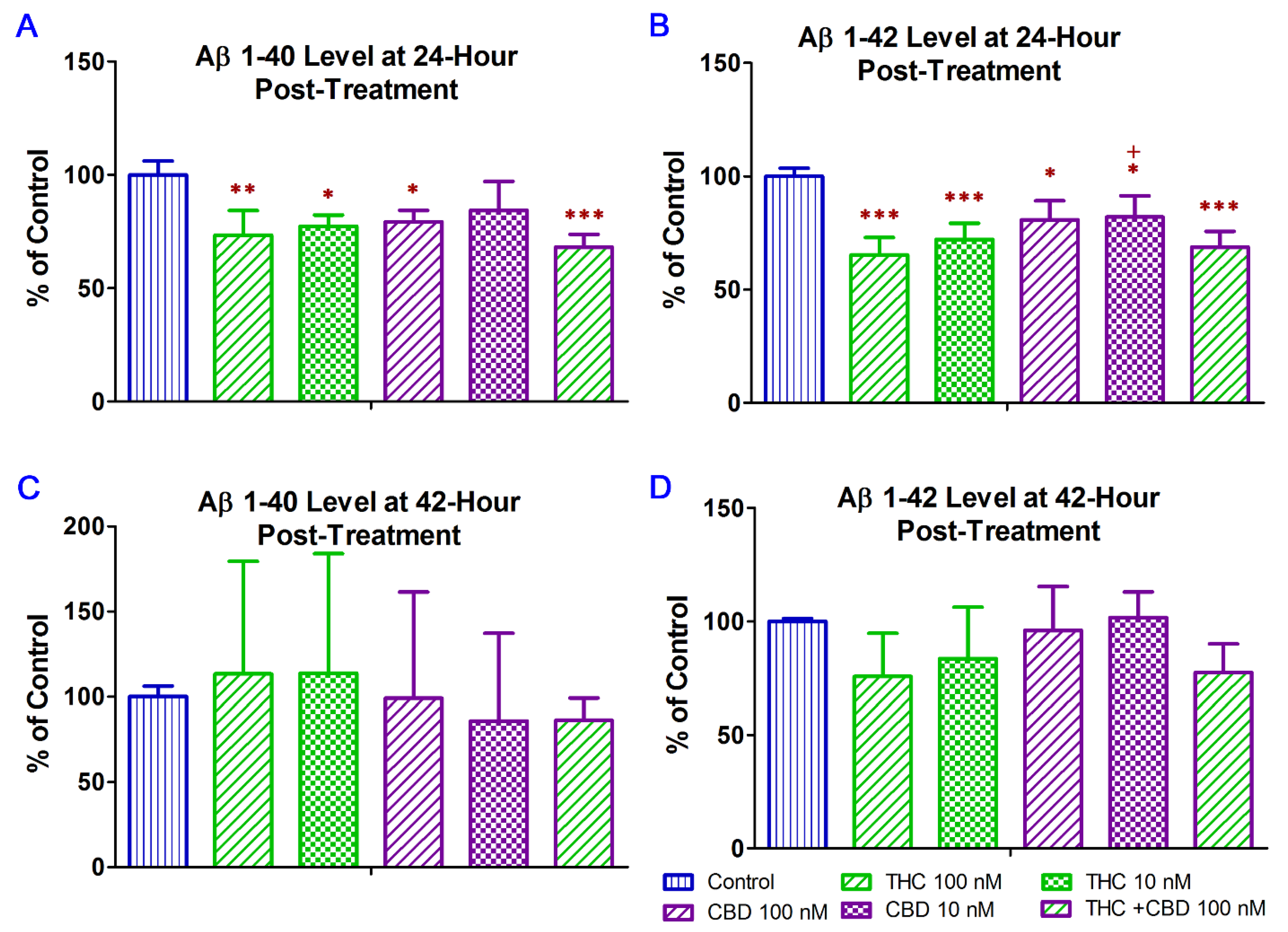
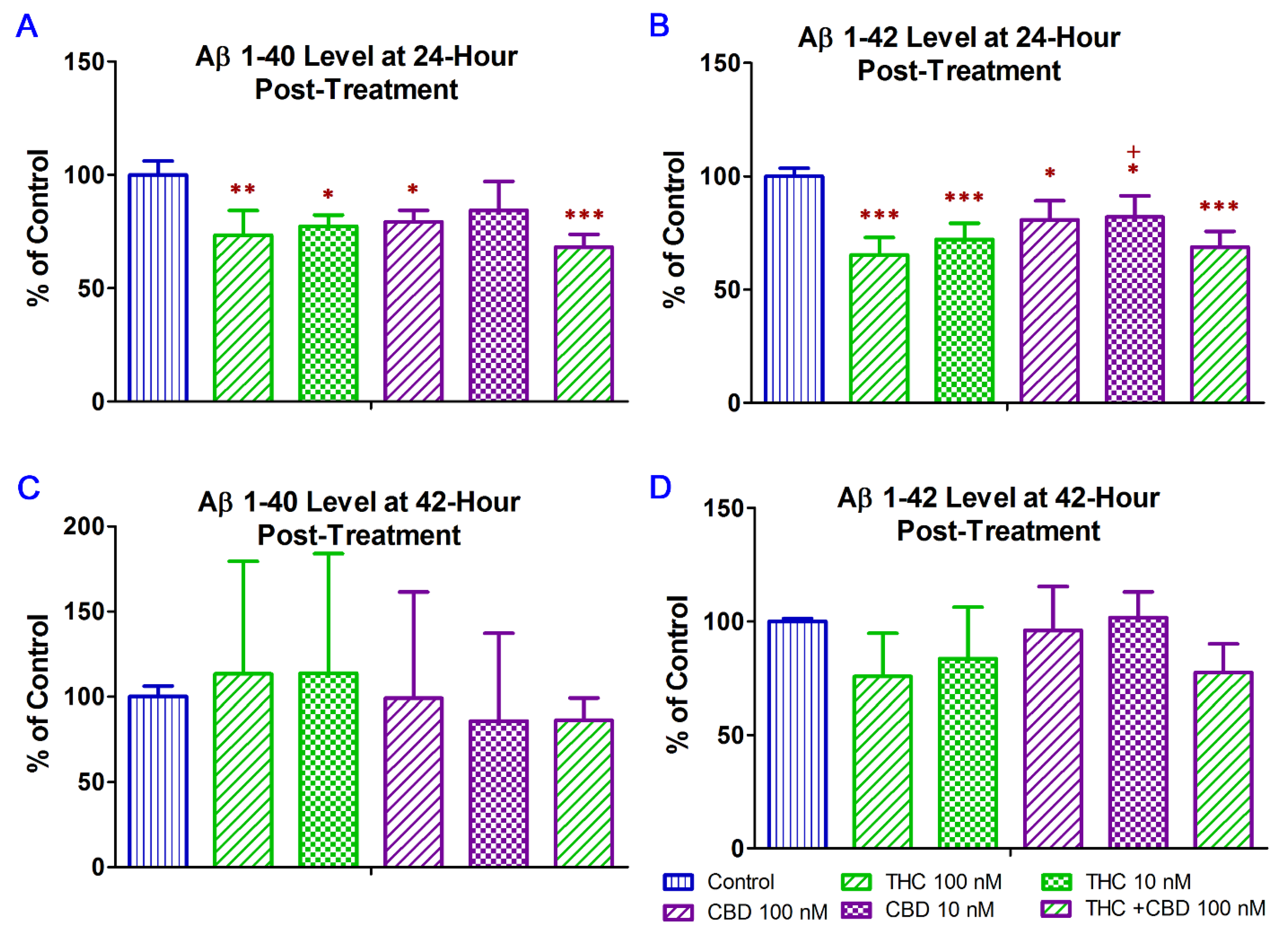
2.2. THC and CBD Reduced the Production of Aβ1–42 Peptide in N2a/APPswe Cells
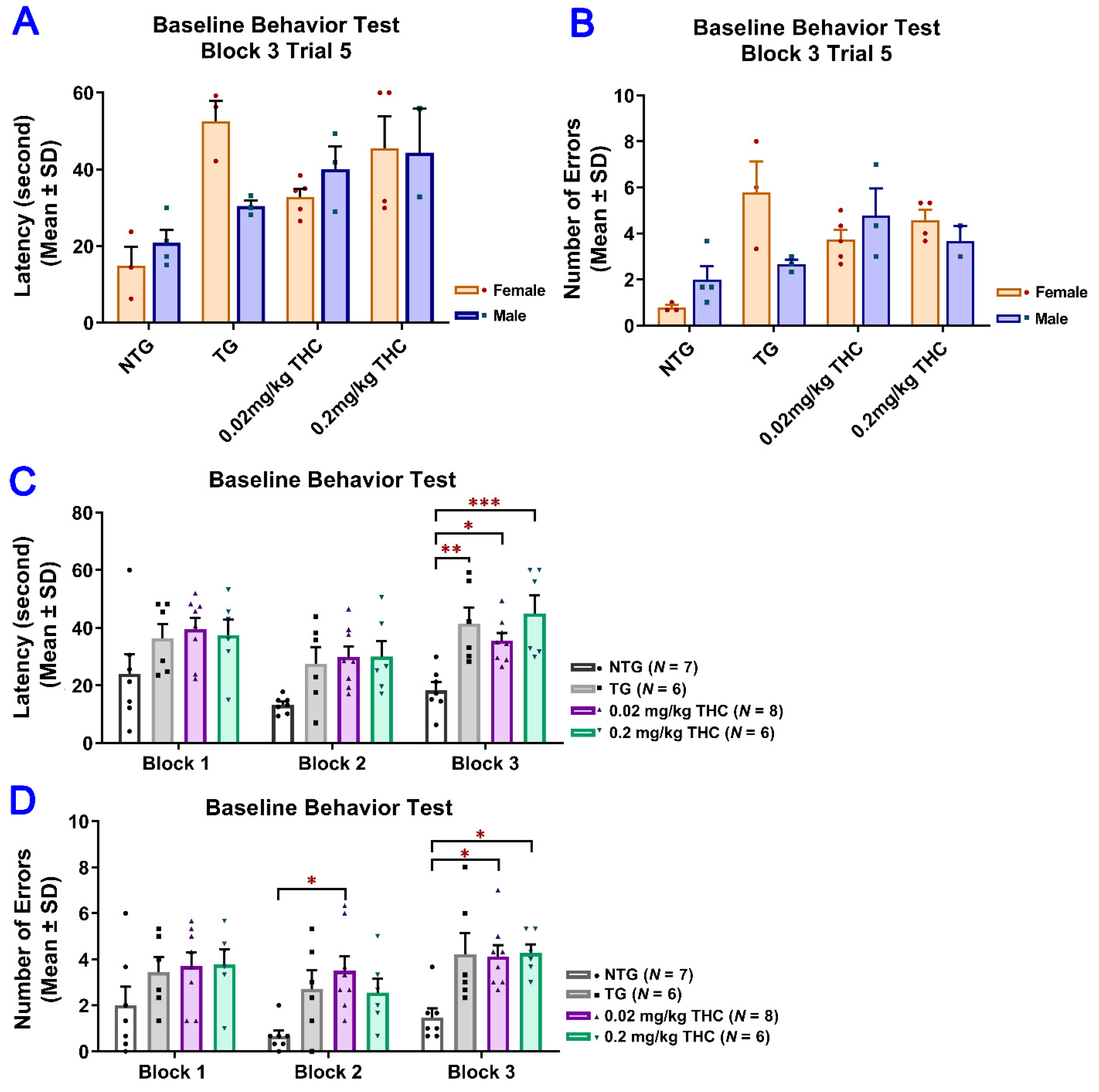
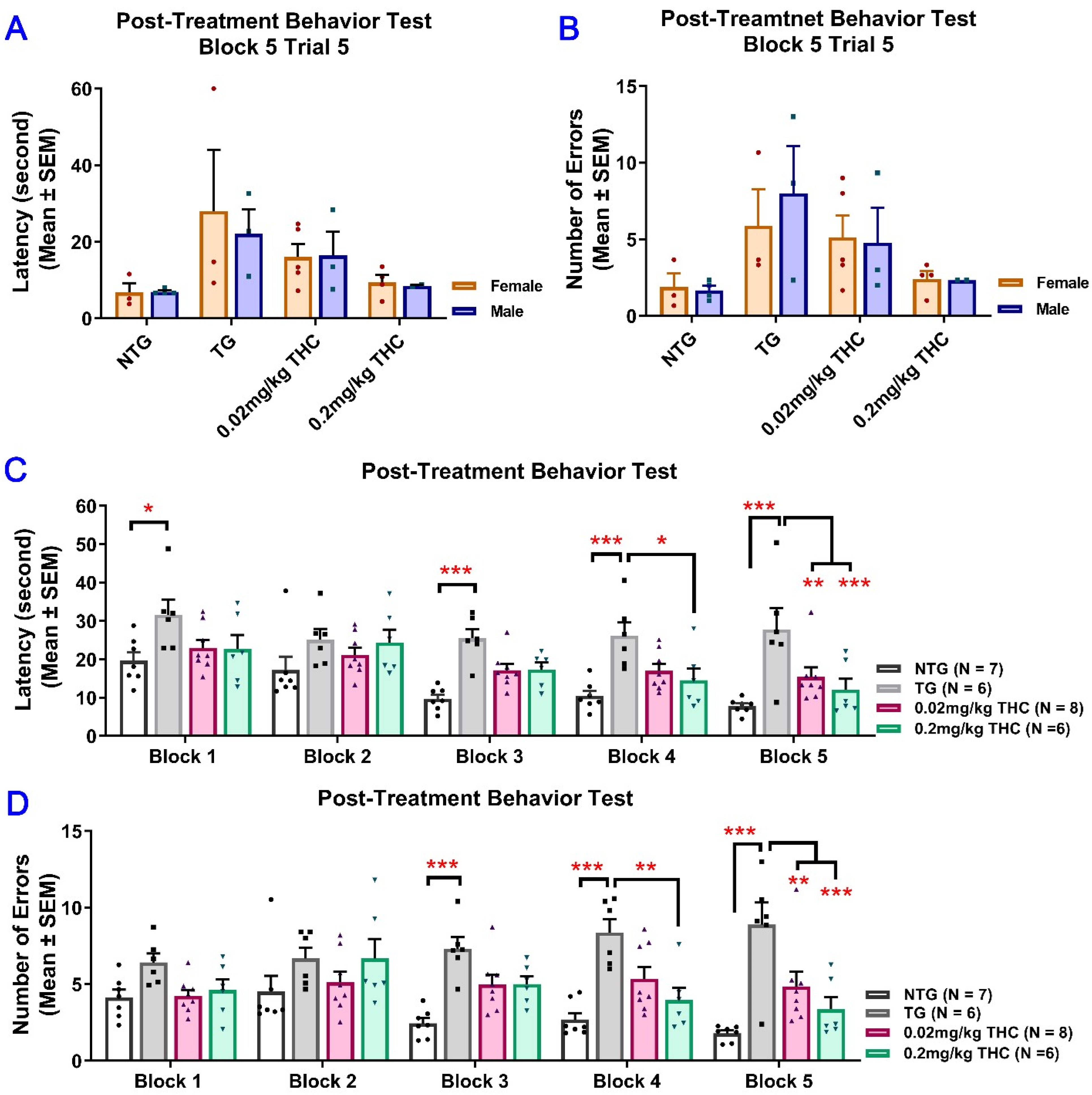
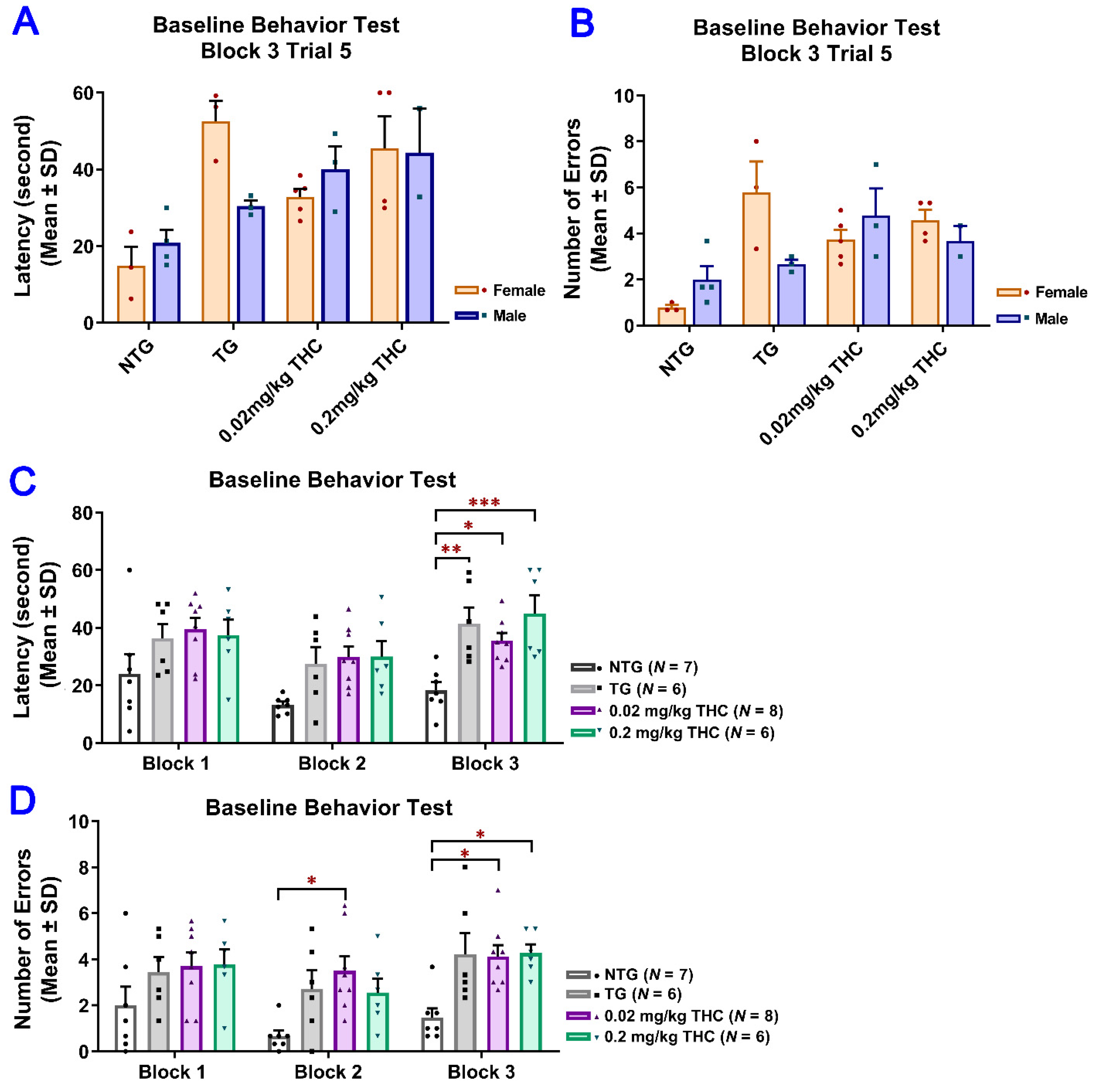
2.3. THC Treatment Improved Impaired Spatial Memory Performance of Aged APP/PS1 Mice
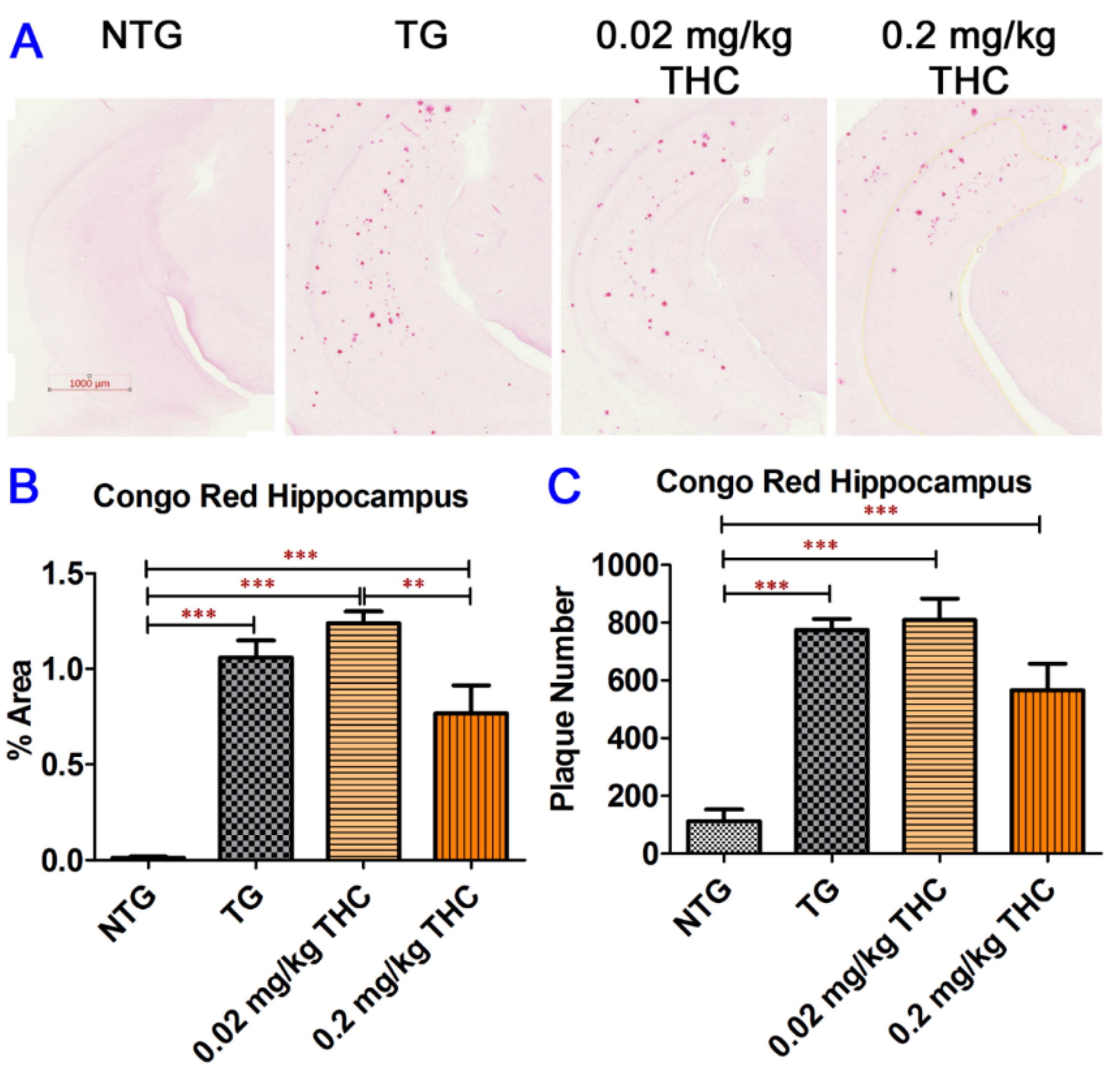
2.4. THC Treatment Resulted in Decreased Oligomeric Aβ Levels in Hippocampi of APP/PS1 Mice
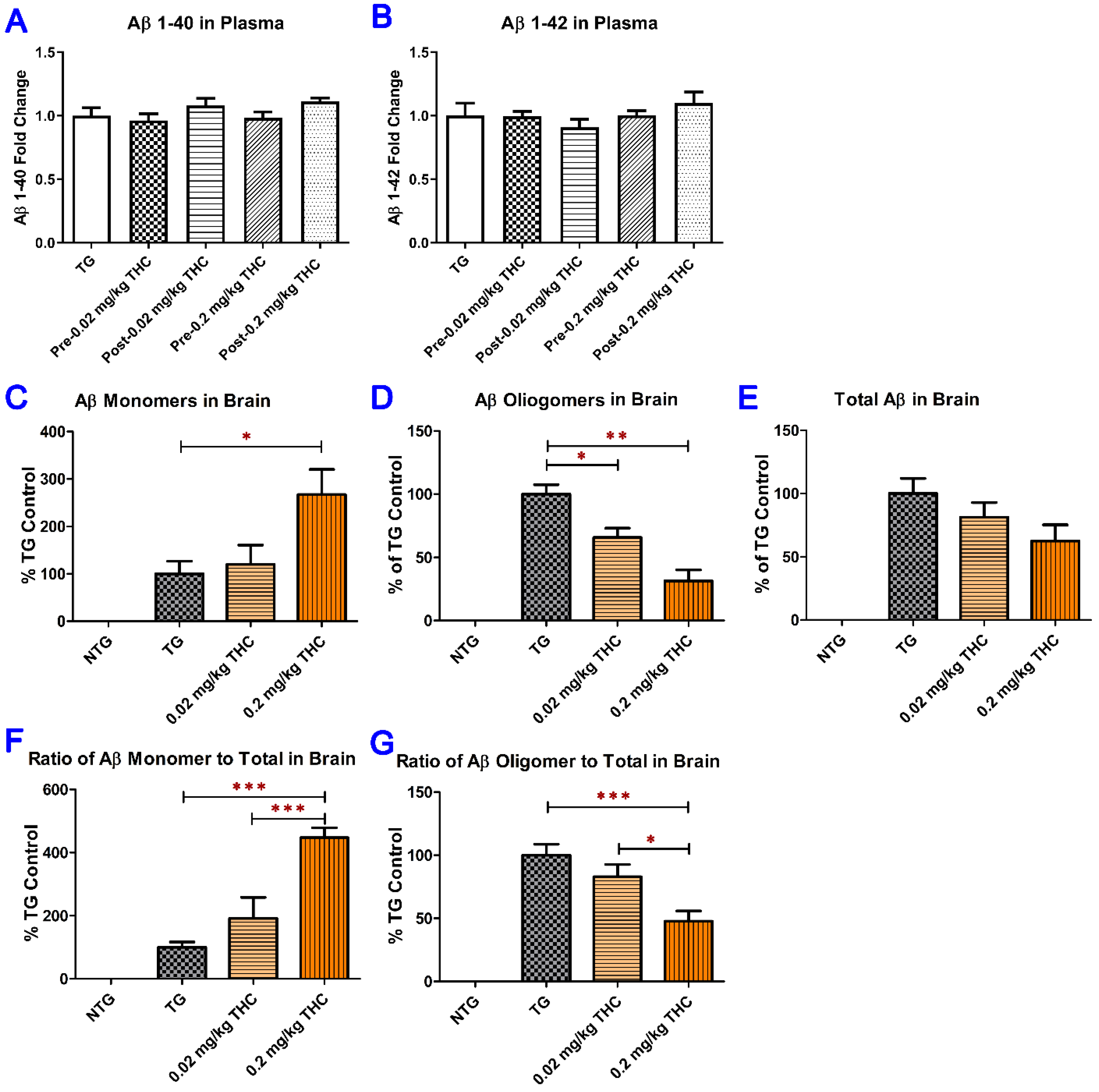
2.5. THC Treatment Reduced Oligomeric Aβ and Increased Monomeric Aβ Levels in Brain Tissues but Had No Significant Effect on Soluble and Insoluble Aβ Peptide Levels in Plasma
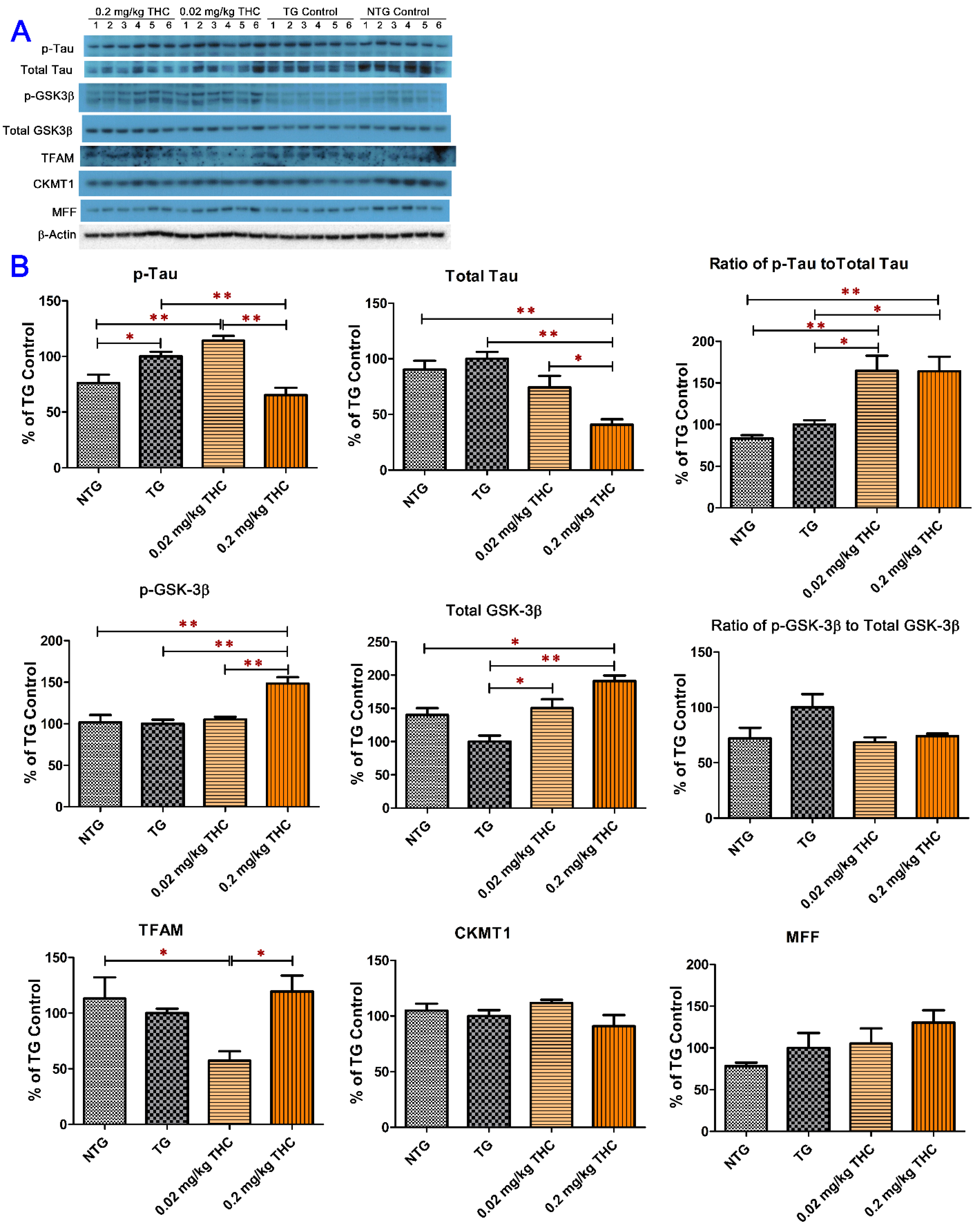
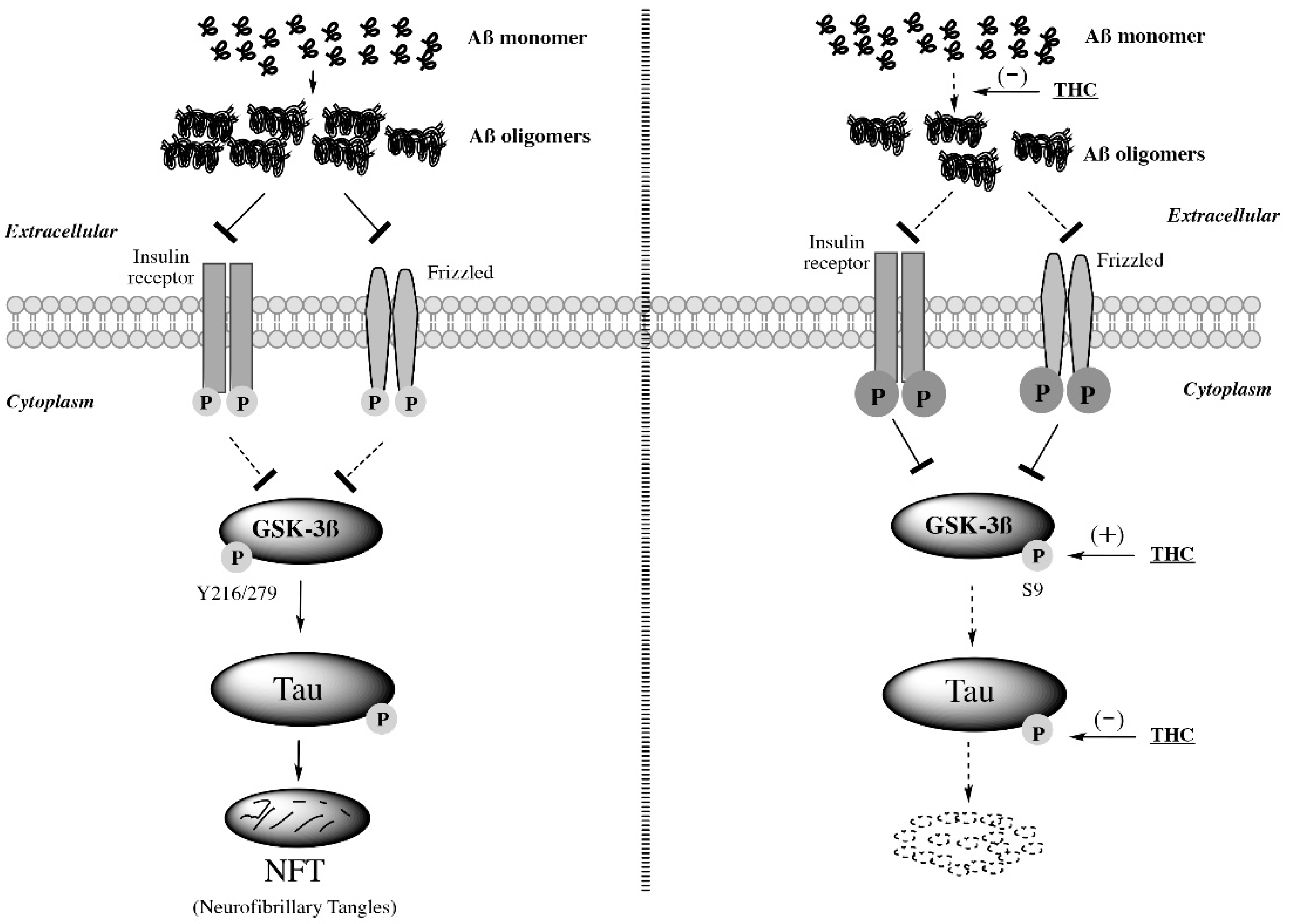
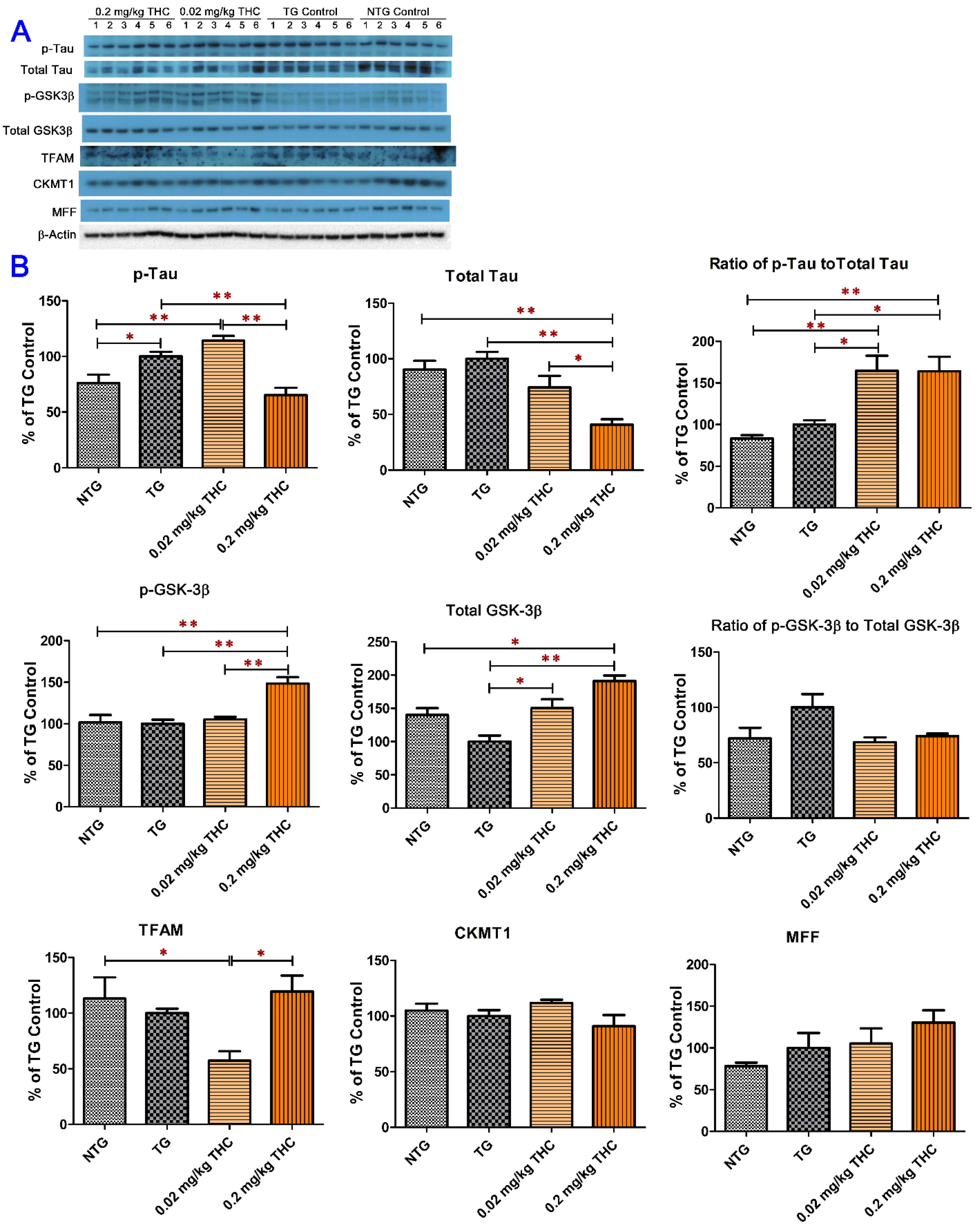
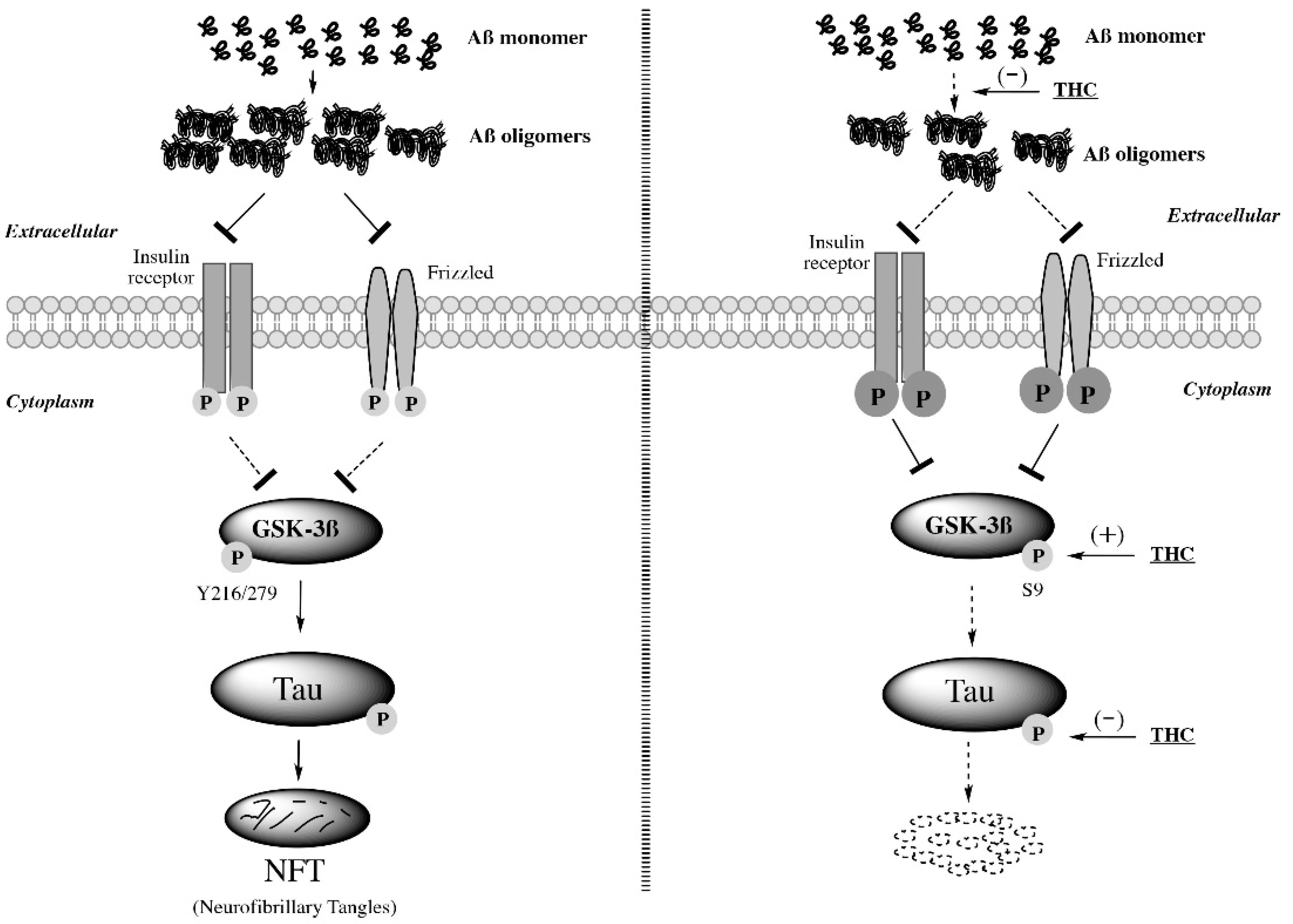
2.6. THC Treatment Reduced Expression of Phospho-Tau and Total Tau and Decreased the Activity of GSK-3β through Increasing Expression of Phospho-GSK3β at Ser9 in APP/PS1 Brain Tissues
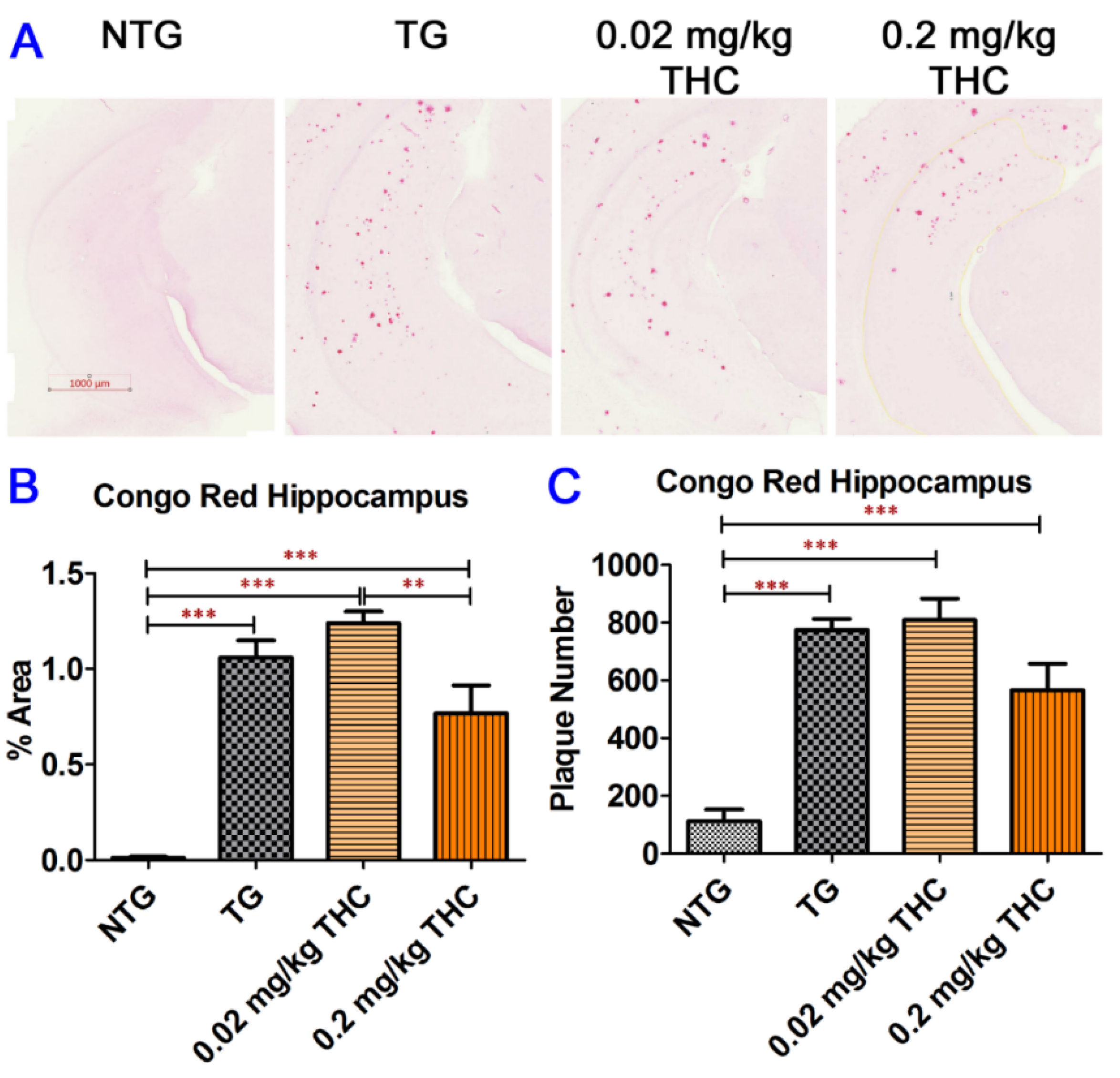
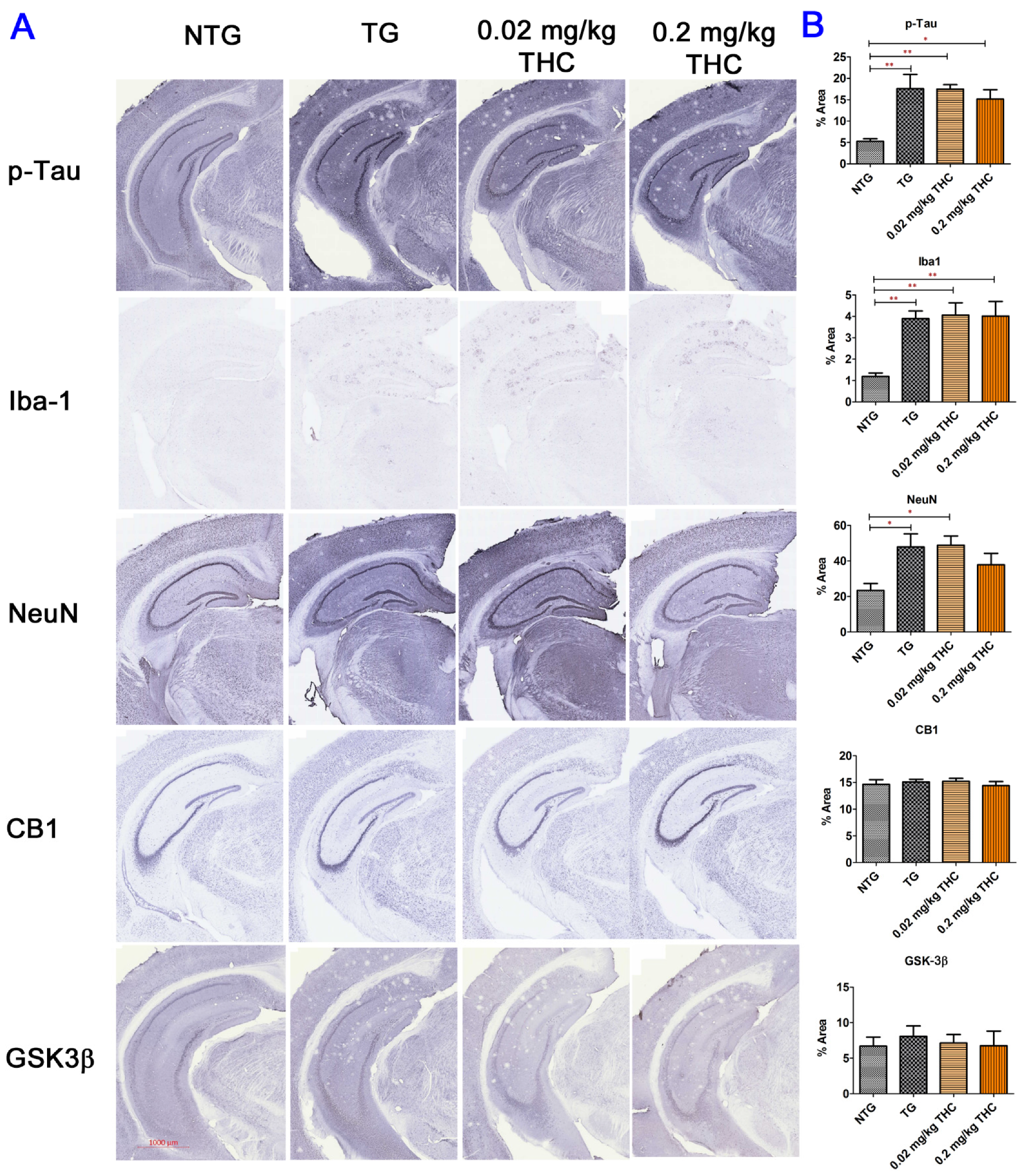
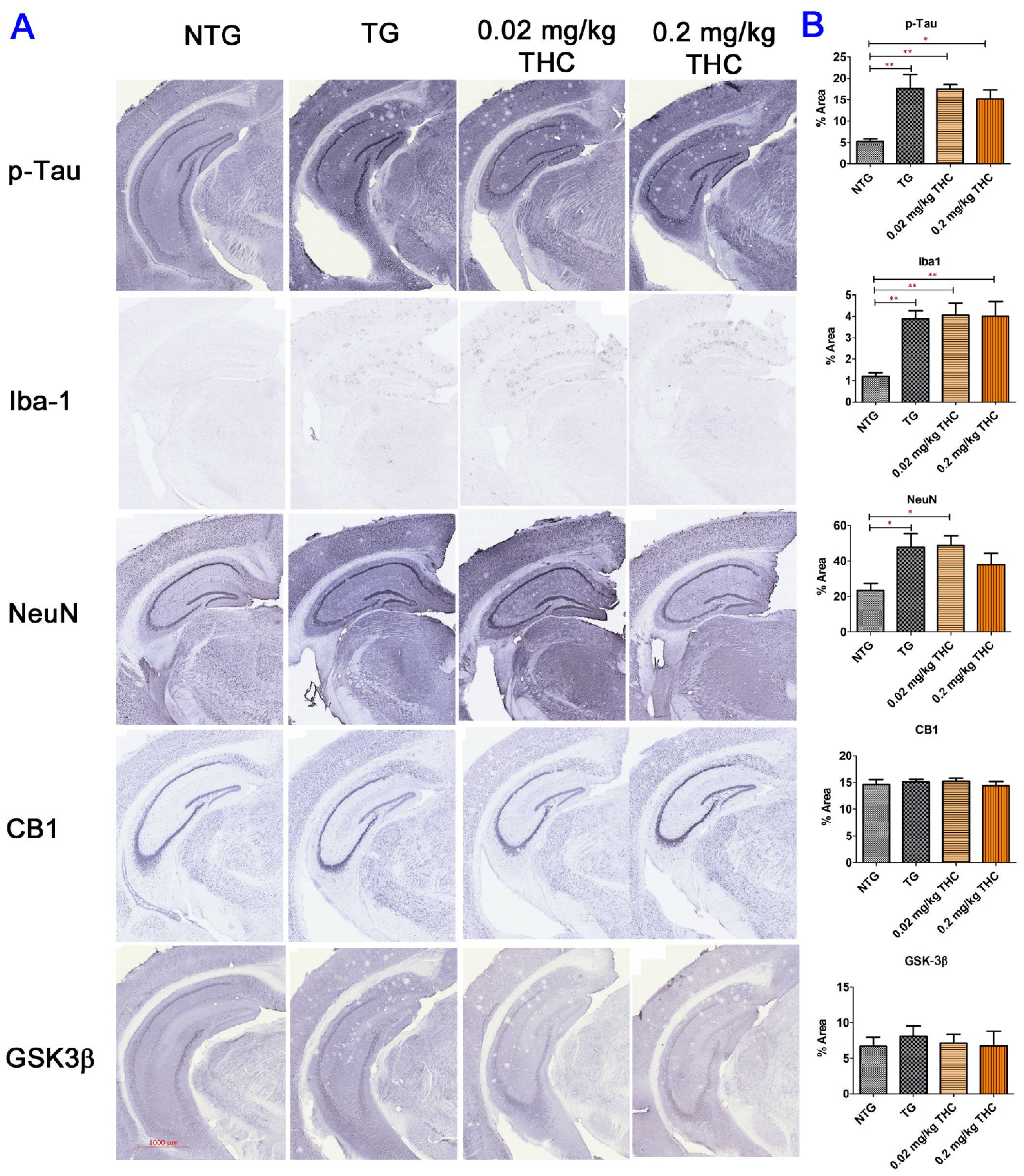
2.7. THC Treatment Had Little Effect on Neuropathologic Changes in the Hippocampi of APP/PS1 Mice
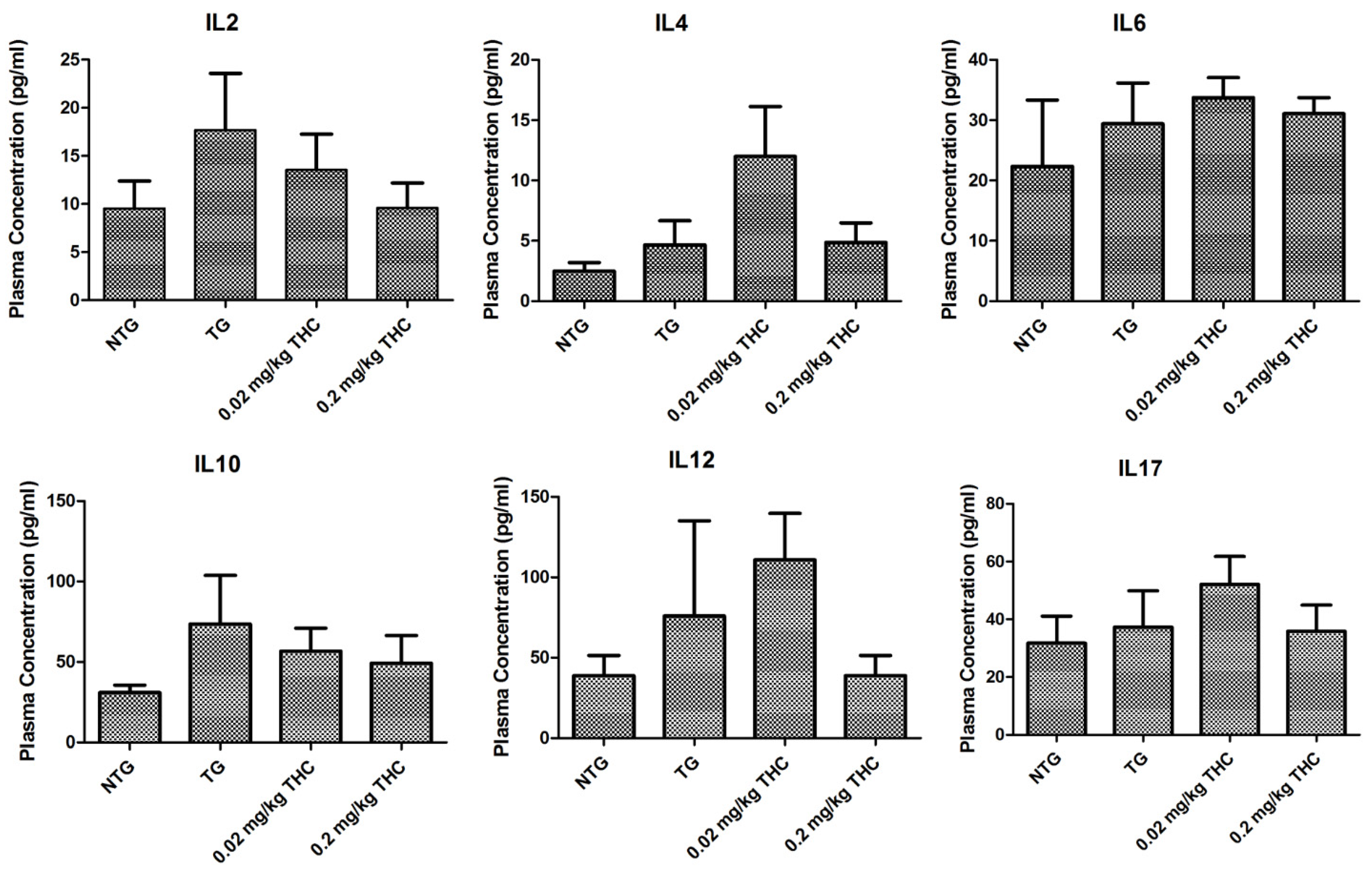
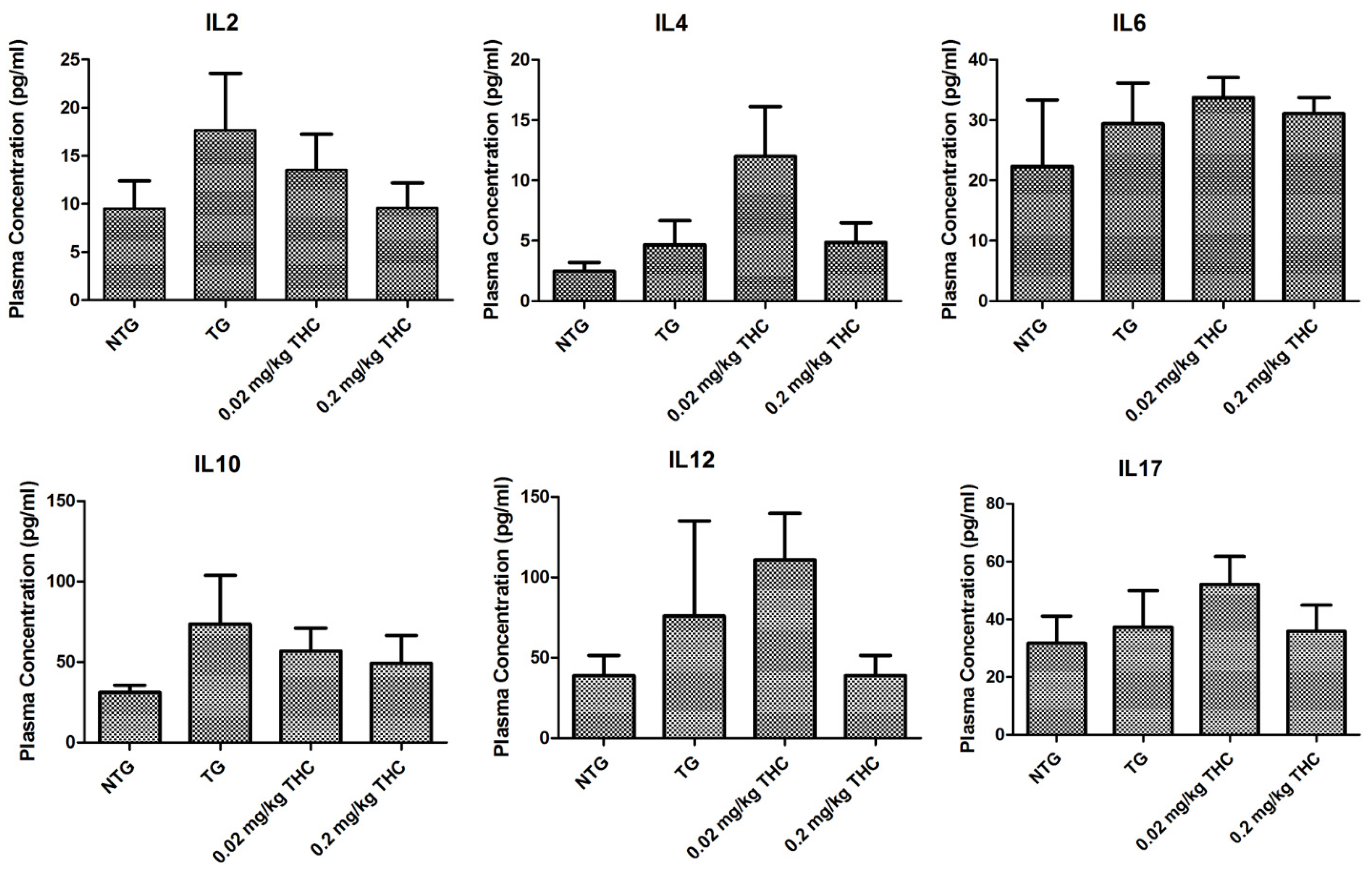
2.8. THC Treatment Had Little Effect on Cytokine Levels in Plasma
3. Discussion
4. Materials and Methods
4.1. Materials, Cell Line, and Animals
4.2. In Vitro Cytotoxicity Assay
4.3. Determination of Aβ Level Using Sandwich Enzyme Linked Immunosorbent Assay (ELISA)
4.4. In Vivo Treatment Protocol
4.5. Radial Arm Water Maze
4.6. Protein and Peptide Extraction
4.7. Semi-Quantitative Western Blotting Analysis of Brain Tissue Homogenate Samples
4.8. Alkaline Congo Red Staining
4.9. Immunohistochemistry (IHC)
4.10. Measurement of Plasma Cytokine Levels Using Multiplex Assay
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mechoulam, R.; Hanuš, L.O.; Pertwee, R.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Bisson, J.; Singh, G.; Graham, J.G.; Chen, S.-N.; Friesen, J.B.; Dahlin, J.L.; Niemitz, M.; Walters, M.A.; Pauli, G.F. The Essential Medicinal Chemistry of Cannabidiol (CBD). J. Med. Chem. 2020, 63, 12137–12155. [Google Scholar] [CrossRef]
