
Nitro-Deficient Niclosamide Confers Reduced Genotoxicity and Retains Mitochondrial Uncoupling Activity for Cancer Therapy

 ,
,
Abstract
:1. Introduction
2. Results
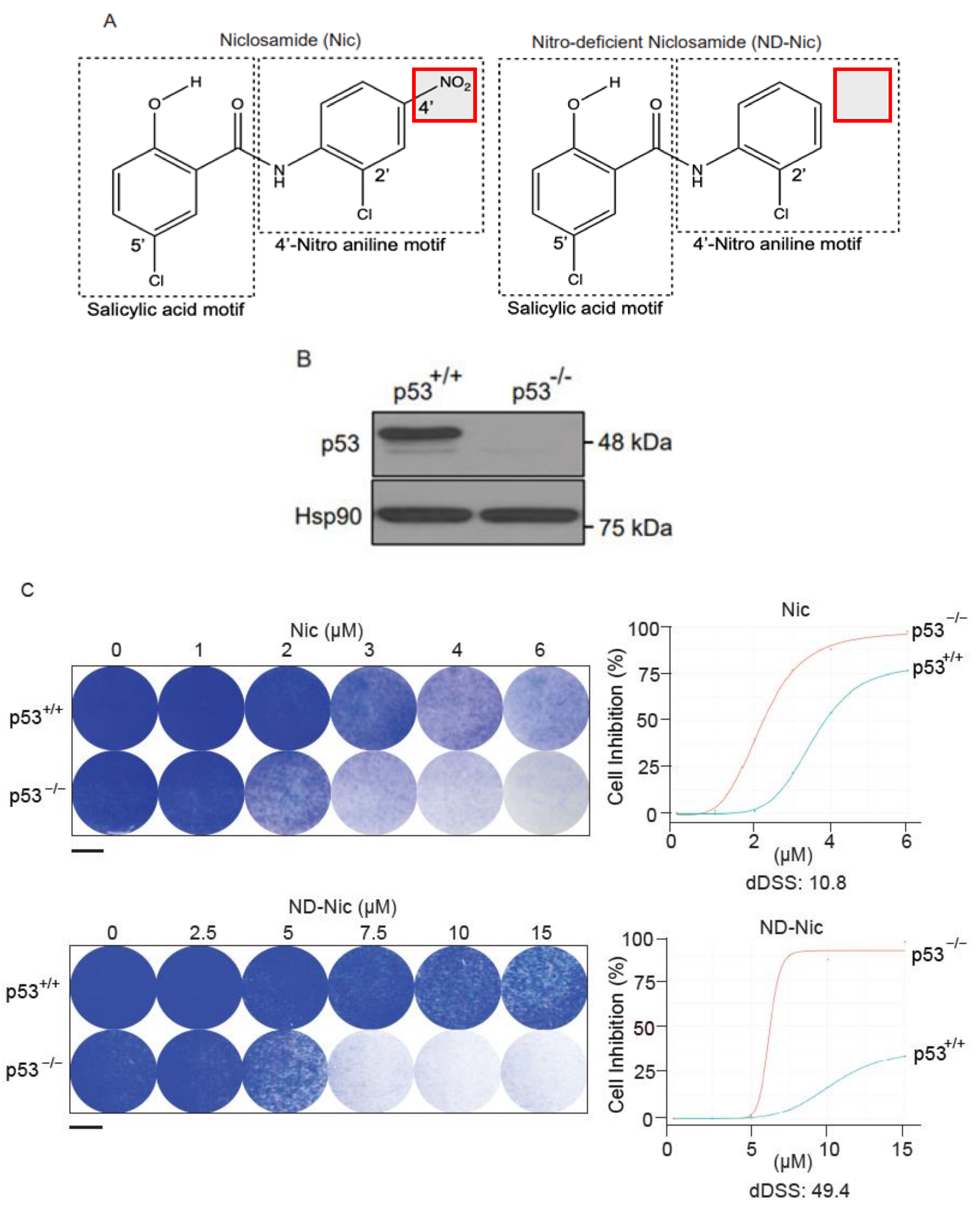
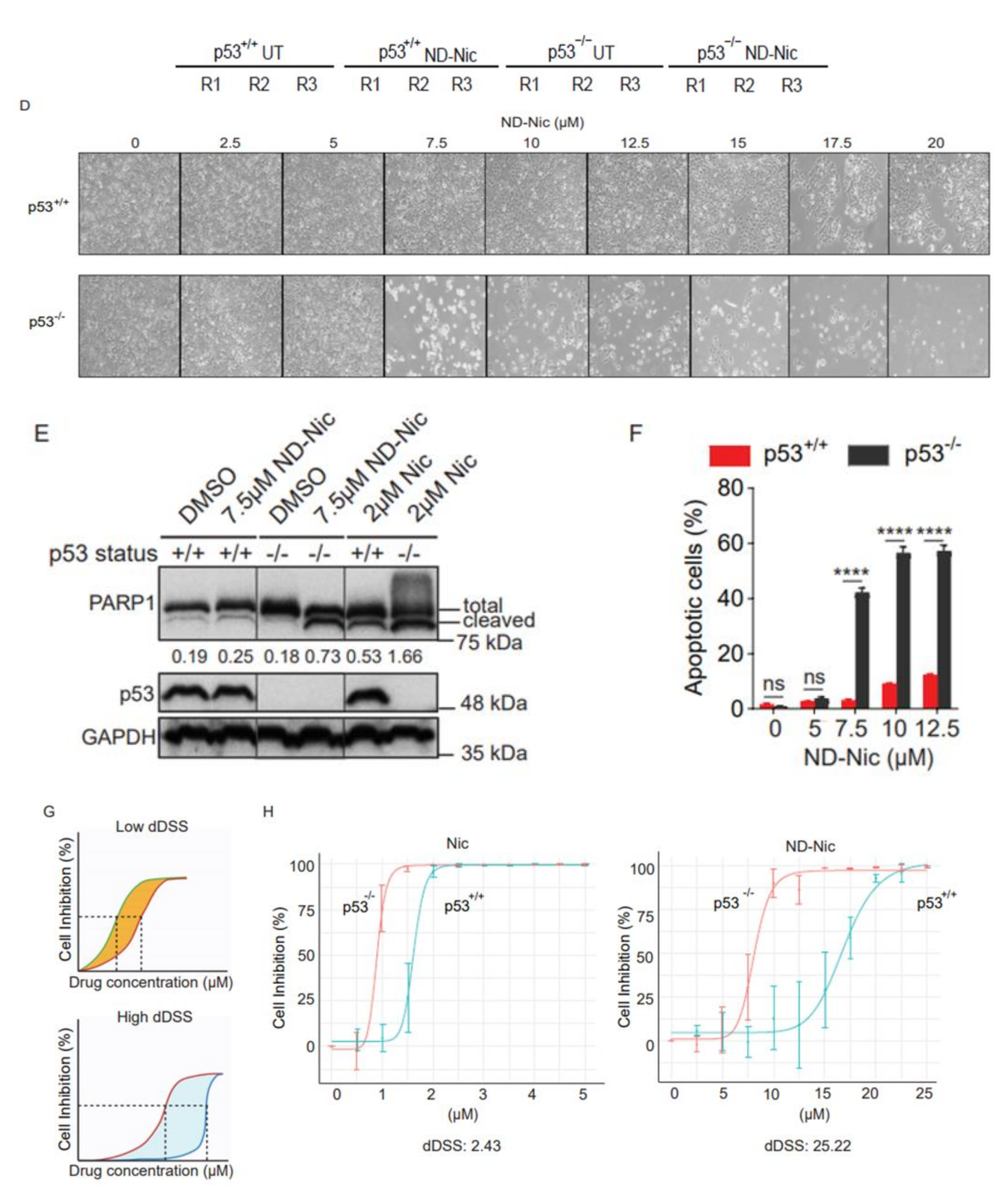
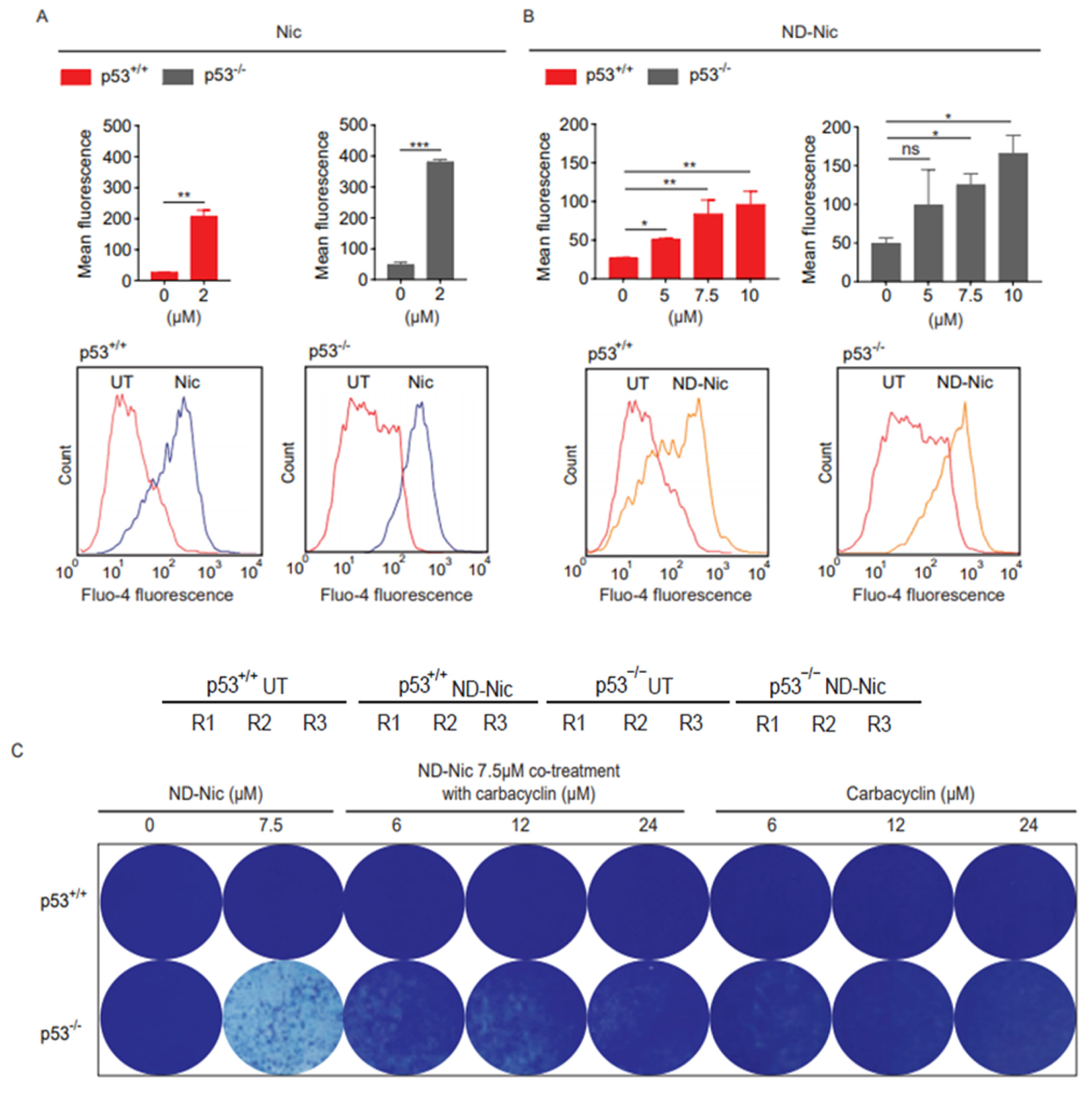
2.1. p53 Status Is a Determinant of Sensitivity to ND-Nic
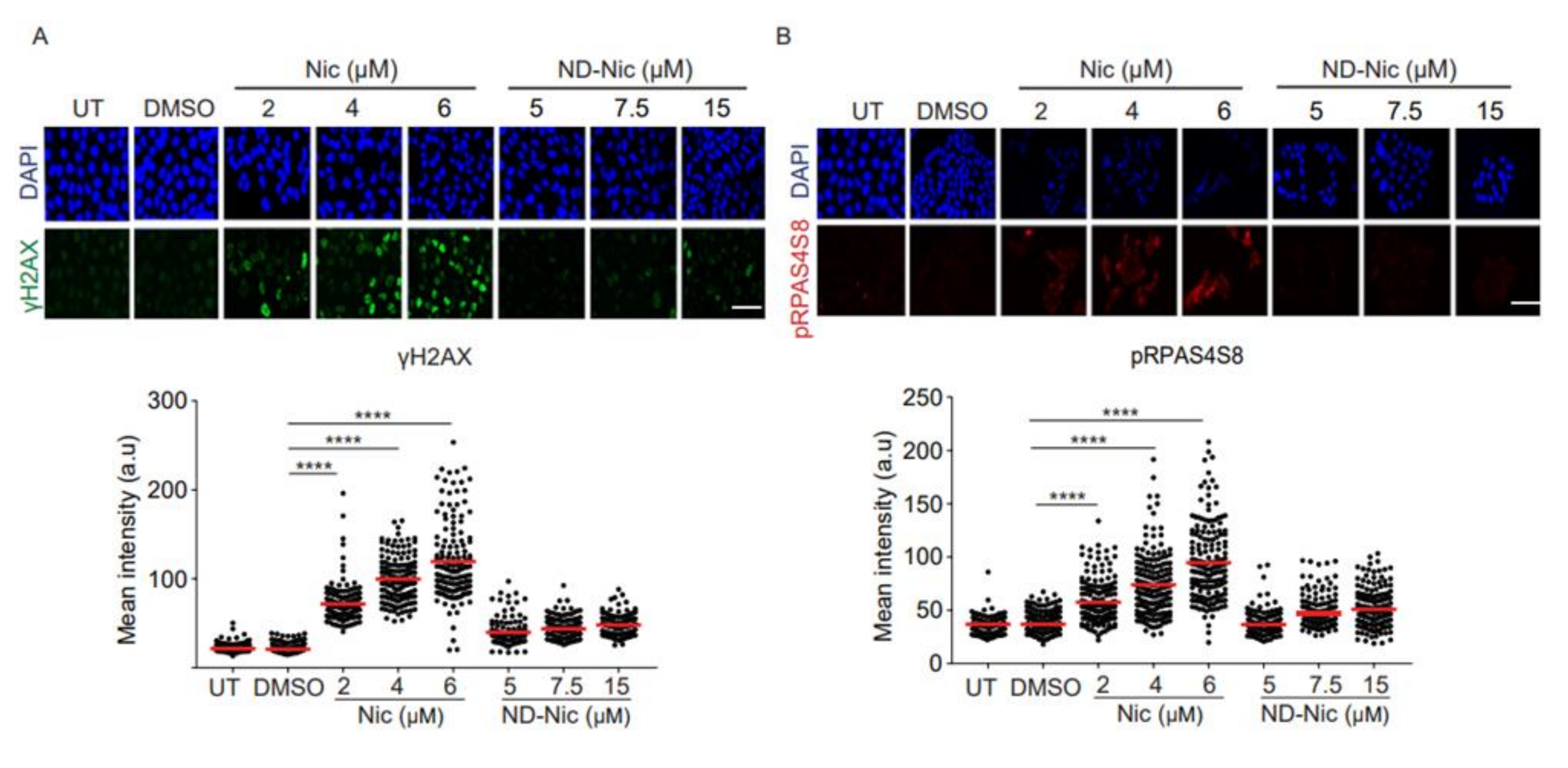
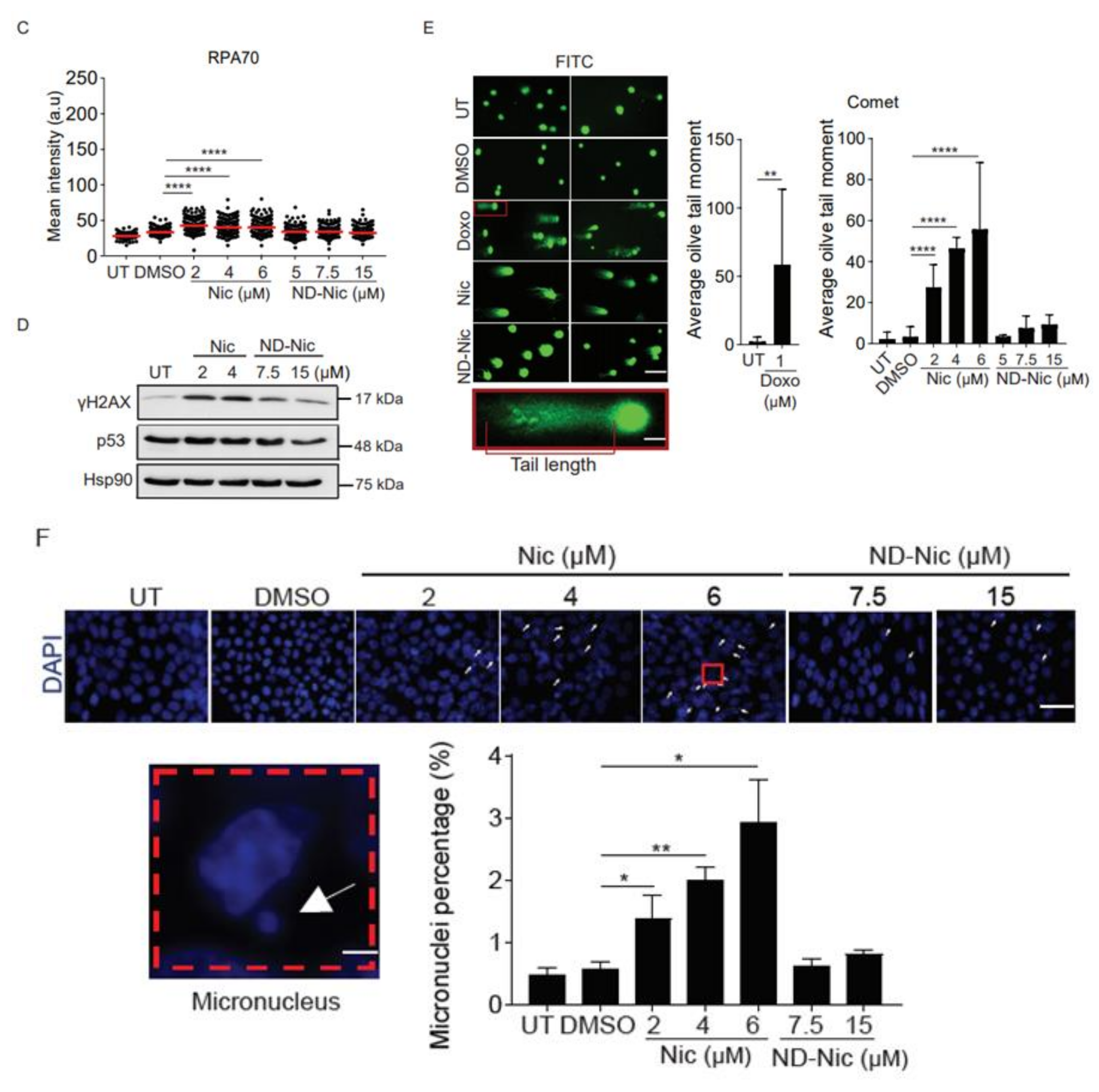
2.2. Niclosamide Induces Genome-Wide DNA Damage in Cancer Cells That Is Attenuated by the Elimination of Its Aniline 4′-NO2 Group
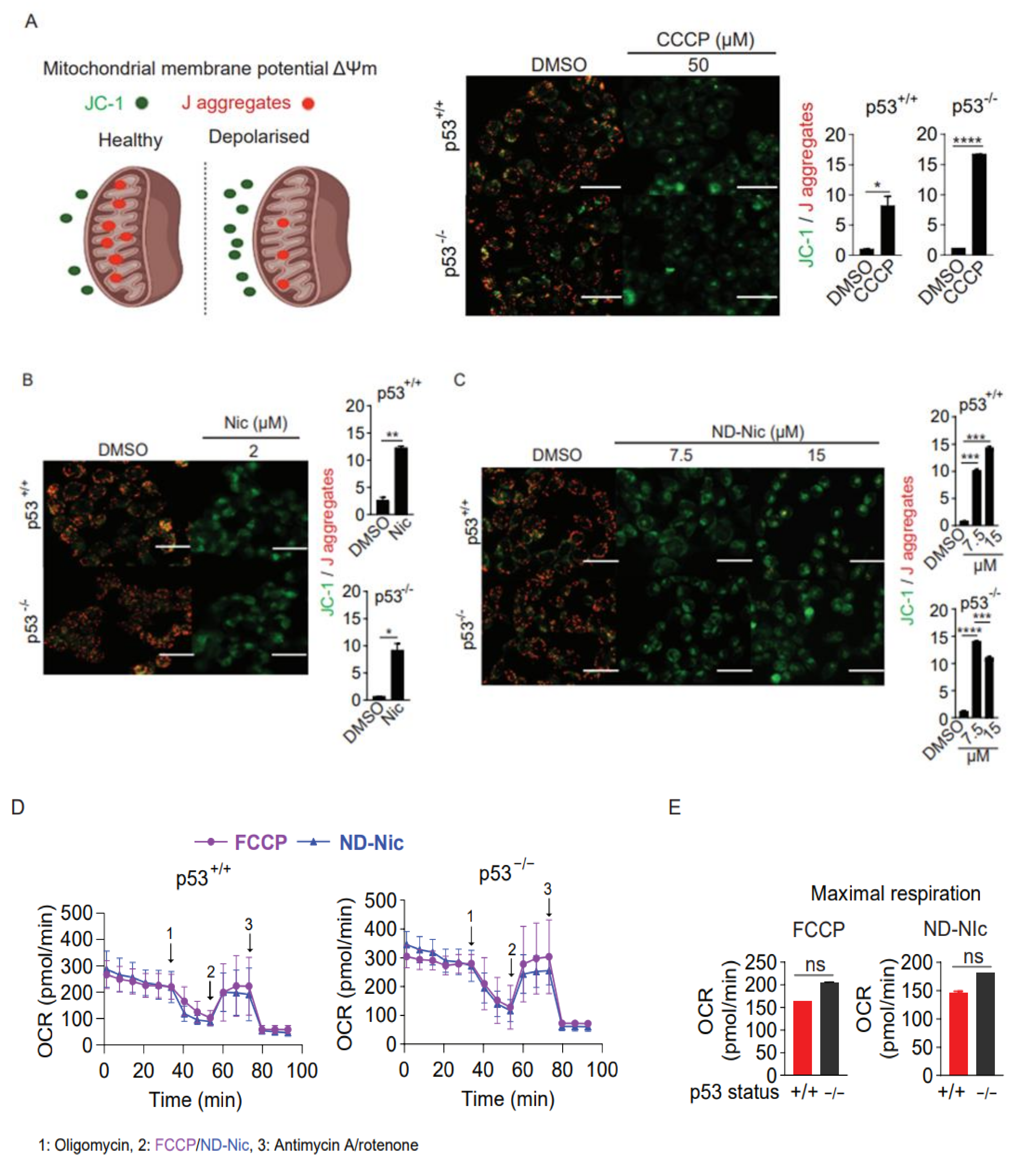
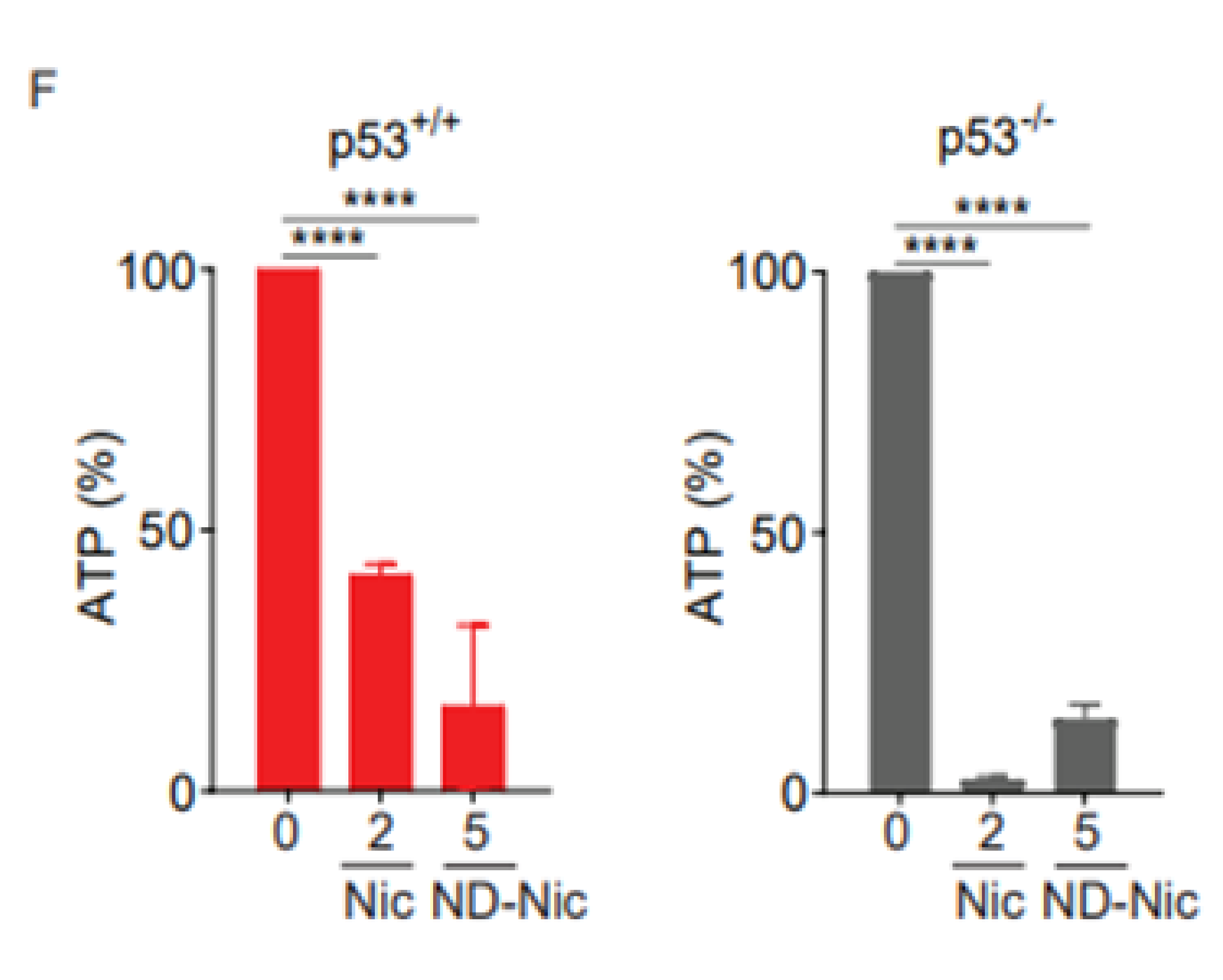
2.3. ND-Nic Induces Calcium Fluxes That Potentiate Its Antitumour Mechanism
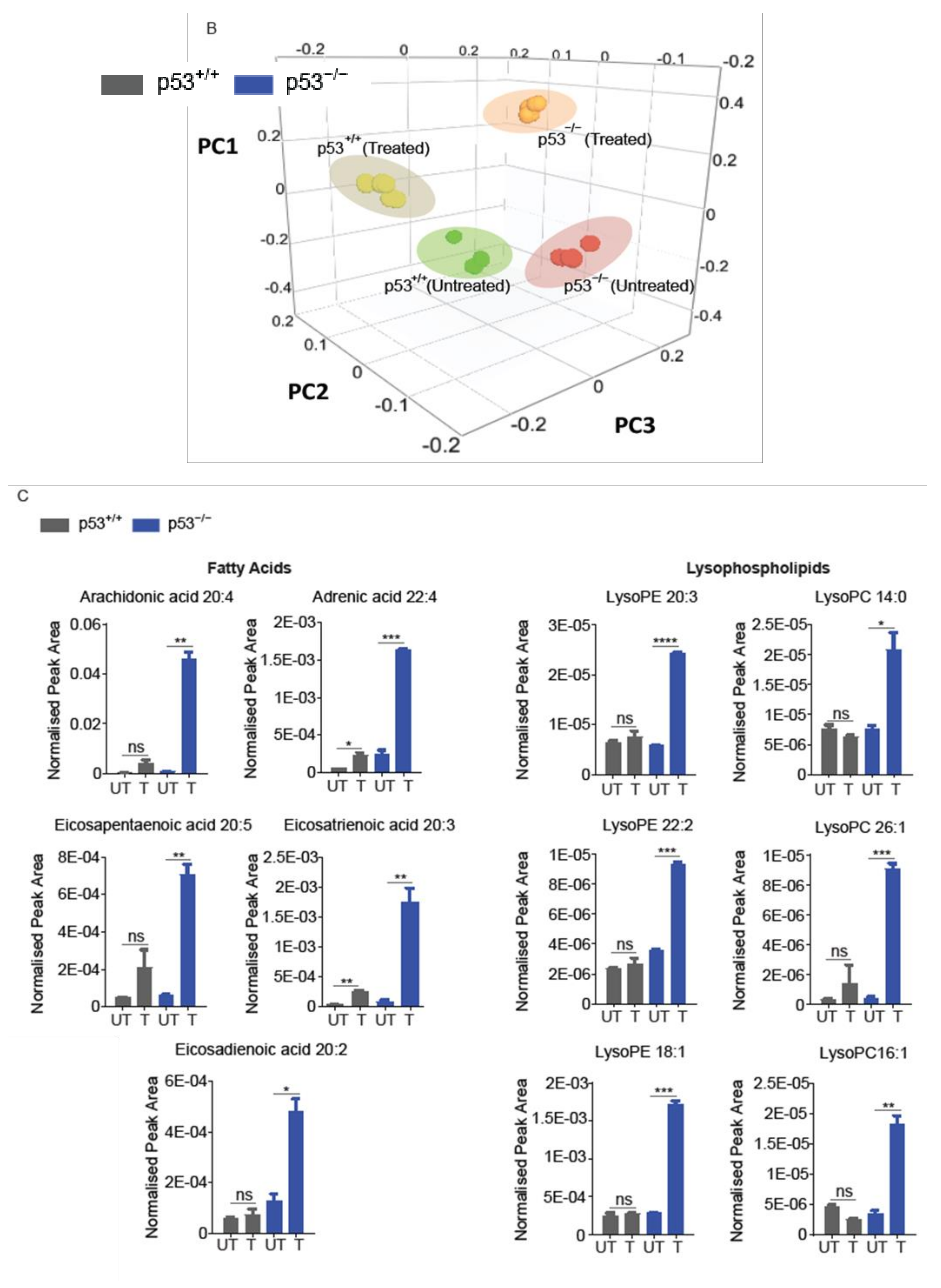
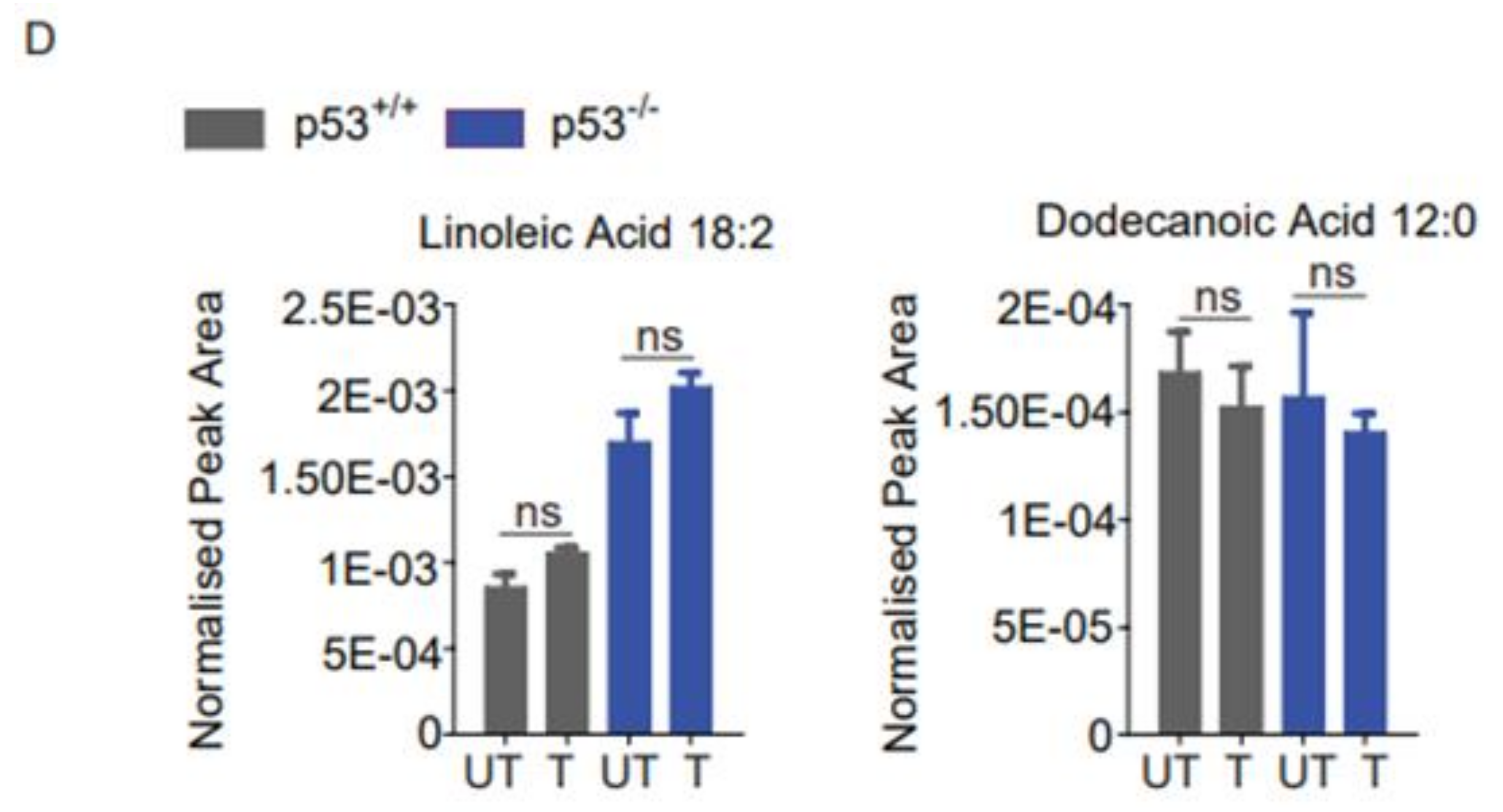
2.4. Lipidomics Profiling Revealed That ND-Nic Induces Accumulation of Long-Chain Fatty Acids and Arachidonic Acid in p53-Deficient Cancer Cells
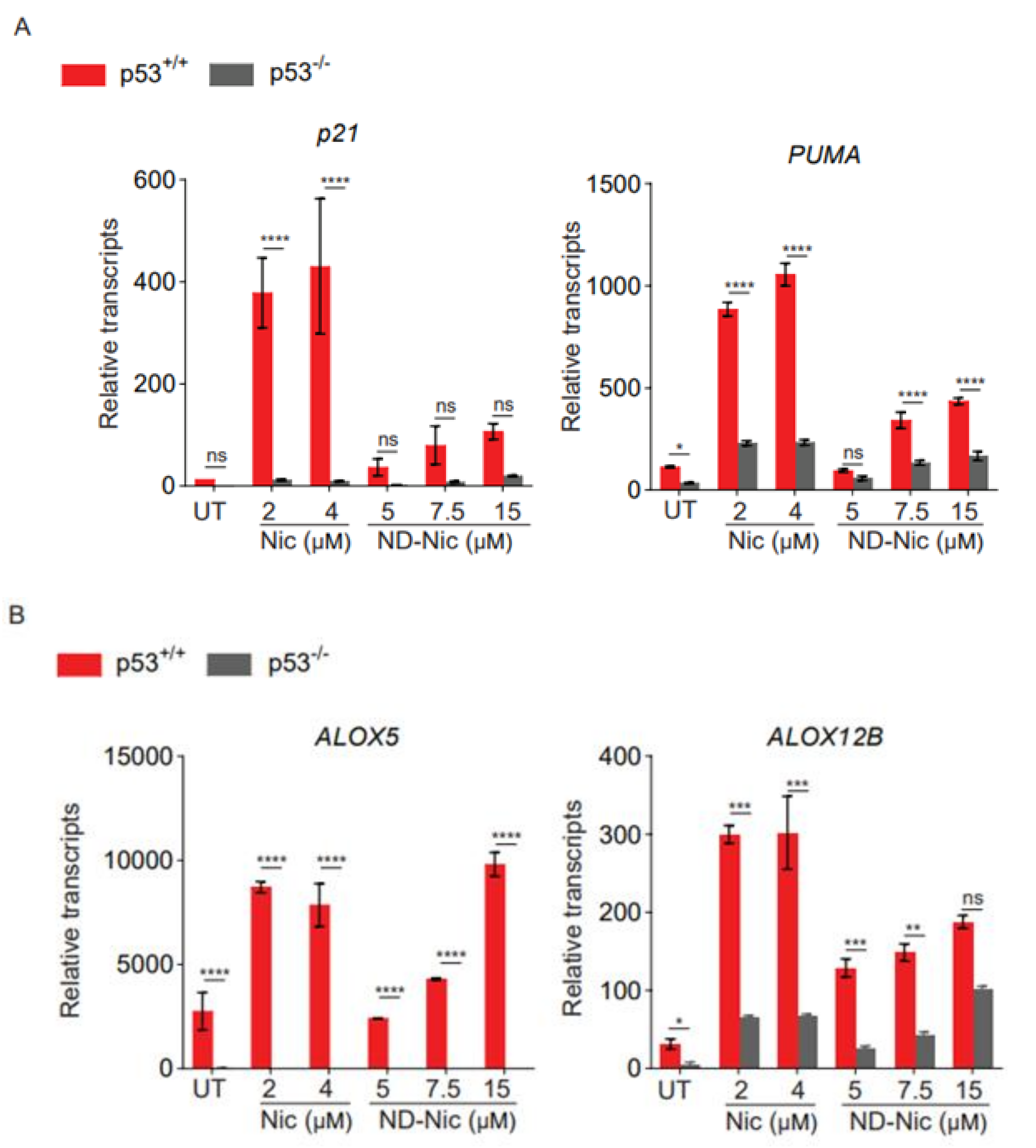
2.5. ND-Nic Activates the p53-Dependent Transcriptional Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. WST-1 Cell Proliferation Assay
4.3. Colony Forming Assay
4.4. ATP Level Measurement
4.5. Fluo-4, AM Calcium Measurement
4.6. Western Blot
4.7. Quantitative Real-Time PCR
4.8. Immunofluorescence
4.9. Comet Assay
4.10. Mitochondrial Uncoupling Assay
4.11. Drug Sensitivity Score Analysis
4.12. Lipidomics Profiling
4.13. Mitochondrial Cellular Respiration
4.14. Annexin-V Apoptosis Assay
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Hadiya, B.M. Niclosamide: Comprehensive profile. Profiles Drug Subst. Excip. Relat. Methodol. 2005, 32, 67–96. [Google Scholar] [PubMed]
- World Health Organization. WHO Technical Report Series: The Selection and Use of Essential Medicines; WHO: Geneva, Switzerland, 2007. [Google Scholar]
- Alasadi, A.; Chen, M.; Swapna, G.; Tao, H.; Guo, J.; Collantes, J.; Fadhil, N.; Montelione, G.T.; Jin, S. Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death Dis. 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frayha, G.J.; Smyth, J.; Gobert, J.G.; Savel, J. The mechanisms of action of antiprotozoal and anthelmintic drugs in man. Gen. Pharmacol. Vasc. Syst. 1997, 28, 273–299. [Google Scholar] [CrossRef]
