
Efficient Isolation and Functional Characterization of Niche Cells from Human Corneal Limbus

 ,
, 
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Localization of Limbal Niche Cells In Situ
2.2. Flow Sorting of Limbal Niche Cells
2.3. Characteristic Features of Sorted Cells
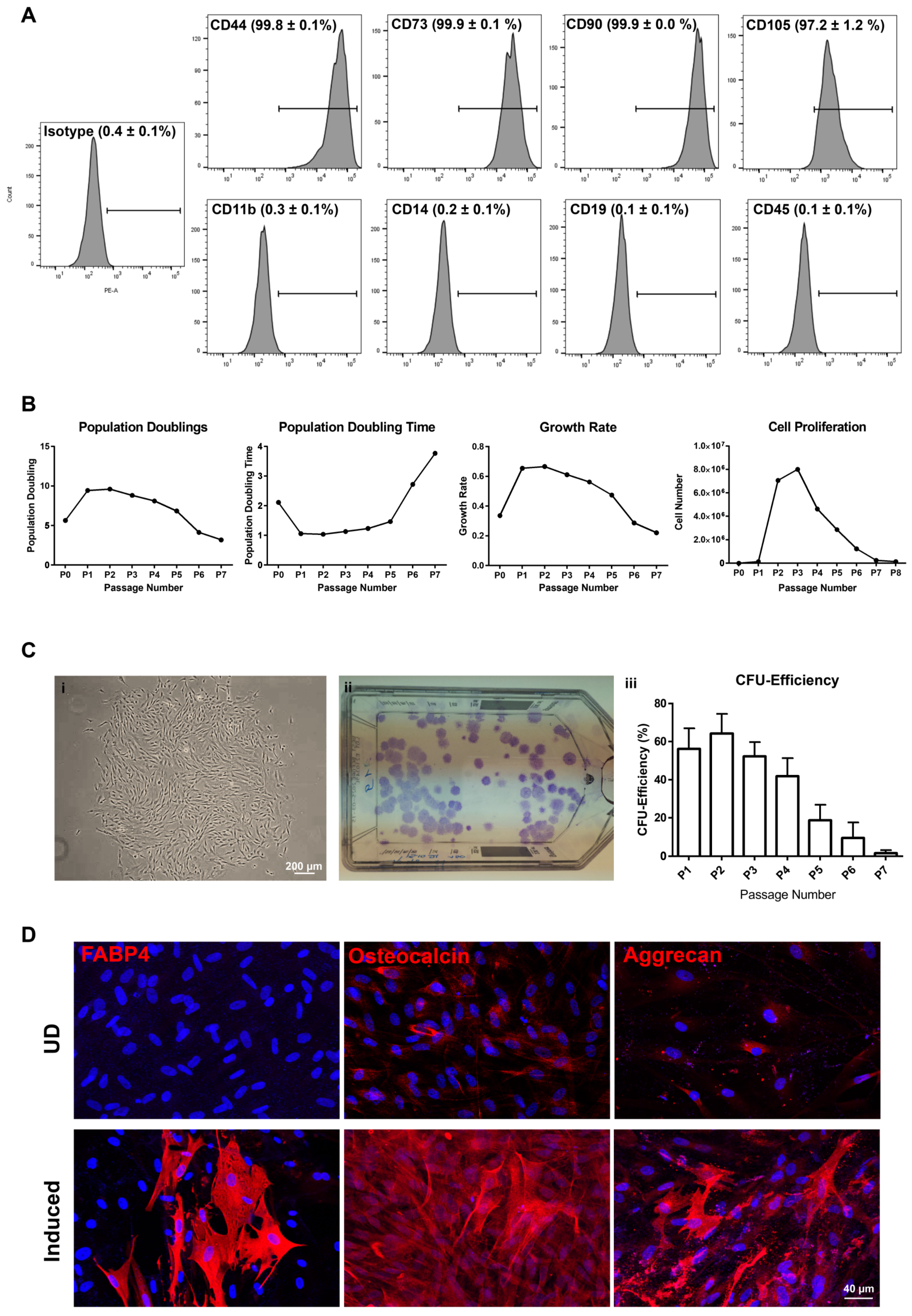
2.3.1. CD90+CD117− Cells (LMSC)
2.3.2. CD90−CD117+ Cells (LM)
2.3.3. CD90−CD117− Cells (LEPC)
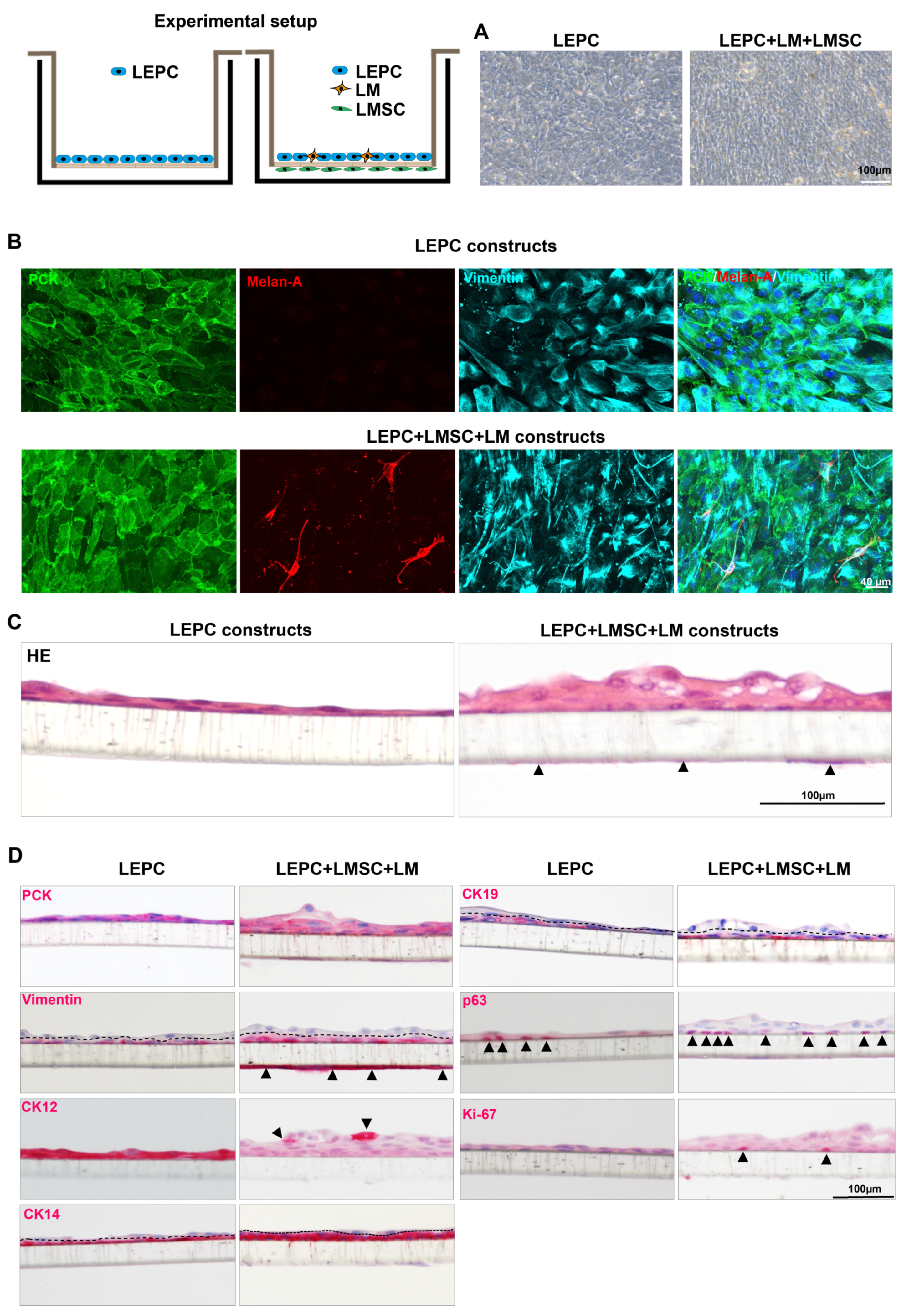
2.3.4. The 3D Co-Cultures
3. Discussion
4. Materials and Methods
4.1. Cell Isolation
4.2. Fluorescence-Activated Cell Sorting (FACS)
4.3. Flow Cytometry
4.4. Growth Characteristics
4.4.1. Population Doubling Assay
4.4.2. Colony-Forming Unit Assay of LMSC and LM
4.5. Trilineage Differentiation
4.6. Melanin Production
4.7. Co-Culture Experiments
4.7.1. Colony-Forming Unit Assay of LEPC
4.7.2. The 3D Co-Cultures
4.8. Histology and Immunohistochemistry—Paraffin
4.9. Immunohistochemistry—Frozen and Immunocytochemistry
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shortt, A.J.; Secker, G.A.; Munro, P.M.; Khaw, P.T.; Tuft, S.J.; Daniels, J.T. Characterization of the Limbal Epithelial Stem Cell Niche: Novel Imaging Techniques Permit in Vivo Observation and Targeted Biopsy of Limbal Epithelial Stem Cells. Stem Cells 2007, 25, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, P.; Di Girolamo, N. Limbal Epithelial Stem Cells: Role of the Niche Microenvironment. Stem Cells 2012, 30, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Polisetti, N.; Zenkel, M.; Menzel-Severing, J.; Kruse, F.E.; Schlötzer-Schrehardt, U. Cell Adhesion Molecules and Stem Cell-Niche-Interactions in the Limbal Stem Cell Niche. Stem Cells 2016, 34, 203–219. [Google Scholar] [CrossRef]
- Mei, H.; Gonzalez, S.; Deng, S.X. Extracellular Matrix Is an Important Component of Limbal Stem Cell Niche. J. Funct. Biomater. 2012, 3, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Dziasko, M.A.; Tuft, S.J.; Daniels, J.T. Limbal Melanocytes Support Limbal Epithelial Stem Cells in 2D and 3D Microenvironments. Exp. Eye Res. 2015, 138, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Dziasko, M.A.; Armer, H.E.; Levis, H.J.; Shortt, A.J.; Tuft, S.; Daniels, J.T. Localisation of Epithelial Cells Capable of Holoclone Formation in Vitro and Direct Interaction with Stromal Cells in the Native Human Limbal Crypt. PLoS ONE 2014, 9, e94283. [Google Scholar] [CrossRef]