- Ashton, C.H. Pharmacology and effects of cannabis: A brief review. Br. J. Psychiatry J. Ment. Sci. 2001, 178, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.; Iuvone, T. The marijuana component cannabidiol inhibits β-amyloid-induced tau protein hyperphosphorylation through Wnt/β-catenin pathway rescue in PC12 cells. Klin. Wochenschr. 2006, 84, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Maroon, J.; Bost, J. Review of the neurological benefits of phytocannabinoids. Surg. Neurol. Int. 2018, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Corrada, M.M.; Brookmeyer, R.; Paganini-Hill, A.; Berlau, D.; Kawas, C.H. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010, 67, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanco, J.C.; Li, C.; Bodea, L.-G.; Mármol, R.M.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Ewers, M.; Sperling, R.A.; Klunk, W.E.; Weiner, M.W.; Hampel, H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 2011, 34, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The Murine Version of BAN2401 (mAb158) Selectively Reduces Amyloid-β Protofibrils in Brain and Cerebrospinal Fluid of tg-ArcSwe Mice. J. Alzheimer’s Dis. 2014, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Watt, G.; Karl, T. In vivo Evidence for Therapeutic Properties of Cannabidiol (CBD) for Alzheimer’s Disease. Front. Pharmacol. 2017, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janefjord, E.; Maag, J.L.; Harvey, B.S.; Smid, S.D. Cannabinoid Effects on β Amyloid Fibril and Aggregate Formation, Neuronal and Microglial-Activated Neurotoxicity In Vitro. Cell. Mol. Neurobiol. 2013, 34, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol Promotes Amyloid Precursor Protein Ubiquitination and Reduction of Beta Amyloid Expression in SHSY5Y APP+ Cells Through PPARγ Involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Maiuri, M.C.; De Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in β-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-κB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and Other Cannabinoids Reduce Microglial Activation In Vitro and In Vivo: Relevance to Alzheimer’s Disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Harvey, B.S.; Ohlsson, K.S.; Maag, J.; Musgrave, I.; Smid, S. Contrasting protective effects of cannabinoids against oxidative stress and amyloid-β evoked neurotoxicity in vitro. NeuroToxicology 2012, 33, 138–146. [Google Scholar] [CrossRef]
