
Conformational Dynamics of the Soluble and Membrane-Bound Forms of Interleukin-1 Receptor Type-1: Insights into Linker Flexibility and Domain Orientation


Abstract
:1. Introduction
2. Results
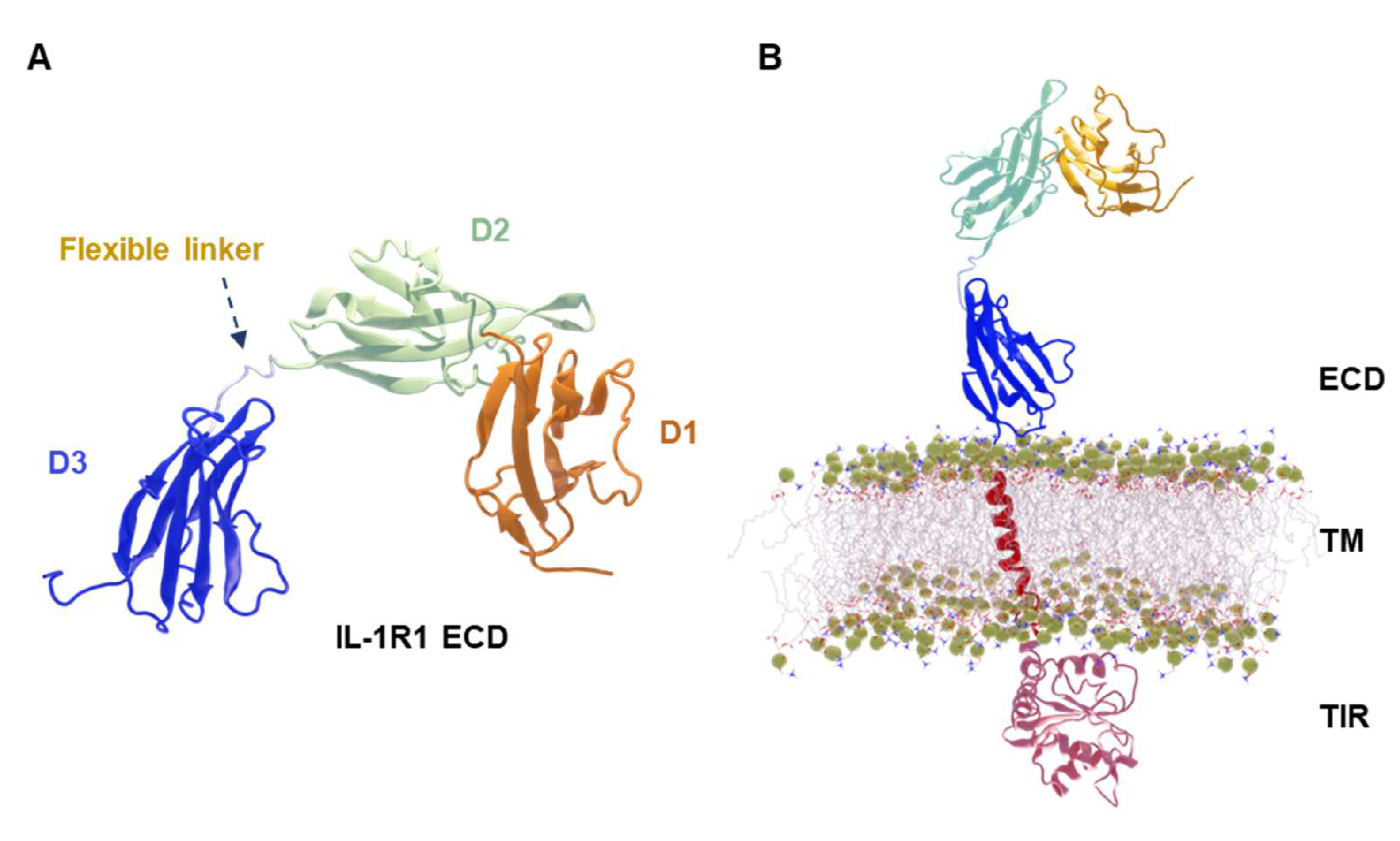
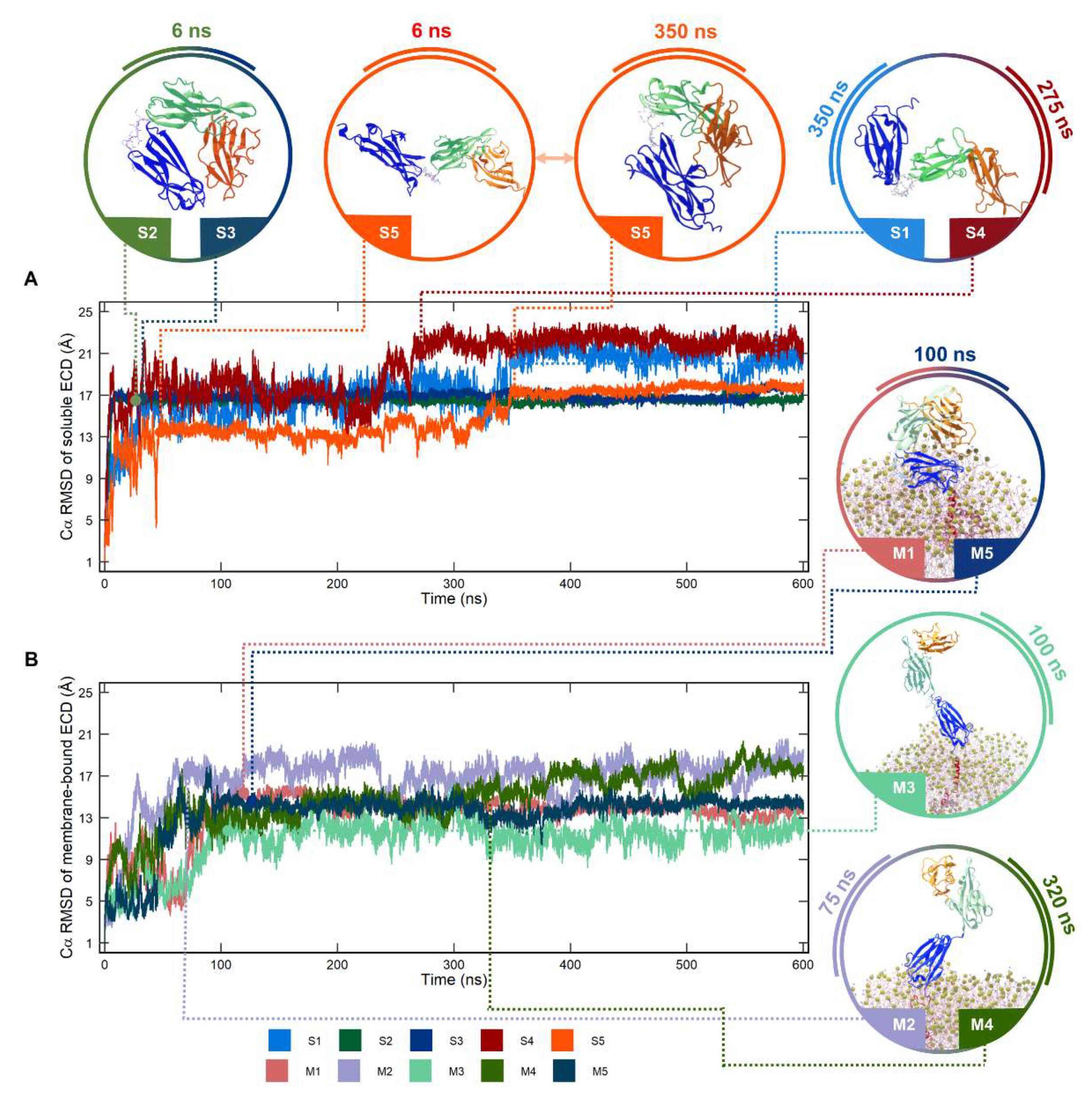
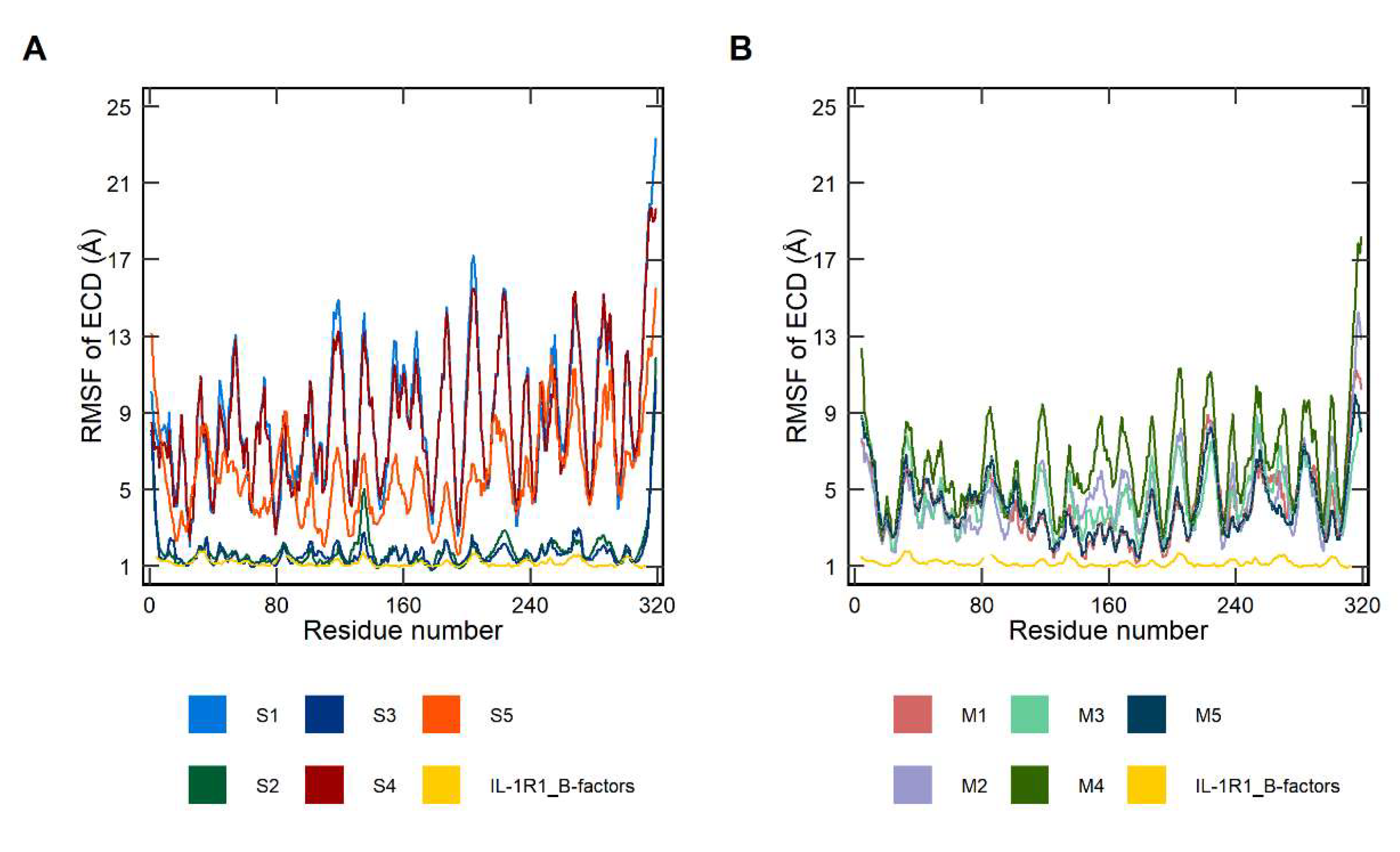
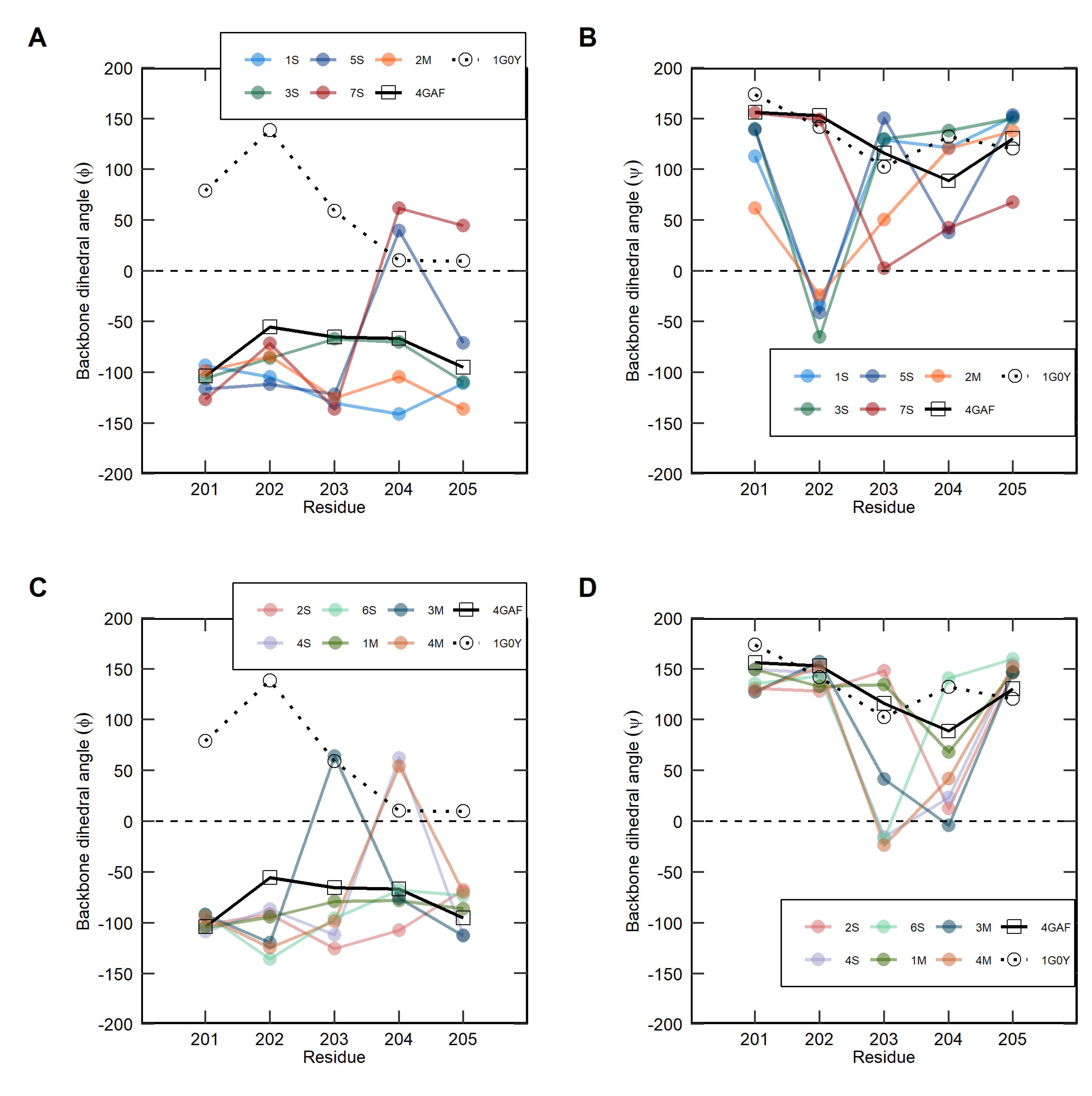
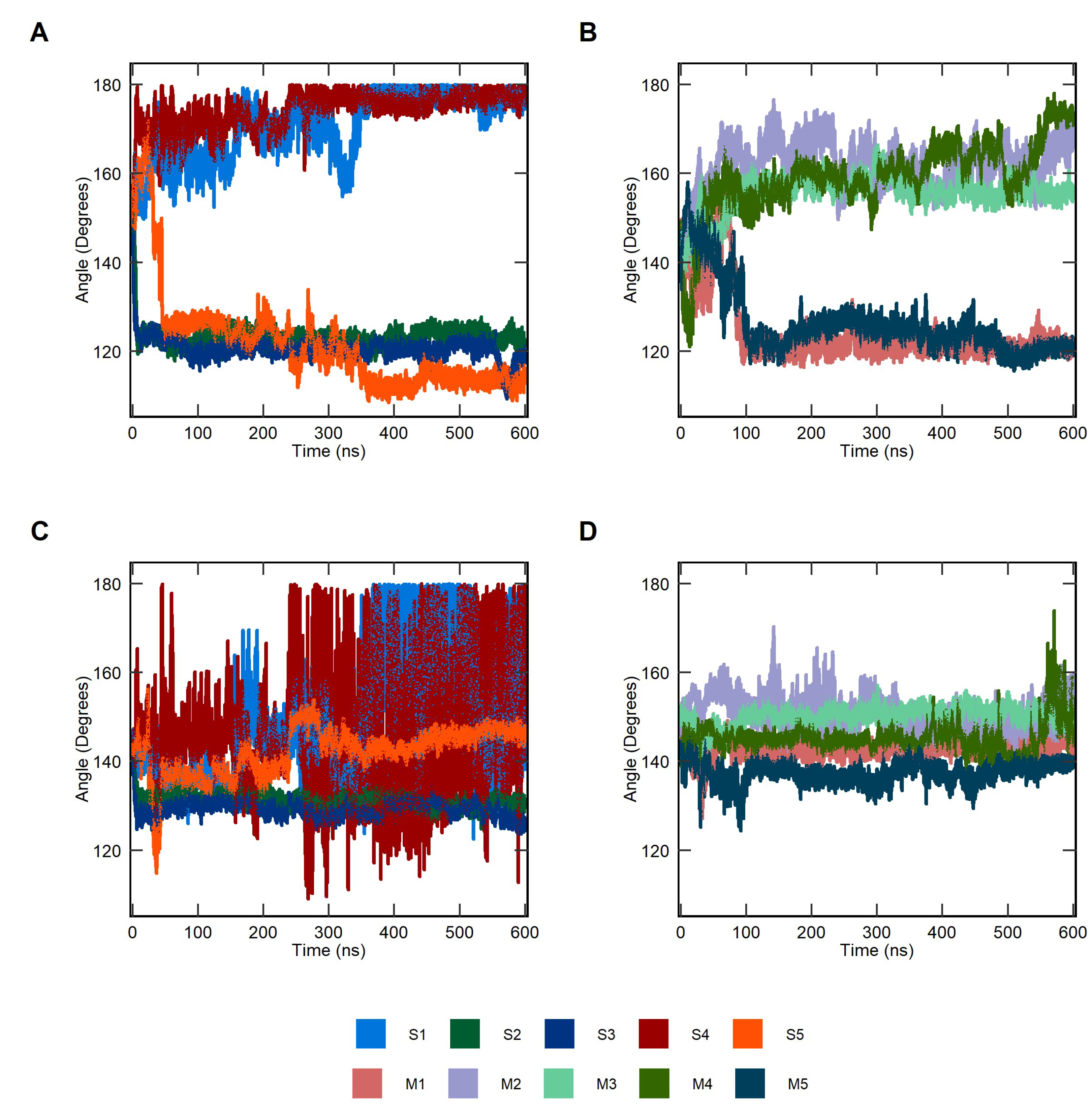
2.1. Conformational Dynamics of Soluble and Membrane-Bound IL-1R1-ECDs
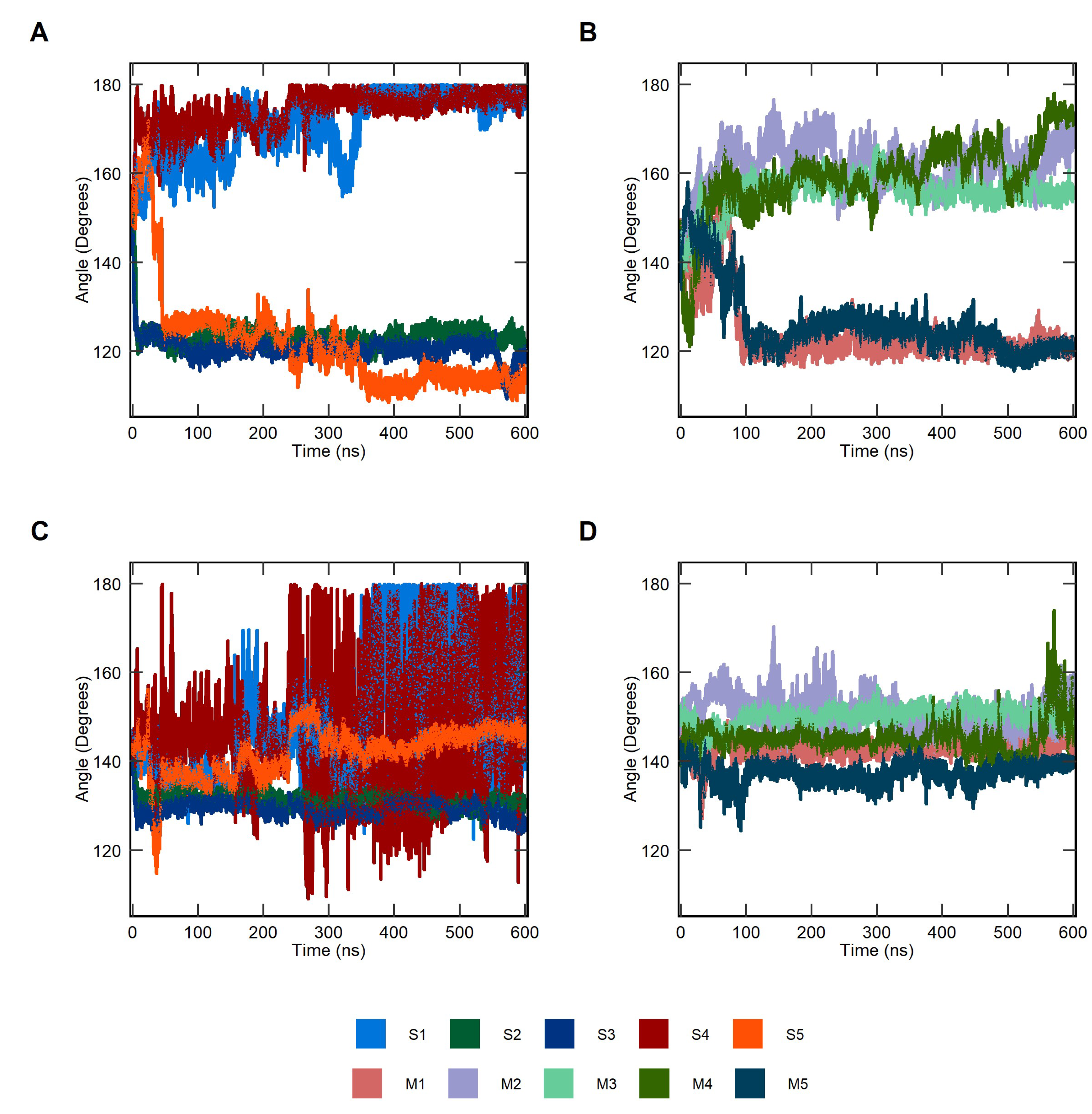
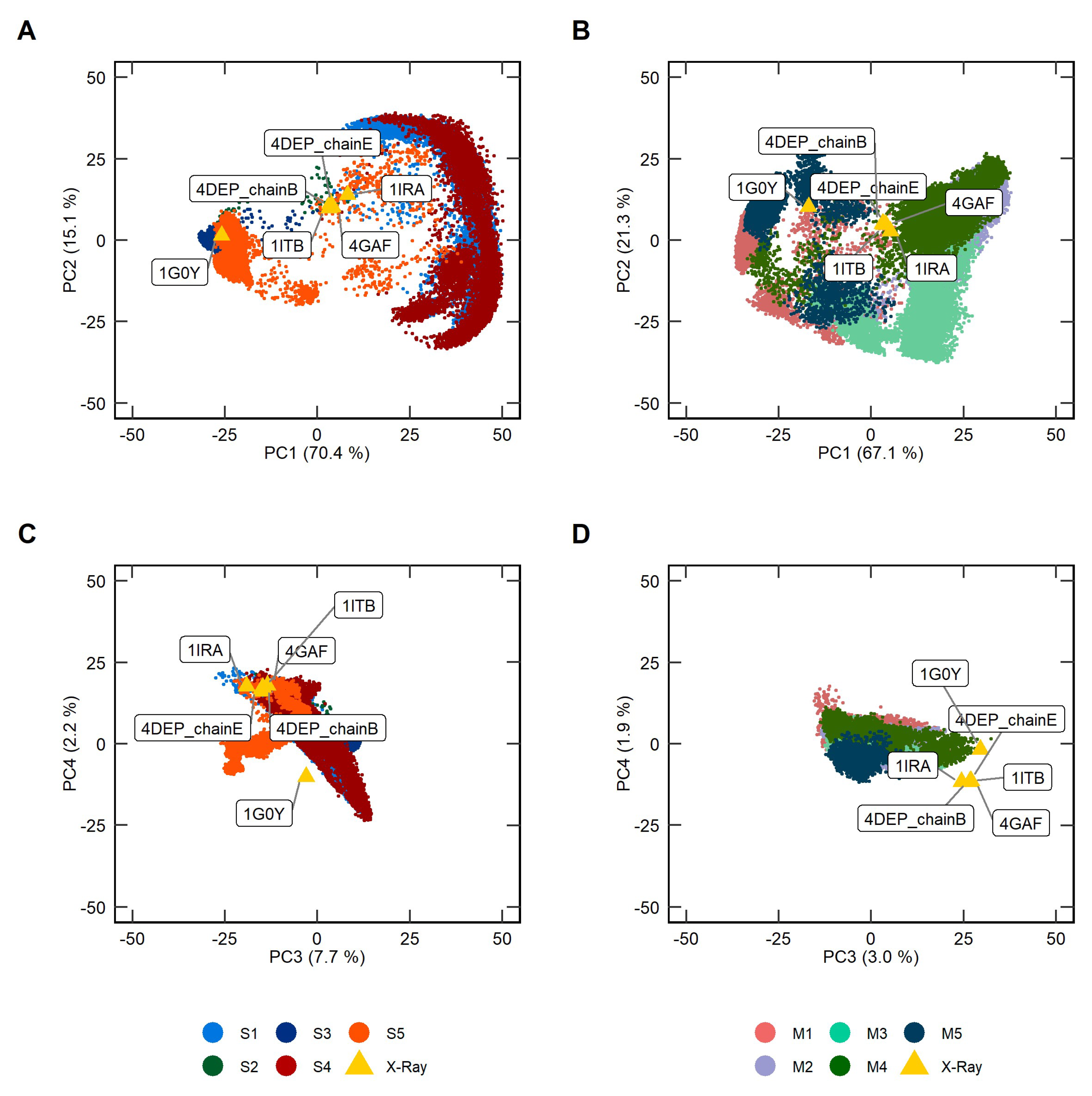
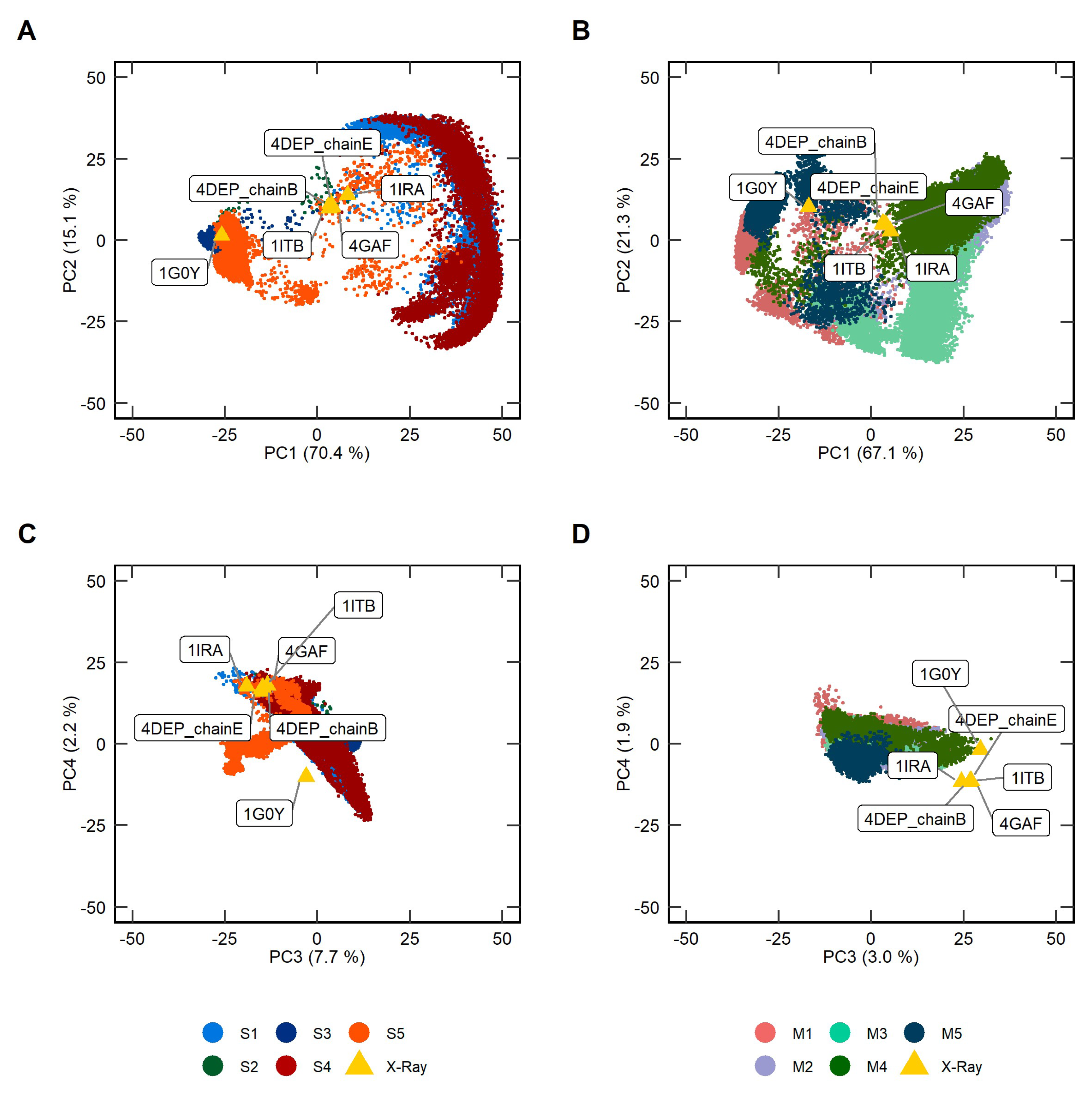
2.2. Principal Component Analysis of Soluble and Membrane-Bound IL-1R1-ECDs MD Trajectories
2.3. Clustering Analysis of Soluble and Membrane-Bound IL-1R1-ECDs Conformations
3. Discussion
4. Materials and Methods
4.1. Protein Structure of Soluble IL-1R1 (ECD)
4.2. Structural Assembly and Refinement of a Full-Length IL-1R1
