
Mitochondrial Proteins as Source of Cancer Neoantigens

{kind=link}
Abstract
:1. Genetics of Mammalian Mitochondria
2. MtDNA Mutations and Cancer
3. Mitochondrial Mutated Proteins and Cancer Immunotherapy
4. Post-Translational Modifications: A Different Way to Generate Neoepitopes?
4.1. Oxidation
4.2. Phosphorylation
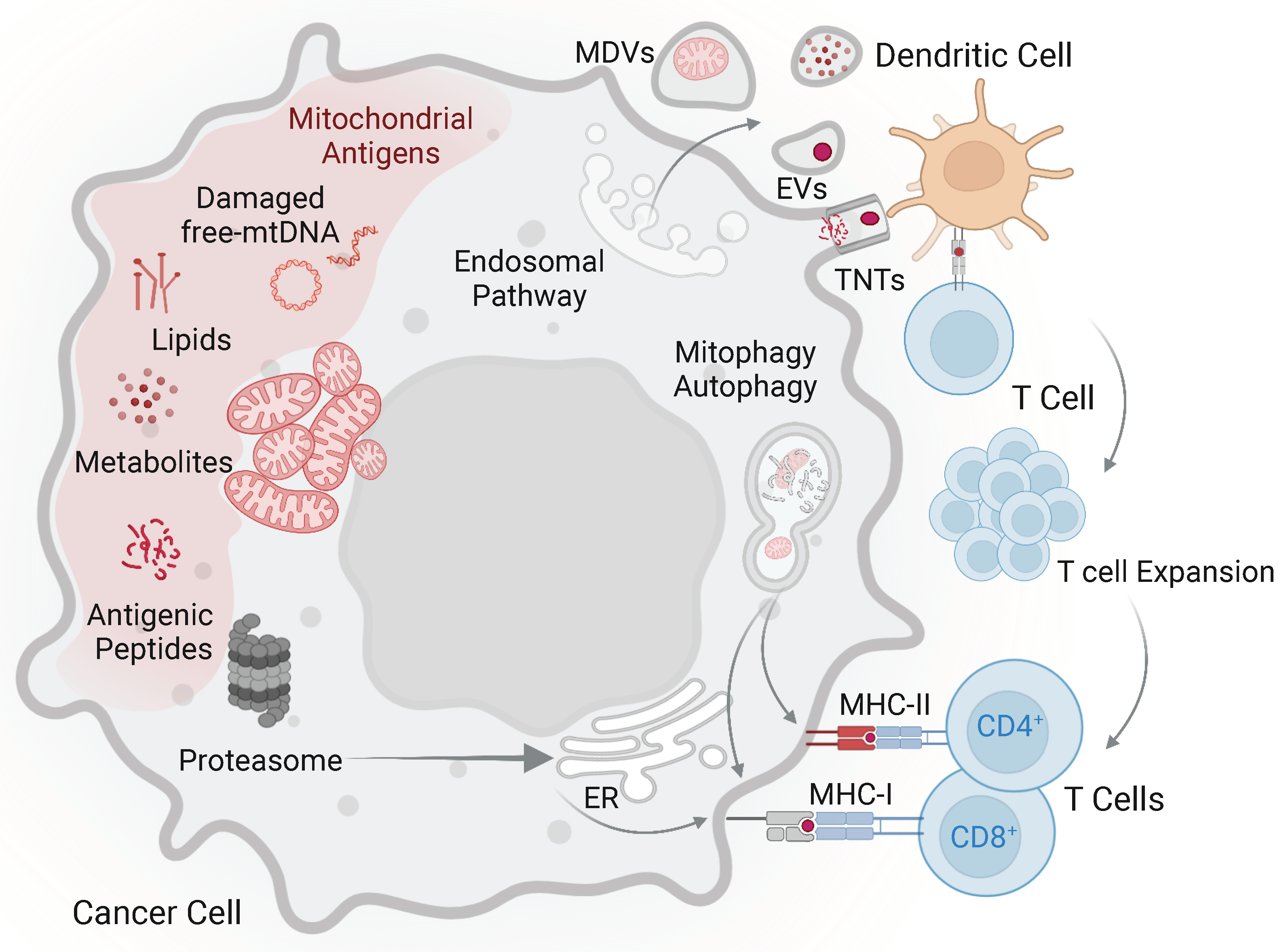
5. Mechanism of Mitochondrial Antigen Direct and Cross-Presentation in Cancer
6. Immune Response to Mitochondrial Proteins in Diseases Other Than Cancer
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chandel, N.S. Evolution of mitochondria as signaling organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wu, M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 2015, 5, 7949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, C. The role of mitochondria in the oncogenic signal transduction. Int. J. Biochem. Cell Biol. 2014, 48, 11–17. [Google Scholar] [CrossRef]
- Wiese, M.; Bannister, A.J. Two genomes, one cell: Mitochondrial-nuclear coordination via epigenetic pathways. Mol. Metab. 2020, 38, 100942. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [Green Version]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Wachsmuth, M.; Hubner, A.; Li, M.; Madea, B.; Stoneking, M. Age-related and heteroplasmy-related variation in human mtDNA copy number. PLoS Genet. 2016, 12, e1005939. [Google Scholar] [CrossRef] [Green Version]
- Kukat, C.; Wurm, C.A.; Spahr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sanchez-Cabo, F.; Torroja, C.; Acin-Perez, R.; Calvo, E.; Aix, E.; Gonzalez-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, K.L.; Dasgupta, S. The landscape of mtDNA modifications in cancer: A tale of two cities. Front. Oncol 2017, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front. Genet. 2021, 12, 627050. [Google Scholar] [CrossRef] [PubMed]
- Vithayathil, S.A.; Ma, Y.; Kaipparettu, B.A. Transmitochondrial cybrids: Tools for functional studies of mutant mitochondria. Methods Mol. Biol. 2012, 837, 219–230. [Google Scholar] [CrossRef]
- Marco-Brualla, J.; Al-Wasaby, S.; Soler, R.; Romanos, E.; Conde, B.; Justo-Mendez, R.; Enriquez, J.A.; Fernandez-Silva, P.; Martinez-Lostao, L.; Villalba, M.; et al. Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells. Cancers 2019, 11, 1027. [Google Scholar] [CrossRef] [Green Version]
- Kaneva, K.; O’Halloran, K.; Triska, P.; Liu, X.; Merkurjev, D.; Bootwalla, M.; Ryutov, A.; Cotter, J.A.; Ostrow, D.; Biegel, J.A.; et al. The spectrum of mitochondrial DNA (mtDNA) mutations in pediatric CNS tumors. Neurooncol. Adv. 2021, 3, vdab074. [Google Scholar] [CrossRef]
- Perez-Amado, C.J.; Tovar, H.; Gomez-Romero, L.; Beltran-Anaya, F.O.; Bautista-Pina, V.; Dominguez-Reyes, C.; Villegas-Carlos, F.; Tenorio-Torres, A.; Alfaro-Ruiz, L.A.; Hidalgo-Miranda, A.; et al. Mitochondrial DNA Mutation Analysis in Breast Cancer: Shifting From Germline Heteroplasmy Toward Homoplasmy in Tumors. Front. Oncol. 2020, 10, 572954. [Google Scholar] [CrossRef]
