
A Redox-Silent Analogue of Tocotrienol May Break the Homeostasis of Proteasomes in Human Malignant Mesothelioma Cells by Inhibiting STAT3 and NRF1


Abstract
:1. Introduction
2. Results
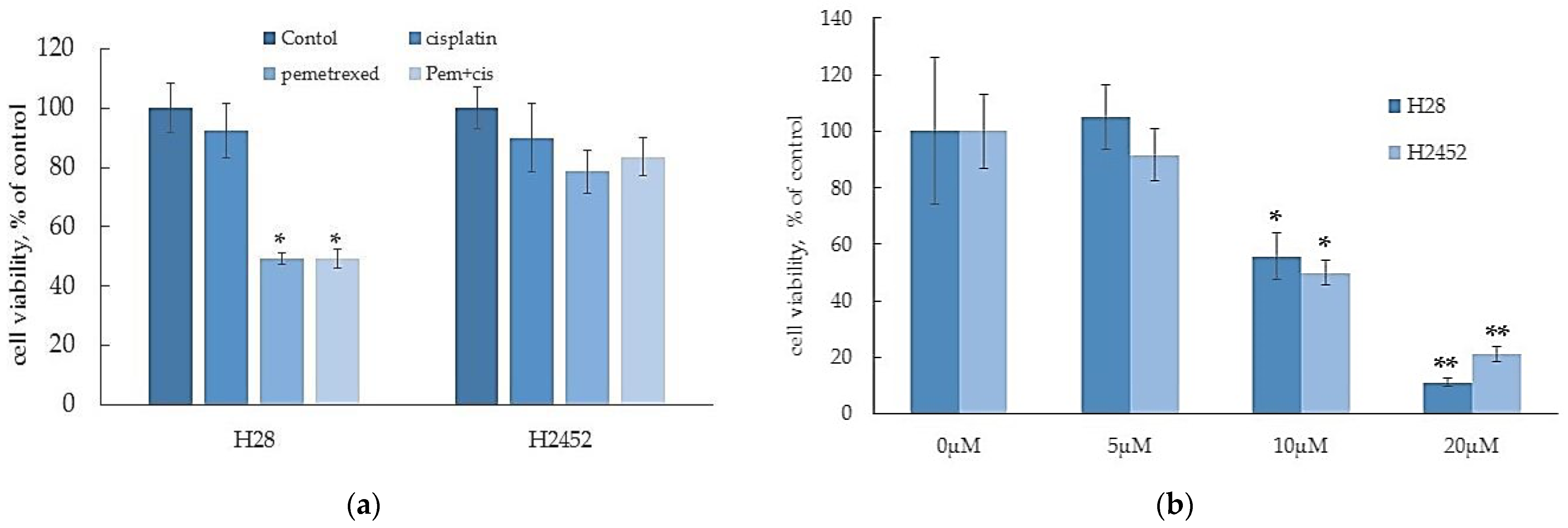
2.1. α-T3E Induces Cell Death in Human MM Cells
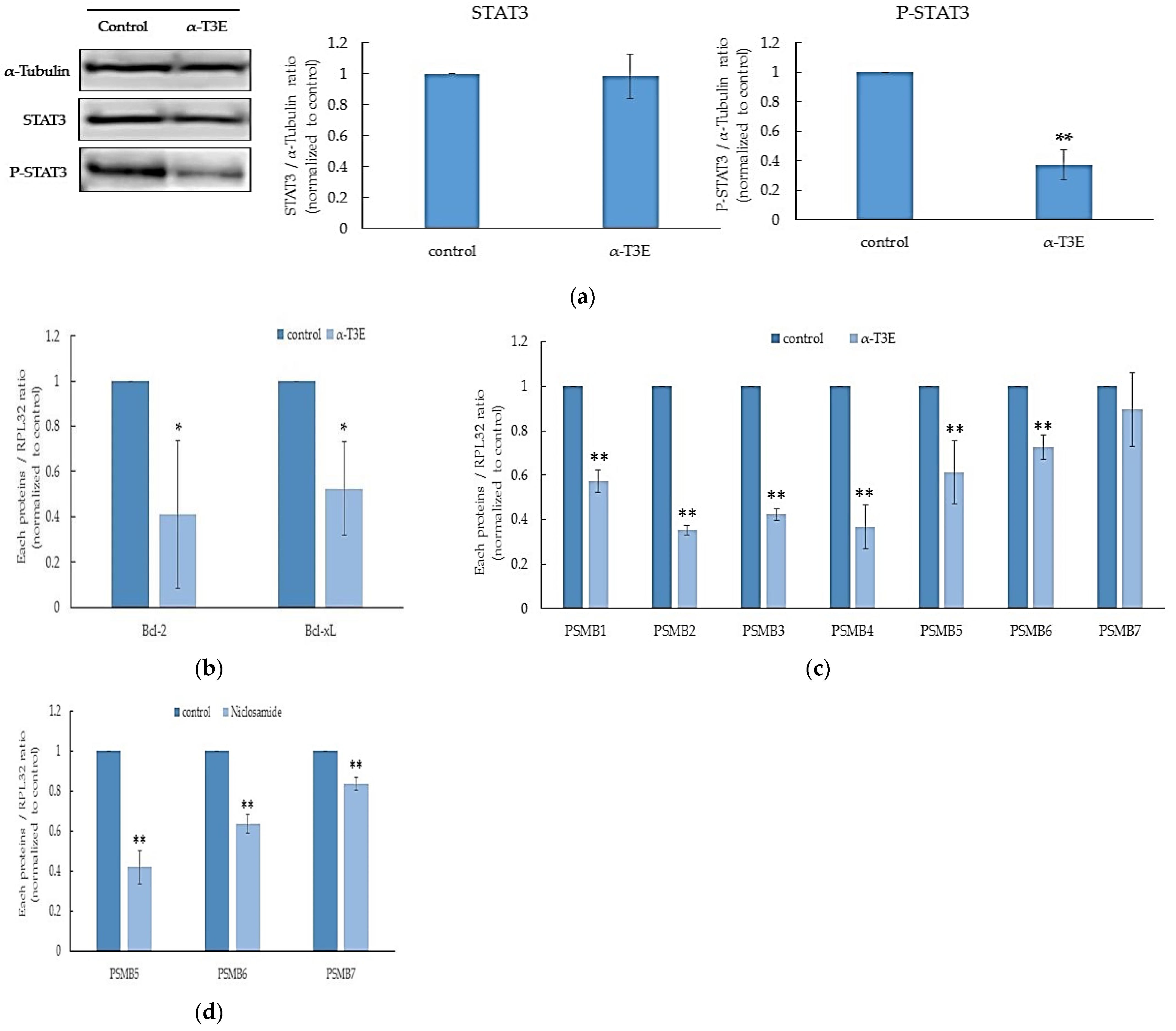
2.2. α-T3E Inhibits Proteasome Activity in Human MM Cells
2.3. The Inactivation of STAT3 by α-T3E Contributes to the Inhibition of Proteasome Activity in Human MM Cells
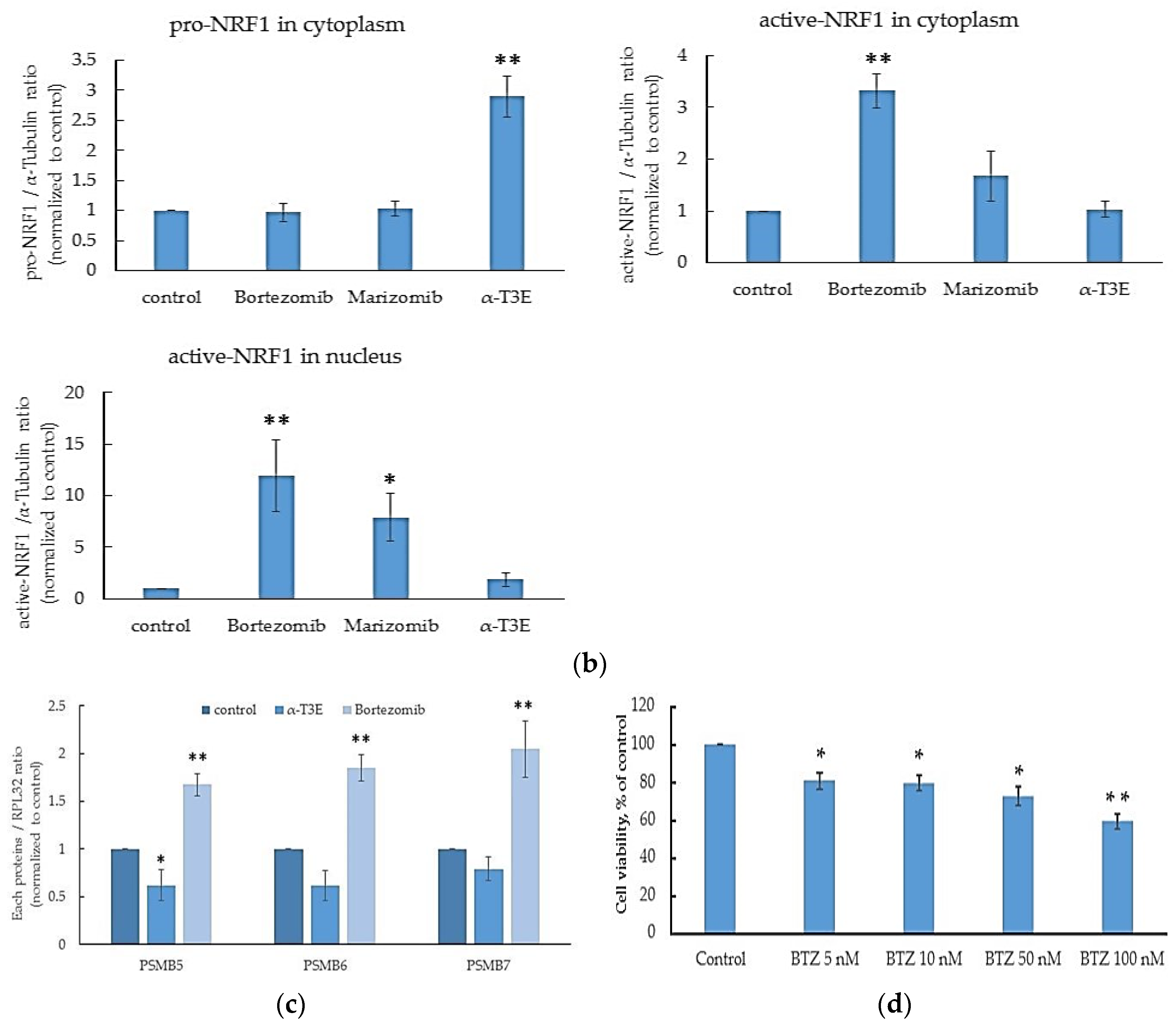
2.4. α-T3E-Dependent Inactivation of NRF1 Functions Contributes to the α-T3E-Dependent Inhibition of Proteasome Functions in Human MM Cells
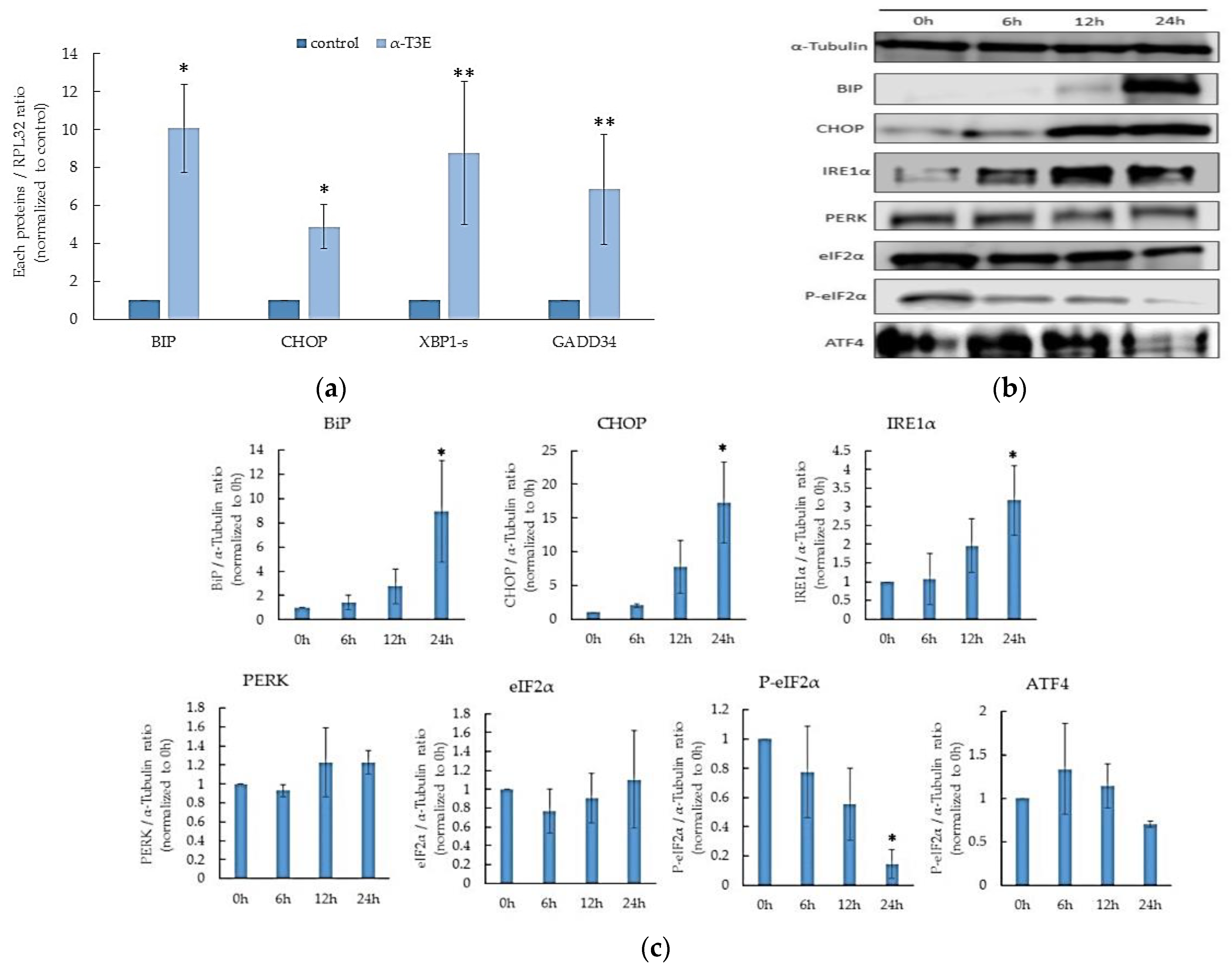
2.5. α-T3E Induces ER Stress, Leading to Cell Death in Human MM Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. α-T3E Synthesis
4.3. Cell Culture
4.4. Cell Viability
4.5. Proteasome Activity
4.6. Isolation of mRNA and Real-Time Quantitative PCR
4.7. Immunoblotting
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donahoe, L.; Cho, J.; de Perrot, M. Novel induction therapies for pleural mesothelioma. Semin. Thorac. Cardiovasc. Surg. 2014, 26, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Tabot, D.C.; Blyth, K.G.; Maskell, K.G.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Meul, T.; Meiners, S. Exploring the proteasome system: A novel concept of proteasome inhibition and regulation. Pharmacol. Ther. 2020, 211, 107526. [Google Scholar] [CrossRef] [PubMed]