- Cheng, D.; Spiro, A.S.; Jenner, A.M.; Garner, B.; Karl, T. Long-Term Cannabidiol Treatment Prevents the Development of Social Recognition Memory Deficits in Alzheimer’s Disease Transgenic Mice. J. Alzheimer’s Dis. 2014, 42, 1383–1396. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L., Jr.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L. Cannabidiol in vivo blunts β-amyloid induced neuroinflammation by suppressing IL-1β and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol Reduces Aβ-Induced Neuroinflammation and Promotes Hippocampal Neurogenesis through PPARγ Involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Aso, E.; Sánchez-Pla, A.; Vegas-Lozano, E.; Maldonado, R.; Ferrer, I. Cannabis-Based Medicine Reduces Multiple Pathological Processes in AβPP/PS1 Mice. J. Alzheimer’s Dis. 2014, 43, 977–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aso, E.; Andrés-Benito, P.; Ferrer, I. Delineating the Efficacy of a Cannabis-Based Medicine at Advanced Stages of Dementia in a Murine Model. J. Alzheimer’s Dis. 2016, 54, 903–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casarejos, M.J.; Perucho, J.; Gomez, A.; Muñoz, M.P.; Fernandez-Estevez, M.; Sagredo, O.; Ruiz, J.F.; Guzman, M.; de Yebenes, J.G.; Mena, M.A. Natural Cannabinoids Improve Dopamine Neurotransmission and Tau and Amyloid Pathology in a Mouse Model of Tauopathy. J. Alzheimer’s Dis. 2013, 35, 525–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.; Li, Y.; Liu, H.; Bai, G.; Mayl, J.; Lin, X.; Sutherland, K.; Nabar, N.; Cai, J. The Potential Therapeutic Effects of THC on Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 42, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Bilkei-Gorzo, A.; Albayram, O.; Draffehn, A.; Michel, K.; Piyanova, A.; Oppenheimer, H.; Dvir-Ginzberg, M.; Rácz, I.; Ulas, T.; Imbeault, S.; et al. A chronic low dose of Δ9-tetrahydrocannabinol (THC) restores cognitive function in old mice. Nat. Med. 2017, 23, 782–787. [Google Scholar] [CrossRef]
- Nidadavolu, P.; Bilkei-Gorzo, A.; Krämer, M.; Schürmann, B.; Palmisano, M.; Beins, E.C.; Madea, B.; Zimmer, A. Efficacy of Δ9-Tetrahydrocannabinol (THC) Alone or in Combination with a 1:1 Ratio of Cannabidiol (CBD) in Reversing the Spatial Learning Deficits in Old Mice. Front. Aging Neurosci. 2021, 13, 560. [Google Scholar] [CrossRef] [PubMed]