4.3. Construction of the Full-Length IL-1R1 Membrane System
4.4. MD Simulations of Full-Length IL-1R1 Membrane System
4.5. MD Simulations of Soluble IL-1R1 (ECD)
4.6. Principal Component Analysis
4.7. Clustering Based on the Backbone Dihedral Angles of the Flexible Linker
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Krumm, B.; Xiang, Y.; Deng, J. Structural biology of the IL-1 superfamily: Key cytokines in the regulation of immune and inflammatory responses. Protein Sci. 2014, 23, 526–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural basis of IL-1 family cytokine signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Bazan, J.F.; Garcia, K.C. Structure of the activating IL-1 receptor signaling complex. Nat. Struct. Mol. Biol. 2012, 19, 455–457. [Google Scholar] [CrossRef]
- Auron, P.E. The interleukin 1 receptor: Ligand interactions and signal transduction. Cytokine Growth Factor Rev. 1998, 9, 221–237. [Google Scholar] [CrossRef]
- Vigers, G.P.A.; Anderson, L.J.; Caffes, P.; Brandhuber, B.J. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1β. Nature 1997, 386, 190–194. [Google Scholar] [CrossRef]
- Vigers, G.P.A.; Dripps, D.J.; Edwards, C.K.; Brandhuber, B.J. X-ray crystal structure of a small antagonist peptide bound to interleukin-1 receptor type 1. J. Biol. Chem. 2000, 275, 36927–36933. [Google Scholar] [CrossRef] [Green Version]
- Günther, S.; Deredge, D.; Bowers, A.L.; Luchini, A.; Bonsor, D.A.; Beadenkopf, R.; Liotta, L.; Wintrode, P.L.; Sundberg, E.J. IL-1 Family Cytokines Use Distinct Molecular Mechanisms to Signal through Their Shared Co-receptor. Immunity 2017, 47, 510–523.e4. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.; Remesh, S.G.; Hammel, M.; Pan, S.; Mahan, A.D.; Wang, S.; Wang, X. Functional Relevance of Interleukin-1 Receptor Inter-domain Flexibility for Cytokine Binding and Signaling. Structure 2019, 27, 1296–1307.e5. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y. Identification of potential small molecule allosteric modulator sites on IL-1R1 ectodomain using accelerated conformational sampling method. PLoS One 2015, 10, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y. Comparative Analyses of the Conformational Dynamics Between the Soluble and Membrane-Bound Cytokine Receptors. Sci. Rep. 2020, 10, 7399. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Shahane, G.; Ding, W.; Palaiokostas, M.; Orsi, M. Physical properties of model biological lipid bilayers: Insights from all-atom molecular dynamics simulations. J. Mol. Model. 2019, 25, 76. [Google Scholar] [CrossRef] [Green Version]
- Poger, D.; Caron, B.; Mark, A.E. Validating lipid force fields against experimental data: Progress, challenges and perspectives. Biochim. Biophys. Acta - Biomembr. 2016, 1858, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.M.; Coreta-Gomes, F.; Ollila, O.H.S.; Moreno, M.J.; Vaz, W.L.C.; Topgaard, D. Cholesterol and POPC segmental order parameters in lipid membranes: Solid state 1 H– 13 C NMR and MD simulation studies. Phys. Chem. Chem. Phys. 2013, 15, 1976–1989. [Google Scholar] [CrossRef]
- Poger, D.; Mark, A.E. On the Validation of Molecular Dynamics Simulations of Saturated and cis -Monounsaturated Phosphatidylcholine Lipid Bilayers: A Comparison with Experiment. J. Chem. Theory Comput. 2010, 6, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Piggot, T.J.; Allison, J.R.; Sessions, R.B.; Essex, J.W. On the Calculation of Acyl Chain Order Parameters from Lipid Simulations. J. Chem. Theory Comput. 2017, 13, 5683–5696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- RStudio Team RStudio: Integrated Development for R. Available online: https://www.rstudio.com (accessed on 23 December 2021).