- Weinbach, E.C.; Garbus, J. Mechanism of action of reagents that uncouple oxidative phosphorylation. Nature 1969, 221, 1016–1018. [Google Scholar] [CrossRef]
- Nguyen, C.; Pandey, S. Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells. Cancers 2019, 11, 916. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, R.; Johnson, E.; Byrne, F.L. Exploring the therapeutic potential of mitochondrial uncouplers in cancer. Mol. Metab. 2021, 51, 101222. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Moskaleva, E.Y.; Perevozchikova, V.; Zhirnik, A.; Severin, S. Molecular mechanisms of antitumor activity of niclosamide. Biochem. Suppl. Ser. B Biomed. Chem. 2015, 9, 312–324. [Google Scholar]
- Arend, R.C.; Londoño-Joshi, A.I.; Samant, R.S.; Li, Y.; Conner, M.; Hidalgo, B.; Alvarez, R.D.; Landen, C.N.; Straughn, J.M.; Buchsbaum, D.J. Inhibition of Wnt/β-catenin pathway by niclosamide: A therapeutic target for ovarian cancer. Gynecol. Oncol. 2014, 134, 112–120. [Google Scholar] [CrossRef]
- Ye, T.; Xiong, Y.; Yan, Y.; Xia, Y.; Song, X.; Liu, L.; Li, D.; Wang, N.; Zhang, L.; Zhu, Y. The anthelmintic drug niclosamide induces apoptosis, impairs metastasis and reduces immunosuppressive cells in breast cancer model. PLoS ONE 2014, 9, e85887. [Google Scholar]
- Yo, Y.-T.; Lin, Y.-W.; Wang, Y.-C.; Balch, C.; Huang, R.-L.; Chan, M.W.; Sytwu, H.-K.; Chen, C.-K.; Chang, C.-C.; Nephew, K.P. Growth inhibition of ovarian tumor–initiating cells by niclosamide. Mol. Cancer Ther. 2012, 11, 1703–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Kobelt, D.; Lemm, M.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J. Natl. Cancer Inst. 2011, 103, 1018–1036. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Coronel, L.; Somalanka, B.; Raju, A.; Aning, O.; An, O.; Ho, Y.; Chen, S.; Mak, S.; Hor, P. Mitochondrial uncoupling reveals a novel therapeutic opportunity for p53-defective cancers. Nat. Commun. 2018, 9, 3931. [Google Scholar] [CrossRef] [Green Version]