- Nakatsu, M.N.; González, S.; Mei, H.; Deng, S.X. Human Limbal Mesenchymal Cells Support the Growth of Human Corneal Epithelial Stem/Progenitor Cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6953–6959. [Google Scholar] [CrossRef][Green Version]
- Li, G.; Zhang, Y.; Cai, S.; Sun, M.; Wang, J.; Li, S.; Li, X.; Tighe, S.; Chen, S.; Xie, H.; et al. Human Limbal Niche Cells Are a Powerful Regenerative Source for the Prevention of Limbal Stem Cell Deficiency in a Rabbit Model. Sci. Rep. 2018, 8, 6566. [Google Scholar] [CrossRef]
- Polisetti, N.; Gießl, A.; Zenkel, M.; Heger, L.; Dudziak, D.; Naschberger, E.; Stich, L.; Steinkasserer, A.; Kruse, F.E.; Schlötzer-Schrehardt, U. Melanocytes as Emerging Key Players in Niche Regulation of Limbal Epithelial Stem Cells. Ocul. Surf. 2021, 22, 172–189. [Google Scholar] [CrossRef]
- Ghareeb, A.E.; Lako, M.; Figueiredo, F.C. Recent Advances in Stem Cell Therapy for Limbal Stem Cell Deficiency: A Narrative Review. Ophthalmol. Ther. 2020, 9, 809–831. [Google Scholar] [CrossRef]
- Rama, P.; Ferrari, G.; Pellegrini, G. Cultivated Limbal Epithelial Transplantation. Curr. Opin. Ophthalmol. 2017, 28, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Funderburgh, J.L.; Funderburgh, M.L.; Du, Y. Stem Cells in the Limbal Stroma. Ocul. Surf. 2016, 14, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Veréb, Z.; Póliska, S.; Albert, R.; Olstad, O.K.; Boratkó, A.; Csortos, C.; Moe, M.C.; Facskó, A.; Petrovski, G. Role of Human Corneal Stroma-Derived Mesenchymal-Like Stem Cells in Corneal Immunity and Wound Healing. Sci. Rep. 2016, 6, 26227. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaibaji, O.; Swioklo, S.; Connon, C.J. Mesenchymal Stromal Cells for Ocular Surface Repair. Expert Opin. Biol. Ther. 2019, 19, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Hayashida, Y.; Chen, M.-Y.; Xie, H.T.; Tseng, S.C.G. A New Isolation Method of Human Limbal Progenitor Cells by Maintaining Close Association with Their Niche Cells. Tissue Eng. Part C Methods 2011, 17, 537–548. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Han, B.; Zhu, Y.-T.; Mahabole, M.; Huang, J.; Beebe, D.C.; Tseng, S.C.G. HC-HA/PTX3 Purified From Amniotic Membrane Promotes BMP Signaling in Limbal Niche Cells to Maintain Quiescence of Limbal Epithelial Progenitor/Stem Cells. Stem Cells 2015, 33, 3341–3355. [Google Scholar] [CrossRef]