- Jayaweera, S.P.E.; Wanigasinghe, K.S.P.; Rajalingam, D.; Silva, G.N. Carfilzomib: A Promising Proteasome Inhibitor for the Treatment of Relapsed and Refractory Multiple Myeloma. Front Oncol. 2021, 11, 740796. [Google Scholar] [CrossRef]
- Walter, R.F.H.; Sydow, S.R.; Berg, E.; Kollmeier, J.; Christoph, D.C.; Christoph, S.; Eberhardt, W.E.E.; Mairinger, T.; Wohlschläger, J.; Schmid, K.W.; et al. Bortezomib sensitivity is tissue dependent and high expression of the 20S proteasome precludes good response in malignant pleural mesothelioma. Cancer Manag. Res. 2019, 11, 8711–8720. [Google Scholar] [CrossRef] [Green Version]
- Vangala, J.R.; Dudem, S.; Jain, N.; Kalivendi, S.V. Regulation of PSMB5 protein and β subunits of mammalian proteasome by constitutively activated signal transducer and activator of transcription 3 (STAT3): Potential role in bortezomib-mediated anticancer therapy. J. Biol. Chem. 2014, 289, 12612–12622. [Google Scholar] [CrossRef] [Green Version]
- Hamazaki, S.; Murata, S. ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond. Int. J. Mol. Sci. 2020, 21, 3683. [Google Scholar] [CrossRef]
- Kashiwagi, K.; Virgona, N.; Harada, K.; Kido, W.; Yano, Y.; Ando, A.; Hagiwara, K.; Yano, T. A redox-silent analogue of tocotrienol acts as a potential cytotoxic agent against human mesothelioma cells. Life Sci. 2009, 84, 650–656. [Google Scholar] [CrossRef]
- Johnson, F.M.; Saigal, B.; Tran, H.; Donato, N.J. Abrogation of signal transducer and activator of transcription 3 reactivation after Src kinase inhibition results in synergistic antitumor effects. Clin. Cancer Res. 2007, 13, 4233–4244. [Google Scholar] [CrossRef] [Green Version]