- Parsons, T.J.; Muniec, D.S.; Sullivan, K.; Woodyatt, N.; Alliston-Greiner, R.; Wilson, M.R.; Berry, D.L.; Holland, K.A.; Weedn, V.W.; Gill, P.; et al. A high observed substitution rate in the human mitochondrial DNA control region. Nat. Genet. 1997, 15, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeronimo, C.; Nomoto, S.; Caballero, O.L.; Usadel, H.; Henrique, R.; Varzim, G.; Oliveira, J.; Lopes, C.; Fliss, M.S.; Sidransky, D. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 2001, 20, 5195–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.B.; Song, J.J.; Hempen, P.M.; Parmigiani, G.; Hruban, R.H.; Kern, S.E. Detection of mitochondrial DNA mutations in pancreatic cancer offers a “mass”-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001, 61, 1299–1304. [Google Scholar]
- Kirches, E.; Krause, G.; Warich-Kirches, M.; Weis, S.; Schneider, T.; Meyer-Puttlitz, B.; Mawrin, C.; Dietzmann, K. High frequency of mitochondrial DNA mutations in glioblastoma multiforme identified by direct sequence comparison to blood samples. Int. J. Cancer 2001, 93, 534–538. [Google Scholar] [CrossRef]
- Coller, H.A.; Khrapko, K.; Bodyak, N.D.; Nekhaeva, E.; Herrero-Jimenez, P.; Thilly, W.G. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat. Genet. 2001, 28, 147–150. [Google Scholar] [CrossRef]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.B.; Alaei-Mahabadi, B.; Sabarinathan, R.; Samuelsson, T.; Gorodkin, J.; Gustafsson, C.M.; Larsson, E. Simultaneous DNA and RNA Mapping of Somatic Mitochondrial Mutations across Diverse Human Cancers. PLoS Genet. 2015, 11, e1005333. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Soares, P.; Radivojac, P.; Li, B.; Samuels, D.C. Comparing phylogeny and the predicted pathogenicity of protein variations reveals equal purifying selection across the global human mtDNA diversity. Am. J. Hum. Genet. 2011, 88, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum. Mol. Genet. 2017, 26, 2912–2922. [Google Scholar] [CrossRef]
- Gorelick, A.N.; Kim, M.; Chatila, W.K.; La, K.; Hakimi, A.A.; Berger, M.F.; Taylor, B.S.; Gammage, P.A.; Reznik, E. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nat. Metab. 2021, 3, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Akimoto, M.; Hayashi, J.I.; Nagase, H.; Takenaga, K. Association of predicted pathogenic mutations in mitochondrial ND genes with distant metastasis in NSCLC and colon cancer. Sci. Rep. 2017, 7, 15535. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Vitiello, A.; Zanetti, M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 2017, 35, 815–817. [Google Scholar] [CrossRef]
- Harel, M.; Ortenberg, R.; Varanasi, S.K.; Mangalhara, K.C.; Mardamshina, M.; Markovits, E.; Baruch, E.N.; Tripple, V.; Arama-Chayoth, M.; Greenberg, E.; et al. Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 2019, 179, 236–250.e18. [Google Scholar] [CrossRef]
- Voo, K.S.; Zeng, G.; Mu, J.B.; Zhou, J.; Su, X.Z.; Wang, R.F. CD4+ T-cell response to mitochondrial cytochrome B in human melanoma. Cancer Res. 2006, 66, 5919–5926. [Google Scholar] [CrossRef] [Green Version]
- Pierini, S.; Fang, C.; Rafail, S.; Facciponte, J.G.; Huang, J.; De Sanctis, F.; Morgan, M.A.; Uribe-Herranz, M.; Tanyi, J.L.; Facciabene, A. A Tumor Mitochondria Vaccine Protects against Experimental Renal Cell Carcinoma. J. Immunol. 2015, 195, 4020–4027. [Google Scholar] [CrossRef] [Green Version]