- Sarne, Y.; Toledano, R.; Rachmany, L.; Sasson, E.; Doron, R. Reversal of age-related cognitive impairments in mice by an extremely low dose of tetrahydrocannabinol. Neurobiol. Aging 2018, 61, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Diamond, D.M.; Gottschall, P.E.; Ugen, K.E.; Dickey, C.A.; Hardy, J.; Duff, K.; Jantzen, P.T.; Dicarlo, G.; Wilcock, D.M.; et al. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 2000, 408, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Gordon, M.N.; Diamond, D.M.; Austin, L.A.; Hatcher, J.M.; Jantzen, P.; Dicarlo, G.; Wilcock, D.; Morgan, D. Behavioral Assessment of Alzheimer’s Transgenic Mice Following Long-Term Aβ Vaccination: Task Specificity and Correlations between Aβ Deposition and Spatial Memory. DNA Cell Biol. 2001, 20, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Takio, K.; Ogawara, M.; Selkoe, D.J. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J. Biol. Chem. 1992, 267, 17082–17086. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The carboxy terminus of the beta. amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Lintner, F.; Jellinger, K.A. Amyloid β peptide 1-42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol. 2004, 107, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Loring, J.F.; Montgomery, J.; Gordon, M.N.; Eastman, P.S.; Morgan, D. Selectively Reduced Expression of Synaptic Plasticity-Related Genes in Amyloid Precursor Protein + Presenilin-1 Transgenic Mice. J. Neurosci. 2003, 23, 5219–5226. [Google Scholar] [CrossRef] [Green Version]
- Ewers, M.; Morgan, D.G.; Gordon, M.N.; Woodruff-Pak, D.S. Associative and motor learning in 12-month-old transgenic APP+PS1 mice. Neurobiol. Aging 2006, 27, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Takashima, A.; Noguchi, K.; Murayama, M.; Sato, M.; Kondo, S.; Saitoh, Y.; Ishiguro, K.; Hoshino, T.; Imahori, K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 2719–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksenov, M.Y.; Aksenova, M.V.; Payne, R.M.; Smith, C.D.; Markesbery, W.R.; Carney, J.M. The Expression of Creatine Kinase Isoenzymes in Neocortex of Patients with Neurodegenerative Disorders: Alzheimer’s and Pick’s Disease. Exp. Neurol. 1997, 146, 458–465. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 57. [Google Scholar] [CrossRef]