- Northrop, A.; Byers, H.A.; Radhakrishnan, S.K. Regulation of NRF1, a master transcription factor of proteasome genes: Implications for cancer and neurodegeneration. Mol. Biol. Cell 2020, 31, 2158–2163. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Raymond, J.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, S.K.; Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. eLIFE 2014, 3, e01856. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.A.; Fumarola, C.; La Monica, S.; Alfieri, R. New therapeutic strategies for malignant pleural mesothelioma. Biochem. Pharmacol. 2017, 123, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Arai, T.; Fusegi, M.; Ando, A.; Yano, T. A Redox-inactive derivative of tocotrienol suppresses tumor growth of mesothelioma cells in a Xenograft model. Biol. Pharm. Bull. 2019, 42, 1034–1037. [Google Scholar] [CrossRef] [Green Version]
- Ramdas, P.; Radhakrishnan, A.K.; Abdu Sani, A.A.; Kumari, M.; Anandha Rao, J.S.; Abdul-Rahman, P.S. Advancing the role of gamma-tocotrienol as proteasomes inhibitor: A quantitative proteomic analysis of MDA-MB-231 human breast cancer cells. Biomolecules 2019, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef]
- Khan, S.; Rammeloo, A.W.; Heikkila, J.J. Withaferin A induces proteasome inhibition, endoplasmic reticulum stress, the heat shock response and acquisition of thermotolerance. PLoS ONE 2012, 7, e50547. [Google Scholar] [CrossRef] [Green Version]