- King, M.L.; Lindberg, M.E.; Stodden, G.R.; Okuda, H.; Ebers, S.D.; Johnson, A.; Montag, A.; Lengyel, E.; MacLean Ii, J.A.; Hayashi, K. WNT7A/beta-catenin signaling induces FGF1 and influences sensitivity to niclosamide in ovarian cancer. Oncogene 2015, 34, 3452–3462. [Google Scholar] [CrossRef] [Green Version]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The NIKOLO trial. BMC Cancer 2018, 18, 297. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Haugk, K.; McKiernan, J.S.; Gulati, R.; Cheng, H.H.; Maes, J.L.; Dumpit, R.F.; Nelson, P.S.; Montgomery, B.; McCune, J.S. Correction: A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE 2018, 13, e0202709. [Google Scholar] [CrossRef] [PubMed]
- Parikh, M.; Liu, C.; Wu, C.-Y.; Evans, C.P.; Dall’Era, M.; Robles, D.; Lara, P.N.; Agarwal, N.; Gao, A.C.; Pan, C.-X. Phase Ib trial of reformulated niclosamide with abiraterone/prednisone in men with castration-resistant prostate cancer. Sci. Rep. 2021, 11, 6377. [Google Scholar] [CrossRef]
- Pearson, R.D.; Hewlett, E.L. Niclosamide therapy for tapeworm infections. In American College of Physicians. Ann. Intern. Med. 1985, 102, 550–551. [Google Scholar] [CrossRef]
- Andrews, P.; Thyssen, J.; Lorke, D. The biology and toxicology of molluscicides, Bayluscide. Pharmacol. Ther. 1982, 19, 245–295. [Google Scholar] [CrossRef]
- Bhagat, H.A.; Compton, S.A.; Musso, D.L.; Laudeman, C.P.; Jackson, K.M.; Yi, N.Y.; Nierobisz, L.S.; Forsberg, L.; Brenman, J.E.; Sexton, J.Z. N-substituted phenylbenzamides of the niclosamide chemotype attenuate obesity related changes in high fat diet fed mice. PLoS ONE 2018, 13, e0204605. [Google Scholar] [CrossRef] [PubMed]
- Shamim, K.; Xu, M.; Hu, X.; Lee, E.M.; Lu, X.; Huang, R.; Shah, P.; Xu, X.; Chen, C.Z.; Shen, M. Application of niclosamide and analogs as small molecule inhibitors of Zika virus and SARS-CoV-2 infection. Bioorganic Med. Chem. Lett. 2021, 40, 127906. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Somanathan, R. Nitroaromatic compounds: Environmental toxicity, carcinogenicity, mutagenicity, therapy and mechanism. J. Appl. Toxicol. 2014, 34, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Basu, A.K. Mutagenicity of nitroaromatic compounds. Chem. Res. Toxicol. 2000, 13, 673–692. [Google Scholar] [CrossRef]