- Motta, M.; Imbesi, R.; Di Rosa, M.; Stivala, F.; Malaguarnera, L. Altered plasma cytokine levels in Alzheimer’s disease: Correlation with the disease progression. Immunol. Lett. 2007, 114, 46–51. [Google Scholar] [CrossRef]
- Crean, R.D.; Crane, N.A.; Mason, B.J. An Evidence-Based Review of Acute and Long-Term Effects of Cannabis Use on Executive Cognitive Functions. J. Addict. Med. 2011, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, P. Toxic Effects of Cannabis and Cannabinoids: Animal Data. Pain Res. Manag. 2005, 10, 23A–26A. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasten, C.R.; Zhang, Y.; Boehm, S.L.I. Acute Cannabinoids Produce Robust Anxiety-Like and Locomotor Effects in Mice, but Long-Term Consequences Are Age- and Sex-Dependent. Front. Behav. Neurosci. 2019, 13, 32. [Google Scholar] [CrossRef] [Green Version]
- Garai, K.; Sureka, R.; Maiti, S. Detecting Amyloid-β Aggregation with Fiber-Based Fluorescence Correlation Spectroscopy. Biophys. J. 2007, 92, L55–L57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Lu, B. Synapses and Dendritic Spines as Pathogenic Targets in Alzheimer’s Disease. Neural Plast. 2012, 2012, 247150. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, P.A.; Kinstrie, R.; Sibbet, G.; Rawjee, T.; Morrice, N.; Cleghon, V. A Chaperone-Dependent GSK3β Transitional Intermediate Mediates Activation-Loop Autophosphorylation. Mol. Cell 2006, 24, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Brennesvik, E.O.; Lai, Y.-C.; Shepherd, P.R. GSK-3β regulation in skeletal muscles by adrenaline and insulin: Evidence that PKA and PKB regulate different pools of GSK-3. Cell. Signal. 2007, 19, 204–210. [Google Scholar] [CrossRef]
- Krishnankutty, A.; Kimura, T.; Saito, T.; Aoyagi, K.; Asada, A.; Takahashi, S.-I.; Ando, K.; Ohara-Imaizumi, M.; Ishiguro, K.; Hisanaga, S.-I. In vivo regulation of glycogen synthase kinase 3β activity in neurons and brains. Sci. Rep. 2017, 7, 8602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdesian, M.H.; Carvalho, M.M.V.F.; Mendes, F.A.; Saraiva, L.M.; Juliano, M.A.; Juliano, L.; Garcia-Abreu, J.; Ferreira, S.T. Amyloid-β Binds to the Extracellular Cysteine-rich Domain of Frizzled and Inhibits Wnt/β-Catenin Signaling. J. Biol. Chem. 2008, 283, 9359–9368. [Google Scholar] [CrossRef] [Green Version]