- Schönthal, A.H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 2013, 85, 653–666. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Yano, T.; Sato, A.; Sekine, M.; Virgona, N.; Ota, M. Redox-inactive analogue of tocotrienol as a potential anti-cancer agent. Anticancer Agents Med. Chem. 2013, 13, 496–501. [Google Scholar] [PubMed]
- Yano, Y.; Satoh, H.; Fukumoto, K.; Kumadaki, I.; Ichikawa, T.; Yamada, K.; Hagiwara, K.; Yano, T. Induction of cytotoxicity in human lung adenocarcinoma cells by 6-O-carboxypropyl-alpha-tocotrienol, a redox-silent derivative of alpha-tocotrienol. Int. J. Cancer 2005, 115, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Northrop, A.; Vangala, J.R.; Feygin, A.; Radhakrishnan, S.K. Disabling the protease DDI2 attenuates the transcriptional activity of NRF1 and potentiates proteasome inhibitor cytotoxicity. Int. J. Mol. Sci. 2020, 21, 327. [Google Scholar] [CrossRef] [Green Version]
- Tomlin, F.M.; Gerling-Driessen, U.; Liu, Y.; Flynn, R.A.; Vangala, J.V.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordoñez-Rueda, D.; et al. Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS. Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Qi, L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Takenokuchi, M.; Miyamoto, K.; Saigo, K.; Taniguchi, T. Bortezomib causes ER stress-related death of acute promyelocytic leukemia cells through excessive accumulation of PML-RARA. Anticancer Res. 2015, 35, 3307–3316. [Google Scholar]
- Luhr, M.; Torgersen, M.L.; Szalai, P.; Hashim, A.; Brech, A.; Staerk, J.; Engedal, N. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J. Biol. Chem. 2019, 294, 8197–8217. [Google Scholar] [CrossRef]