- Beristain-Castillo, E.; Martínez-Vázquez, M.; Camacho-Carranza, R.; Espinosa-Aguirre, J.J. CYP1A1 and Cnr nitroreductase bioactivated niclosamide in vitro. Mutagenesis 2013, 28, 645–651. [Google Scholar] [CrossRef]
- Espinosa-Aguirre, J.; Reyes, R.; de Nava, C.C. Mutagenic activity of 2-chloro-4-nitroaniline and 5-chlorosalicylic acid in Salmonella typhimurium: Two possible metabolites of niclosamide. Mutat. Res. Lett. 1991, 264, 139–145. [Google Scholar] [CrossRef]
- De Nava, C.C.; Espinosa, J.; Garcia, L.; Zapata, A.; Martinez, E. Mutagenicity of antiamebic and anthelmintic drugs in the Salmonella typhimurium microsomal test system. Mutat. Res./Genet. Toxicol. 1983, 117, 79–91. [Google Scholar] [CrossRef]
- Shimizu, M.; Yano, E. Mutagenicity of mono-nitrobenzene derivatives in the Ames test and rec assay. Mutat. Res./Genet. Toxicol. 1986, 170, 11–22. [Google Scholar] [CrossRef]
- Claus, S.P.; Guillou, H.; Ellero-Simatos, S. The gut microbiota: A major player in the toxicity of environmental pollutants? Npj Biofilms Microbiomes 2017, 3, 17001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.N.; Josimovich, J.B.; Tsou, K.C.; Seligman, A.M. Nitroreductase activity of animal tissues and of normal and neoplastic human tissues. Cancer 1956, 9, 176–182. [Google Scholar] [CrossRef]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Duan, L.; He, Q.; Zhang, Z.; Zhou, Y.; Wu, D.; Pan, J.; Pei, D.; Ding, K. Identification of niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. Acs Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Lin, C.; Roberts, M.J.; Waud, W.R.; Piazza, G.A.; Li, Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS ONE 2011, 6, e29290. [Google Scholar] [CrossRef]
- Wang, A.M.; Ku, H.H.; Liang, Y.C.; Chen, Y.C.; Hwu, Y.M.; Yeh, T.S. The autonomous notch signal pathway is activated by baicalin and baicalein but is suppressed by niclosamide in K562 cells. J. Cell. Biochem. 2009, 106, 682–692. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Jungsuwadee, P.; Vore, M.; Butterfield, D.A.; St Clair, D.K. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol. Interv. 2007, 7, 147. [Google Scholar] [CrossRef]