- Takashima, A.; Murayama, M.; Murayama, O.; Kohno, T.; Honda, T.; Yasutake, K.; Nihonmatsu, N.; Mercken, M.; Yamaguchi, H.; Sugihara, S.; et al. Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc. Natl. Acad. Sci. USA 1998, 95, 9637–9641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, F.; Avila, J. The role of glycogen synthase kinase 3 in the early stages of Alzheimers’ disease. FEBS Lett. 2008, 582, 3848–3854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyllaert, D.; Kremer, A.; Jaworski, T.; Borghgraef, P.; Devijver, H.; Croes, S.; Dewachter, I.; Van Leuven, F. Glycogen synthase kinase-3β, or a link between amyloid and tau pathology? Genes, Brain Behav. 2008, 7, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.A.; Sun, Q.; Gamblin, T.C. Tau phosphorylation by GSK-3β promotes tangle-like filament morphology. Mol. Neurodegener. 2007, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Lloret, A.; Badia, M.-C.; Giraldo, E.; Ermak, G.; Alonso, M.-D.; Pallardó, F.V.; Davies, K.J.; Viña, J. Amyloid-β Toxicity and Tau Hyperphosphorylation are Linked Via RCAN1 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 701–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalecka-Franaszek, E.; Chuang, D.-M. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 8745–8750. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Kukat, C.; Larsson, N.-G. mtDNA makes a U-turn for the mitochondrial nucleoid. Trends Cell Biol. 2013, 23, 457–463. [Google Scholar] [CrossRef]
- Tachikawa, M.; Fukaya, M.; Terasaki, T.; Ohtsuki, S.; Watanabe, M. Distinct cellular expressions of creatine synthetic enzyme GAMT and creatine kinases uCK-Mi and CK-B suggest a novel neuron-glial relationship for brain energy homeostasis. Eur. J. Neurosci. 2004, 20, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Hemmer, W. Creatine kinase in non-muscle tissues and cells. Mol. Cell. Biochem. 1994, 133–134, 193–220. [Google Scholar] [CrossRef]
- Lewis, T.L.; Kwon, S.-K.; Lee, A.; Shaw, R.; Polleux, F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 2018, 9, 5008. [Google Scholar] [CrossRef] [Green Version]