- Liu, L.; Ito, S.; Nishio, N.; Sun, Y.; Chen, N.; Tanaka, Y.; Isobe, K. GADD34 facilitates cell death resulting from proteasome inhibition. Anticancer Res. 2015, 35, 5317–5324. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence |
|---|---|---|
| BCL-2 | Forward Primer | GTGTGTGGAGAGCGTCAACC |
| Reverse Primer | CAGCCAGGAGAAATCAAACAGA | |
| BCL-XL | Forward Primer | ACCTGACATCCCAGCTCCAC |
| Reverse Primer | GTCTACGCTTTCCACGCACA | |
| PSMB1 | Forward Primer | TTTCGCCCTACGTTTTCAAC |
| Reverse Primer | TACAGCCCCCTTTCCTTCTT | |
| PSMB2 | Forward Primer | AAGGCCCCGACTATGTTCTT |
| Reverse Primer | AGGTTGGCAGATTCAGGATG | |
| PSMB3 | Forward Primer | GAAGGGGAAGAACTGTGTGG |
| Reverse Primer | CCTGGTGGTGATTTTGTCCT | |
| PSMB4 | Forward Primer | TCAGTCCTCGGCGTTAAGTT |
| Reverse Primer | GCTTAGCACTGGCTGCTTCT | |
| PSMB5 | Forward Primer | CCATACCTGCTAGGCACCAT |
| Reverse Primer | GCACCTCCTGAGTAGGCATC | |
| PSMB6 | Forward Primer | CCTATTCACGACCGCATTTT |
| Reverse Primer | TCCCGGTAGGTAGCATCAAC | |
| PSMB7 | Forward Primer | CGGCTGTGTCGGTGTATG |
| Reverse Primer | GCCAGTTTTCCGGACCTT | |
| BIP | Forward Primer | CGAGGAGGAGGACAAGAAGG |
| Reverse Primer | CACCTTGAACGGCAAGAACT | |
| CHOP | Forward Primer | GGAGGTGGAAGCCTGGTATG |
| Reverse Primer | GTGACCTCTGCTGGTTCTGG | |
| s-XBP1 | Forward Primer | GGTCTGCTGAGTCCGCAGCAGG |
| Reverse Primer | GGGCTTGGTATATATGTGG | |
| GADD34 | Forward Primer | GAAGGAGGAAAAGGCACACA |
| Reverse Primer | CCTCACCCTCCTCTTCATCAC | |
| RPL32 | Forward Primer | AACCCTGTTGTCAATGCCTC |