- Altmeyer, M.; Lukas, J. Guarding against collateral damage during chromatin transactions. Cell 2013, 153, 1431–1434. [Google Scholar] [CrossRef] [Green Version]
- Pinoli, P.; Srihari, S.; Wong, L.; Ceri, S. Identifying collateral and synthetic lethal vulnerabilities within the DNA-damage response. BMC Bioinform. 2021, 22, 250. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Yadav, B.; Pemovska, T.; Szwajda, A.; Kulesskiy, E.; Kontro, M.; Karjalainen, R.; Majumder, M.M.; Malani, D.; Murumägi, A.; Knowles, J. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep. 2014, 4, 5193. [Google Scholar] [CrossRef] [Green Version]
- Aning, O.A.; Cheok, C.F. Drugging in the absence of p53. J. Mol. Cell Biol. 2019, 11, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.; El-Osta, A.; Karagiannis, T. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Cheok, C.F.; Dey, A.; Lane, D.P. Cyclin-dependent kinase inhibitors sensitize tumor cells to nutlin-induced apoptosis: A potent drug combination. Mol. Cancer Res. 2007, 5, 1133–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. In DNA Repair Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 613–626. [Google Scholar]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, Y.; Yang, C. Evaluating in vitro DNA damage using comet assay. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Kwon, M.; Leibowitz, M.L.; Lee, J.-H. Small but mighty: The causes and consequences of micronucleus rupture. Exp. Mol. Med. 2020, 52, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Cveticanin, J.; Joksic, G.; Leskovac, A.; Petrovic, S.; Sobot, A.V.; Neskovic, O. Using carbon nanotubes to induce micronuclei and double strand breaks of the DNA in human cells. Nanotechnology 2009, 21, 015102. [Google Scholar] [CrossRef]
- Bhuyan, B.; Zimmer, D.; Mazurek, J.; Trzos, R.; Harbach, P.; Shu, V.; Johnson, M. Comparative genotoxicity of adriamycin and menogarol, two anthracycline antitumor agents. Cancer Res. 1983, 43, 5293–5297. [Google Scholar]