- Prota, G.; Gileadi, U.; Rei, M.; Lechuga-Vieco, A.V.; Chen, J.L.; Galiani, S.; Bedard, M.; Lau, V.W.C.; Fanchi, L.F.; Artibani, M.; et al. Enhanced Immunogenicity of Mitochondrial-Localized Proteins in Cancer Cells. Cancer Immunol. Res. 2020, 8, 685–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Tykodi, S.S.; Braciale, T.J. Recognition of a sequestered self peptide by influenza virus-specific CD8+ cytolytic T lymphocytes. J. Immunol. 2000, 164, 1669–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasada, T.; Ghendler, Y.; Neveu, J.M.; Lane, W.S.; Reinherz, E.L. A naturally processed mitochondrial self-peptide in complex with thymic MHC molecules functions as a selecting ligand for a viral-specific T cell receptor. J. Exp. Med. 2001, 194, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, S.; Van de Water, J.; Ansari, A.; Nakamura, M.; Ishibashi, H.; Coppel, R.L.; Lake, J.; Keeffe, E.B.; Roche, T.E.; Gershwin, M.E. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J. Clin. Invest. 1998, 102, 1831–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, S.; Kita, H.; He, X.S.; Ansari, A.A.; Lian, Z.X.; Van De Water, J.; Yamamoto, K.; Tsuji, T.; Coppel, R.L.; Kaplan, M.; et al. Comprehensive mapping of HLA-A0201-restricted CD8 T-cell epitopes on PDC-E2 in primary biliary cirrhosis. Hepatology 2002, 36, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 90. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Petersen, J.; Wurzbacher, S.J.; Williamson, N.A.; Ramarathinam, S.H.; Reid, H.H.; Nair, A.K.N.; Zhao, A.Y.; Nastovska, R.; Rudge, G.; Rossjohn, J.; et al. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc. Natl. Acad. Sci. USA 2009, 106, 2776–2781. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, F.; Stones, D.H.; Zarling, A.L.; Willcox, C.R.; Shabanowitz, J.; Cummings, K.L.; Hunt, D.F.; Cobbold, M.; Engelhard, V.H.; Willcox, B.E. The antigenic identity of human class I MHC phosphopeptides is critically dependent upon phosphorylation status. Oncotarget 2017, 8, 54160–54172. [Google Scholar] [CrossRef] [Green Version]
- Theivendran, S.; Tang, J.; Lei, C.; Yang, Y.; Song, H.; Gu, Z.; Wang, Y.; Yang, Y.; Jin, L.; Yu, C. Post translational modification-assisted cancer immunotherapy for effective breast cancer treatment. Chem. Sci. 2020, 11, 10421–10430. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fulton, K.M.; Twine, S.M.; Li, J. Identification of MHC Peptides Using Mass Spectrometry for Neoantigen Discovery and Cancer Vaccine Development. Mass Spectrom. Rev. 2021, 40, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Schwanninger, A.; Weinberger, B.; Almanzar, G.; Parson, W.; Buus, S.; Lindner, H.; Grubeck-Loebenstein, B. Oxidative stress can alter the antigenicity of immunodominant peptides. J. Leukoc. Biol. 2010, 87, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Depontieu, F.R.; Qian, J.; Zarling, A.L.; McMiller, T.L.; Salay, T.M.; Norris, A.; English, A.M.; Shabanowitz, J.; Engelhard, V.H.; Hunt, D.F.; et al. Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc. Natl. Acad. Sci. USA 2009, 106, 12073–12078. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, L.G.; Metheringham, R.L.; Brentville, V.A. Autophagy, citrullination and cancer. Autophagy 2016, 12, 1055–1056. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhong, L.; Guo, R. The Role of Posttranslational Modification and Mitochondrial Quality Control in Cardiovascular Diseases. Oxid. Med. Cell Longev. 2021, 2021, 6635836. [Google Scholar] [CrossRef]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.L.; Ledermann, J.A.; Aitkens, E.; Benjamin, E.; Katz, D.R.; Chain, B.M. Oxidation of ovarian epithelial cancer cells by hypochlorous acid enhances immunogenicity and stimulates T cells that recognize autologous primary tumor. Clin. Cancer Res. 2008, 14, 4898–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemi, N.M.; Pagliarini, D.J. The extensive and functionally uncharacterized mitochondrial phosphoproteome. J. Biol. Chem 2021, 297, 100880. [Google Scholar] [CrossRef]
- Gorska, M.; Marino Gammazza, A.; Zmijewski, M.A.; Campanella, C.; Cappello, F.; Wasiewicz, T.; Kuban-Jankowska, A.; Daca, A.; Sielicka, A.; Popowska, U.; et al. Geldanamycin-induced osteosarcoma cell death is associated with hyperacetylation and loss of mitochondrial pool of heat shock protein 60 (hsp60). PLoS ONE 2013, 8, e71135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhard, V.H.; Obeng, R.C.; Cummings, K.L.; Petroni, G.R.; Ambakhutwala, A.L.; Chianese-Bullock, K.A.; Smith, K.T.; Lulu, A.; Varhegyi, N.; Smolkin, M.E.; et al. MHC-restricted phosphopeptide antigens: Preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 2020, 8, e000262. [Google Scholar] [CrossRef] [PubMed]
- Penny, S.A.; Abelin, J.G.; Malaker, S.A.; Myers, P.T.; Saeed, A.Z.; Steadman, L.G.; Bai, D.L.; Ward, S.T.; Shabanowitz, J.; Hunt, D.F.; et al. Tumor Infiltrating Lymphocytes Target HLA-I Phosphopeptides Derived From Cancer Signaling in Colorectal Cancer. Front. Immunol. 2021, 12, 723566. [Google Scholar] [CrossRef]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Cruz, F.M.; Colbert, J.D.; Merino, E.; Kriegsman, B.A.; Rock, K.L. The Biology and Underlying Mechanisms of Cross-Presentation of Exogenous Antigens on MHC-I Molecules. Annu. Rev. Immunol. 2017, 35, 149–176. [Google Scholar] [CrossRef] [Green Version]
- Pinto, G.; Brou, C.; Zurzolo, C. Tunneling Nanotubes: The Fuel of Tumor Progression? Trends Cancer 2020, 6, 874–888. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2022, 17, 98–106. [Google Scholar] [CrossRef]
- Ma, F.; Vayalil, J.; Lee, G.; Wang, Y.; Peng, G. Emerging role of tumor-derived extracellular vesicles in T cell suppression and dysfunction in the tumor microenvironment. J. Immunother. Cancer 2021, 9, e003217. [Google Scholar] [CrossRef]