- Xie, H.-T.; Chen, S.-Y.; Li, G.-G.; Tseng, S.C.G. Isolation and Expansion of Human Limbal Stromal Niche Cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 279–286. [Google Scholar] [CrossRef]
- Li, G.-G.; Zhu, Y.-T.; Xie, H.-T.; Chen, S.-Y.; Tseng, S.C.G. Mesenchymal Stem Cells Derived from Human Limbal Niche Cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5686–5697. [Google Scholar] [CrossRef]
- Li, Y.; Inoue, T.; Takamatsu, F.; Kobayashi, T.; Shiraishi, A.; Maeda, N.; Ohashi, Y.; Nishida, K. Differences between Niche Cells and Limbal Stromal Cells in Maintenance of Corneal Limbal Stem Cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1453–1462. [Google Scholar] [CrossRef]
- González, S.; Deng, S.X. Presence of Native Limbal Stromal Cells Increases the Expansion Efficiency of Limbal Stem/Progenitor Cells in Culture. Exp. Eye Res. 2013, 116, 169–176. [Google Scholar] [CrossRef]
- Polisetty, N.; Fatima, A.; Madhira, S.L.; Sangwan, V.S.; Vemuganti, G.K. Mesenchymal Cells from Limbal Stroma of Human Eye. Mol. Vis. 2008, 14, 431–442. [Google Scholar] [PubMed]
- Xiao, Y.-T.; Qu, J.-Y.; Xie, H.-T.; Zhang, M.-C.; Zhao, X.-Y. A Comparison of Methods for Isolation of Limbal Niche Cells: Maintenance of Limbal Epithelial Stem/Progenitor Cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 16. [Google Scholar] [CrossRef] [PubMed]
- Polisetti, N.; Gießl, A.; Li, S.; Sorokin, L.; Kruse, F.E.; Schlötzer-Schrehardt, U. Laminin-511-E8 Promotes Efficient in Vitro Expansion of Human Limbal Melanocytes. Sci. Rep. 2020, 10, 11074. [Google Scholar] [CrossRef] [PubMed]
- Polisetti, N.; Schlötzer-Schrehardt, U.; Reinhard, T.; Schlunck, G. Isolation and Enrichment of Melanocytes from Human Corneal Limbus Using CD117 (c-Kit) as Selection Marker. Sci. Rep. 2020, 10, 17588. [Google Scholar] [CrossRef] [PubMed]