- Aydemir, N.; Bilaloğlu, R. Genotoxicity of two anticancer drugs, gemcitabine and topotecan, in mouse bone marrow in vivo. 2003, 537, 43–51. Mutat Res. 2003, 537, 43–51. [Google Scholar] [CrossRef]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors. Proc. Natl. Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.; Surralles, J.; Crott, J.; Parry, J.; Norppa, H.; Eastmond, D.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, H.; Neary, G.; Williamson, F. The relative biological efficiency of single doses of fast neutrons and gamma-rays on Vicia faba roots and the effect of oxygen: Part II. Chromosome damage: The production of micronuclei. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1959, 1, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Stich, H.; Rosin, M. Micronuclei in exfoliated human cells as a tool for studies in cancer risk and cancer intervention. Cancer Lett. 1984, 22, 241–253. [Google Scholar] [CrossRef]
- Terradas, M.; Martín, M.; Tusell, L.; Genescà, A. Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat. Res. Rev. Mutat. Res. 2010, 705, 60–67. [Google Scholar] [CrossRef]
- Chan, S.L.; Liu, D.; Kyriazis, G.A.; Bagsiyao, P.; Ouyang, X.; Mattson, M.P. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J. Biol. Chem. 2006, 281, 37391–37403. [Google Scholar] [CrossRef] [Green Version]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Scorrano, L.; Penzo, D.; Petronilli, V.; Pagano, F.; Bernardi, P. Arachidonic acid causes cell death through the mitochondrial permeability transition: Implications for tumor necrosis factor-α apoptotic signaling. J. Biol. Chem. 2001, 276, 12035–12040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tertyshnikova, S.; Fein, A. Inhibition of inositol 1, 4, 5-trisphosphate-induced Ca2+ release by cAMP-dependent protein kinase in a living cell. Proc. Natl. Acad. Sci. USA 1998, 95, 1613–1617. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G.I.; Jin, S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med. 2014, 20, 1263–1269. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Acuna, U.M.; Fernandes, N.F.; Chettiar, S.; Li, P.-K.; De Blanco, E.C. Structure–Activity Relationship of Niclosamide Derivatives. Anticancer Res. 2017, 37, 2839–2843. [Google Scholar]