- Headley, M.B.; Bins, A.; Nip, A.; Roberts, E.W.; Looney, M.R.; Gerard, A.; Krummel, M.F. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 2016, 531, 513–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choong, C.J.; Okuno, T.; Ikenaka, K.; Baba, K.; Hayakawa, H.; Koike, M.; Yokota, M.; Doi, J.; Kakuda, K.; Takeuchi, T.; et al. Alternative mitochondrial quality control mediated by extracellular release. Autophagy 2021, 17, 2962–2974. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, H.; Tanaka, M.; Nagoya, M.; Fujimaki, H.; Sato, K.; Yago, T.; Nagata, T.; Minami, M. Epitope selection in major histocompatibility complex class I-mediated pathway is affected by the intracellular localization of an antigen. Eur. J. Immunol. 1997, 27, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 2016, 272, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.; English, L.; Boulais, J.; Chemali, M.; Caron-Lizotte, O.; Desjardins, M.; Thibault, P. Quantitative proteomics reveals the induction of mitophagy in tumor necrosis factor-alpha-activated (TNFalpha) macrophages. Mol. Cell Proteom. 2013, 12, 2394–2407. [Google Scholar] [CrossRef] [Green Version]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Fahmy, A.M.; Boulais, J.; Desjardins, M.; Matheoud, D. Mitochondrial antigen presentation: A mechanism linking Parkinson’s disease to autoimmunity. Curr. Opin. Immunol. 2019, 58, 31–37. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Kelley, R.E.; Olson, M.S.; Pinckard, R.N. Characterization of anti-heart mitochondria autoantibodies produced in dogs following myocardial infarction. Circ. Res. 1974, 35, 862–870. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Duvvuri, B.; Grigull, J.; Jamnik, R.; Wither, J.E.; Wu, G.E. Experimental evidence that mutated-self peptides derived from mitochondrial DNA somatic mutations have the potential to trigger autoimmunity. Hum. Immunol. 2014, 75, 873–879. [Google Scholar] [CrossRef]
- Loveland, B.; Wang, C.-R.; Yonekawa, H.; Hermel, E.; Lindahl, K.F. Maternally transmitted histocompatibility antigen of mice: A hydrophobic peptide of a mitochondrially encoded protein. Cell 1990, 60, 971–980. [Google Scholar] [CrossRef]
- Lindahl, K.F.; Hausmann, B.; Robinson, P.J.; Guénet, J.L.; Wharton, D.C.; Winking, H. Mta, the maternally transmitted antigen, is determined jointly by the chromosomal Hmt and the extrachromosomal Mtf genes. J. Exp. Med. 1986, 163, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Deuse, T.; Hu, X.; Agbor-Enoh, S.; Koch, M.; Spitzer, M.H.; Gravina, A.; Alawi, M.; Marishta, A.; Peters, B. De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat. Biotechnol. 2019, 37, 1137–1144. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prota, G.; Lechuga-Vieco, A.V.; De Libero, G. Mitochondrial Proteins as Source of Cancer Neoantigens. Int. J. Mol. Sci. 2022, 23, 2627. https://doi.org/10.3390/ijms23052627
Prota G, Lechuga-Vieco AV, De Libero G. Mitochondrial Proteins as Source of Cancer Neoantigens. International Journal of Molecular Sciences. 2022; 23(5):2627. https://doi.org/10.3390/ijms23052627
Chicago/Turabian StyleProta, Gennaro, Ana Victoria Lechuga-Vieco, and Gennaro De Libero. 2022. "Mitochondrial Proteins as Source of Cancer Neoantigens" International Journal of Molecular Sciences 23, no. 5: 2627. https://doi.org/10.3390/ijms23052627
APA StyleProta, G., Lechuga-Vieco, A. V., & De Libero, G. (2022). Mitochondrial Proteins as Source of Cancer Neoantigens. International Journal of Molecular Sciences, 23(5), 2627. https://doi.org/10.3390/ijms23052627