- Bojic, S.; Hallam, D.; Alcada, N.; Ghareeb, A.; Queen, R.; Pervinder, S.; Buck, H.; Amitai Lange, A.; Figueiredo, G.; Rooney, P.; et al. CD200 Expression Marks a Population of Quiescent Limbal Epithelial Stem Cells with Holoclone Forming Ability. Stem Cells 2018, 36, 1723–1735. [Google Scholar] [CrossRef]
- Truong, T.; Huynh, K.; Nakatsu, M.; Deng, S. SSEA4 Is a Potential Negative Marker for the Enrichment of Human Corneal Epithelial Stem/Progenitor Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6315–6320. [Google Scholar] [CrossRef]
- Hayashi, R.; Yamato, M.; Saito, T.; Oshima, T.; Okano, T.; Tano, Y.; Nishida, K. Enrichment of Corneal Epithelial Stem/Progenitor Cells Using Cell Surface Markers, Integrin A6 and CD71. Biochem. Biophys. Res. Commun. 2008, 367, 256–263. [Google Scholar] [CrossRef]
- Norrick, A.; Esterlechner, J.; Niebergall-Roth, E.; Dehio, U.; Sadeghi, S.; Schröder, H.M.; Ballikaya, S.; Stemler, N.; Ganss, C.; Dieter, K.; et al. Process Development and Safety Evaluation of ABCB5+ Limbal Stem Cells as Advanced-Therapy Medicinal Product to Treat Limbal Stem Cell Deficiency. Stem Cell Res. Ther. 2021, 12, 194. [Google Scholar] [CrossRef]
- Hayashi, R.; Yamato, M.; Sugiyama, H.; Sumide, T.; Yang, J.; Okano, T.; Tano, Y.; Nishida, K. N-Cadherin Is Expressed by Putative Stem/Progenitor Cells and Melanocytes in the Human Limbal Epithelial Stem Cell Niche. Stem Cells 2007, 25, 289–296. [Google Scholar] [CrossRef]
- Chang, C.-Y.A.; McGhee, J.J.; Green, C.R.; Sherwin, T. Comparison of Stem Cell Properties in Cell Populations Isolated from Human Central and Limbal Corneal Epithelium. Cornea 2011, 30, 1155–1162. [Google Scholar] [CrossRef]
- Polisetti, N.; Agarwal, P.; Khan, I.; Kondaiah, P.; Sangwan, V.S.; Vemuganti, G.K. Gene Expression Profile of Epithelial Cells and Mesenchymal Cells Derived from Limbal Explant Culture. Mol. Vis. 2010, 16, 1227–1240. [Google Scholar] [PubMed]
- Branch, M.J.; Hashmani, K.; Dhillon, P.; Jones, D.R.E.; Dua, H.S.; Hopkinson, A. Mesenchymal Stem Cells in the Human Corneal Limbal Stroma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5109–5116. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Cheng, A.M.S.; Zhang, Y.; Zhu, Y.-T.; He, H.; Mahabole, M.; Tseng, S.C.G. Pax 6 Controls Neural Crest Potential of Limbal Niche Cells to Support Self-Renewal of Limbal Epithelial Stem Cells. Sci. Rep. 2019, 9, 9763. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The Development of Fibroblast Colonies in Monolayer Cultures of Guinea-Pig Bone Marrow and Spleen Cells. Cell Prolif. 1970, 3, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Godwin, L.S.; Castle, J.T.; Kohli, J.S.; Goff, P.S.; Cairney, C.J.; Keith, W.N.; Sviderskaya, E.V.; Bennett, D.C. Isolation, Culture, and Transfection of Melanocytes. Curr. Protoc. Cell Biol. 2014, 63, 1.8.1–1.8.20. [Google Scholar] [CrossRef]
- Zhang, T.; Choi, J.; Kovacs, M.A.; Shi, J.; Xu, M.; NISC Comparative Sequencing Program; Melanoma Meta-Analysis Consortium; Goldstein, A.M.; Trower, A.J.; Bishop, D.T.; et al. Cell-Type-Specific EQTL of Primary Melanocytes Facilitates Identification of Melanoma Susceptibility Genes. Genome Res. 2018, 28, 1621–1635. [Google Scholar] [CrossRef]
- Willemsen, M.; Luiten, R.M.; Teunissen, M.B.M. Instant Isolation of Highly Purified Human Melanocytes from Freshly Prepared Epidermal Cell Suspensions. Pigment. Cell Melanoma Res. 2020, 33, 763–766. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Green, H. Serial Cultivation of Strains of Human Epidermal Keratinocytes: The Formation of Keratinizing Colonies from Single Cells. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Meyer-Blazejewska, E.A.; Kruse, F.E.; Bitterer, K.; Meyer, C.; Hofmann-Rummelt, C.; Wünsch, P.H.; Schlötzer-Schrehardt, U. Preservation of the Limbal Stem Cell Phenotype by Appropriate Culture Techniques. Investig. Ophthalmol. Vis. Sci. 2010, 51, 765–774. [Google Scholar] [CrossRef]
- Ghoubay-Benallaoua, D.; Basli, E.; Goldschmidt, P.; Pecha, F.; Chaumeil, C.; Laroche, L.; Borderie, V. Human Epithelial Cell Cultures from Superficial Limbal Explants. Mol. Vis. 2011, 17, 341–354. [Google Scholar]
- Mei, H.; González, S.; Nakatsu, M.N.; Baclagon, E.R.; Chen, F.V.; Deng, S.X. Human Adipose-Derived Stem Cells Support the Growth of Limbal Stem/Progenitor Cells. PLoS ONE 2017, 12, e0186238. [Google Scholar] [CrossRef] [PubMed]