- Demine, S.; Reddy, N.; Renard, P.; Raes, M.; Arnould, T. Unraveling biochemical pathways affected by mitochondrial dysfunctions using metabolomic approaches. Metabolites 2014, 4, 831–878. [Google Scholar] [CrossRef] [Green Version]
- Nikolakopoulou, Z.; Shaikh, M.H.; Dehlawi, H.; Michael-Titus, A.T.; Parkinson, E.K. The induction of apoptosis in pre-malignant keratinocytes by omega-3 polyunsaturated fatty acids docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) is inhibited by albumin. Toxicol. Lett. 2013, 218, 150–158. [Google Scholar] [CrossRef]
- Dutta, S.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Zebisch, A.; Bullinger, L.; Berghold, A.; Döhner, K.; Sill, H. Functional classification of TP53 mutations in acute myeloid leukemia. Cancers 2020, 12, 637. [Google Scholar] [CrossRef] [Green Version]
- Tuna, M.; Ju, Z.; Yoshihara, K.; Amos, C.I.; Tanyi, J.L.; Mills, G.B. Clinical relevance of TP53 hotspot mutations in high-grade serous ovarian cancers. Br. J. Cancer 2020, 122, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Zhang, L.; Zhang, Y.; Chelluri, R.; Boufraqech, M.; Nilubol, N.; Patel, D.; Shen, M.; Kebebew, E. Identification of niclosamide as a novel anticancer agent for adrenocortical carcinoma. Clin. Cancer Res. 2016, 22, 3458–3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G.I.; Jin, S. Niclosamide ethanolamine improves blood glycemic control and reduces hepatic steatosis in mice. Nat. Med. 2014, 20, 1263. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.H.; Chong, W.P.; Ng, S.W.; Basri, N.; Xu, S.; Lam, K.P. Aberrant presentation of self-lipids by autoimmune B cells depletes peripheral iNKT cells. Cell Rep. 2014, 9, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Jurgeit, A.; McDowell, R.; Moese, S.; Meldrum, E.; Schwendener, R.; Greber, U.F. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog. 2012, 8, e1002976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, K.H.; Turner, N.; Bentley, N.L.; Das, A.; Wu, L.E.; Richani, D.; Bustamante, S.; Gilchrist, R.; Morris, M.; Shepherd, P.R.; et al. Niclosamide reduces glucagon sensitivity via hepatic PKA inhibition in obese mice: Implications for glucose metabolism improvements in type 2 diabetes. Sci. Rep. 2017, 7, 