| Reverse Primer | CATCTCCTTCTCGGCATCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishii, K.; Fusegi, M.; Mori, T.; Teshima, K.; Ninomiya, N.; Kohno, K.; Sato, A.; Ishida, T.; Miyakoshi, Y.; Yano, T. A Redox-Silent Analogue of Tocotrienol May Break the Homeostasis of Proteasomes in Human Malignant Mesothelioma Cells by Inhibiting STAT3 and NRF1. Int. J. Mol. Sci. 2022, 23, 2655. https://doi.org/10.3390/ijms23052655
Ishii K, Fusegi M, Mori T, Teshima K, Ninomiya N, Kohno K, Sato A, Ishida T, Miyakoshi Y, Yano T. A Redox-Silent Analogue of Tocotrienol May Break the Homeostasis of Proteasomes in Human Malignant Mesothelioma Cells by Inhibiting STAT3 and NRF1. International Journal of Molecular Sciences. 2022; 23(5):2655. https://doi.org/10.3390/ijms23052655
Chicago/Turabian StyleIshii, Kyota, Momoka Fusegi, Tatsuki Mori, Kosuke Teshima, Nanako Ninomiya, Kakeru Kohno, Ayami Sato, Tatsuya Ishida, Yuichi Miyakoshi, and Tomohiro Yano. 2022. "A Redox-Silent Analogue of Tocotrienol May Break the Homeostasis of Proteasomes in Human Malignant Mesothelioma Cells by Inhibiting STAT3 and NRF1" International Journal of Molecular Sciences 23, no. 5: 2655. https://doi.org/10.3390/ijms23052655
APA StyleIshii, K., Fusegi, M., Mori, T., Teshima, K., Ninomiya, N., Kohno, K., Sato, A., Ishida, T., Miyakoshi, Y., & Yano, T. (2022). A Redox-Silent Analogue of Tocotrienol May Break the Homeostasis of Proteasomes in Human Malignant Mesothelioma Cells by Inhibiting STAT3 and NRF1. International Journal of Molecular Sciences, 23(5), 2655. https://doi.org/10.3390/ijms23052655