- Polisetti, N.; Schlunck, G.; Reinhard, T.; Kruse, F.E.; Schlötzer-Schrehardt, U. Isolation and Ex Vivo Expansion of Human Limbal Epithelial Progenitor Cells. Bio-Protocol 2020, 10, e3754. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-W.; Hu, F.-R.; Wang, I.-J.; Hou, Y.-C.; Chen, W.-L. Migration of Limbal Melanocytes onto the Central Cornea after Ocular Surface Reconstruction: An in Vivo Confocal Microscopic Case Report. Cornea 2010, 29, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; González, S.; Nakatsu, M.N.; Baclagon, E.R.; Lopes, V.S.; Williams, D.S.; Deng, S.X. A Three-Dimensional Culture Method to Expand Limbal Stem/Progenitor Cells. Tissue Eng. Part C Methods 2014, 20, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Badaloni, A.; Chiara, F.; Stjernberg, J.; Polisetti, N.; Nihlberg, K.; Consalez, G.G.; Sigvardsson, M. Molecular Characterization of Prospectively Isolated Multipotent Mesenchymal Progenitors Provides New Insight into the Cellular Identity of Mesenchymal Stem Cells in Mouse Bone Marrow. Mol. Cell. Biol. 2013, 33, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Wolf, J.; Auw-Haedrich, C.; Schlecht, A.; Boneva, S.; Lapp, T.; Horres, R.; Agostini, H.; Martin, G.; Reinhard, T.; et al. Expression of the COVID-19 Receptor ACE2 in the Human Conjunctiva. J. Med. Virol. 2020, 92, 2081–2086. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polisetti, N.; Sharaf, L.; Schlötzer-Schrehardt, U.; Schlunck, G.; Reinhard, T. Efficient Isolation and Functional Characterization of Niche Cells from Human Corneal Limbus. Int. J. Mol. Sci. 2022, 23, 2750. https://doi.org/10.3390/ijms23052750
Polisetti N, Sharaf L, Schlötzer-Schrehardt U, Schlunck G, Reinhard T. Efficient Isolation and Functional Characterization of Niche Cells from Human Corneal Limbus. International Journal of Molecular Sciences. 2022; 23(5):2750. https://doi.org/10.3390/ijms23052750
Chicago/Turabian StylePolisetti, Naresh, Lyne Sharaf, Ursula Schlötzer-Schrehardt, Günther Schlunck, and Thomas Reinhard. 2022. "Efficient Isolation and Functional Characterization of Niche Cells from Human Corneal Limbus" International Journal of Molecular Sciences 23, no. 5: 2750. https://doi.org/10.3390/ijms23052750
APA StylePolisetti, N., Sharaf, L., Schlötzer-Schrehardt, U., Schlunck, G., & Reinhard, T. (2022). Efficient Isolation and Functional Characterization of Niche Cells from Human Corneal Limbus. International Journal of Molecular Sciences, 23(5), 2750. https://doi.org/10.3390/ijms23052750