- Cha, M.Y.; Chen, H.; Chan, D. Removal of the Mitochondrial Fission Factor Mff Exacerbates Neuronal Loss and Neurological Phenotypes in a Huntington’s Disease Mouse Model. PLoS Curr. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Wolff, V.; Schlagowski, A.-I.; Rouyer, O.; Charles, A.-L.; Singh, F.; Auger, C.; Schini-Kerth, V.; Marescaux, C.; Raul, J.-S.; Zoll, J.; et al. Tetrahydrocannabinol Induces Brain Mitochondrial Respiratory Chain Dysfunction and Increases Oxidative Stress: A Potential Mechanism Involved in Cannabis-Related Stroke. BioMed Res. Int. 2015, 2015, 323706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, V.J.; Mocatta, T.J.; Winterbourn, C.C.; Darlow, B.A.; Volpe, J.J.; E. Inder, T. The Relationship of CSF and Plasma Cytokine Levels to Cerebral White Matter Injury in the Premature Newborn. Pediatr. Res. 2005, 57, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Nam, S.M.; Yoo, D.-Y.; Jung, H.Y.; Kim, I.Y.; Hwang, I.K.; Seong, J.K.; Yoon, Y.S. Comparison of Adult Hippocampal Neurogenesis and Susceptibility to Treadmill Exercise in Nine Mouse Strains. Neural Plast. 2017, 2017, 5863258. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.-M.; Perez-Tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, D.; Hasan, S.; Tam, S.K.E.; Engle, S.J.; Brandon, N.J.; Sharp, T.; Foster, R.G.; Harrison, P.J.; Bannerman, D.; Peirson, S.N. d-amino acid oxidase knockout (Dao−/−) mice show enhanced short-term memory performance and heightened anxiety, but no sleep or circadian rhythm disruption. Eur. J. Neurosci. 2015, 41, 1167–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchett, D.; Taylor, A.M.; Barkus, C.; Engle, S.J.; Brandon, N.; Sharp, T.; Foster, R.G.; Harrison, P.J.; Peirson, S.N.; Bannerman, D. Searching for cognitive enhancement in the Morris water maze: Better and worse performance in D-amino acid oxidase knockout (Dao−/−) mice. Eur. J. Neurosci. 2016, 43, 979–989. [Google Scholar] [CrossRef] [Green Version]
- Alamed, J.; Wilcock, D.M.; Diamond, D.M.; Gordon, M.N.; Morgan, D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc. 2006, 1, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; King, D.L.; Gordon, M.N.; Morgan, D.; Hatcher, J.M.; Hope, C.E.; Diamond, D.M. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001, 891, 42–53. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Rojiani, A.; Rosenthal, A.; Subbarao, S.; Freeman, M.J.; Gordon, M.N.; Morgan, D. Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflammation 2004, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Vorhees, C.V.; Williams, M.T. Assessing Spatial Learning and Memory in Rodents. ILAR J. 2014, 55, 310–332. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D. Water Maze Tasks in Mice: Special Reference to Alzheimer’s Transgenic Mice. In Methods of Behavior Analysis in Neuroscience, 2nd ed.; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009. [Google Scholar]
- Gresack, J.E.; Frick, K.M. Male mice exhibit better spatial working and reference memory than females in a water-escape radial arm maze task. Brain Res. 2003, 982, 98–107. [Google Scholar] [CrossRef]
- Izco, M.; Pesini, P.; Pérez-Grijalba, V.; Fandos, N.; Sarasa, M. Optimized Protocol for Amyloid-β Extraction from the Brain. J. Alzheimer’s Dis. 2013, 34, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Puchtler, H.; Sweat, F. Amidoblack as a stain for hemoglobin. Arch. Pathol. 1962, 73, 245–249. [Google Scholar]
- Adams, J.C. Biotin amplification of biotin and horseradish peroxidase signals in histochemical stains. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1992, 40, 1457–1463. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Hong, Y.; Yan, J.; Brown, B.; Lin, X.; Zhang, X.; Shen, N.; Li, M.; Cai, J.; Gordon, M.; et al. Low-Dose Delta-9-Tetrahydrocannabinol as Beneficial Treatment for Aged APP/PS1 Mice. Int. J. Mol. Sci. 2022, 23, 2757. https://doi.org/10.3390/ijms23052757
Wang Y, Hong Y, Yan J, Brown B, Lin X, Zhang X, Shen N, Li M, Cai J, Gordon M, et al. Low-Dose Delta-9-Tetrahydrocannabinol as Beneficial Treatment for Aged APP/PS1 Mice. International Journal of Molecular Sciences. 2022; 23(5):2757. https://doi.org/10.3390/ijms23052757
Chicago/Turabian StyleWang, Yanhong, Yuzhu Hong, Jiyu Yan, Breanna Brown, Xiaoyang Lin, Xiaolin Zhang, Ning Shen, Minghua Li, Jianfeng Cai, Marcia Gordon, and et al. 2022. "Low-Dose Delta-9-Tetrahydrocannabinol as Beneficial Treatment for Aged APP/PS1 Mice" International Journal of Molecular Sciences 23, no. 5: 2757. https://doi.org/10.3390/ijms23052757
APA StyleWang, Y., Hong, Y., Yan, J., Brown, B., Lin, X., Zhang, X., Shen, N., Li, M., Cai, J., Gordon, M., Morgan, D., Zhou, Q., & Cao, C. (2022). Low-Dose Delta-9-Tetrahydrocannabinol as Beneficial Treatment for Aged APP/PS1 Mice. International Journal of Molecular Sciences, 23(5), 2757. https://doi.org/10.3390/ijms23052757