40159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Gautam, P.; Wennerberg, K.; Aittokallio, T. A normalized drug response metric improves accuracy and consistency of anticancer drug sensitivity quantification in cell-based screening. Commun. Biol. 2020, 3, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Mogi, A.; Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 583929. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | qPCR Primers |
|---|---|
| Alox5 | Forward: 5′–GGTACCTGAAGTACATCACGCTGA–3′ |
| Reverse: 5′–CGTCGGTGTTGCTTGAGAATGTGA–3′ | |
| Alox12B | Forward: 5′–ACGGCCGTATCTACCACTTC–3′ |
| Reverse: 5′–AGTCCTGCTTGGCTCTGATC–3′ | |
| p21 | Forward: 5′–GAAAACGGCGGCAGACCAGC–3′ |
| Reverse: 5′–TGTAGAGCGGGCCTTTGAGG–3′ | |
| PUMA | Forward: 5′–ACGACCTCAACGCACAGTACG–3′ |
| Reverse: 5′–TCCCATGATGAGATTGTACAGGAC–3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngai, T.W.; Elfar, G.A.; Yeo, P.; Phua, N.; Hor, J.H.; Chen, S.; Ho, Y.S.; Cheok, C.F. Nitro-Deficient Niclosamide Confers Reduced Genotoxicity and Retains Mitochondrial Uncoupling Activity for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 10420. https://doi.org/10.3390/ijms221910420
Ngai TW, Elfar GA, Yeo P, Phua N, Hor JH, Chen S, Ho YS, Cheok CF. Nitro-Deficient Niclosamide Confers Reduced Genotoxicity and Retains Mitochondrial Uncoupling Activity for Cancer Therapy. International Journal of Molecular Sciences. 2021; 22(19):10420. https://doi.org/10.3390/ijms221910420
Chicago/Turabian StyleNgai, Tsz Wai, Gamal Ahmed Elfar, Pearlyn Yeo, Nicholas Phua, Jin Hui Hor, Shuwen Chen, Ying Swan Ho, and Chit Fang Cheok. 2021. "Nitro-Deficient Niclosamide Confers Reduced Genotoxicity and Retains Mitochondrial Uncoupling Activity for Cancer Therapy" International Journal of Molecular Sciences 22, no. 19: 10420. https://doi.org/10.3390/ijms221910420
APA StyleNgai, T. W., Elfar, G. A., Yeo, P., Phua, N., Hor, J. H., Chen, S., Ho, Y. S., & Cheok, C. F. (2021). Nitro-Deficient Niclosamide Confers Reduced Genotoxicity and Retains Mitochondrial Uncoupling Activity for Cancer Therapy. International Journal of Molecular Sciences, 22(19), 10420. https://doi.org/10.3390/ijms221910420