- Durrant, J.D. BlendMol: Advanced macromolecular visualization in Blender. Bioinformatics 2019, 35, 2323–2325. [Google Scholar] [CrossRef] [PubMed]
- Kent, B.R. 3D Scientific Visualization with Blender; Morgan & Claypool Publishers: Burlington, MA, USA, 2015; ISBN 978-1-6270-5612-0. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Townson, S.A.; Kovalchin, J.T.; Masci, A.; Kiner, O.; Shu, Y.; King, B.M.; Schirmer, E.; Golden, K.; Thomas, C.; et al. Design of a superior cytokine antagonist for topical ophthalmic use. Proc. Natl. Acad. Sci. USA 2013, 110, 3913–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Sali, A. Make alpha helix - Modeller Wiki. Available online: https://salilab.org/modeller/wiki/Makealphahelix (accessed on 3 July 2021).
- Khan, J.A.; Brint, E.K.; O’Neill, L.A.J.; Tong, L. Crystal Structure of the Toll/Interleukin-1 Receptor Domain of Human IL-1RAPL. J. Biol. Chem. 2004, 279, 31664–31670. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Toshchakov, V.Y.; Neuwald, A.F. A survey of TIR domain sequence and structure divergence. Immunogenetics 2020, 72, 181–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.; van der Spoel, D.; Lindahl, E.; Hess, B.; GROMACS Development Team. GROMACS User Manual version 2019.3. Available online: http://www.gromacs.org (accessed on 23 September 2021).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.G.; Hoefling, M.; Aponte-Santamaría, C.; Grubmüller, H.; Groenhof, G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010, 31, 2169–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinforma. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Berger, O.; Edholm, O.; Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Braga, C.; Travis, K.P. A configurational temperature Nosé-Hoover thermostat. J. Chem. Phys. 2005, 123, 134101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC The PyMOL Molecular Graphics System, Version 2.4.1; Schrödinger LLC: New York, NY, USA, 2020.
- Schreuder, H.; Tardif, C.; Trump-Kallmeyer, S.; Soffientini, A.; Sarubbi, E.; Akeson, A.; Bowlin, T.; Yanofsky, S.; Barrett, R.W. A new cytokine-receptor binding mode revealed by the crystal structure of the IL-1 receptor with an antagonist. Nature 1997, 386, 194–200. [Google Scholar] [CrossRef]
- Reynolds, A.P.; Richards, G.; de la Iglesia, B.; Rayward-Smith, V.J. Clustering Rules: A Comparison of Partitioning and Hierarchical Clustering Algorithms. J. Math. Model. Algorithms 2006, 5, 475–504. [Google Scholar] [CrossRef]
- Kaufman, L.; Rousseeuw, P.J. Finding Groups in Data; Wiley Series in Probability and Statistics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; ISBN 9780470316801. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Average RMSD (Å) | Rg (nm) | # HBintra | SASA (nm2) |
|---|---|---|---|---|
| 4GAF 1 | -- | 3.0 | 229 | 179.4 |
| 1G0Y | 16.8 | 2.2 | 249 | 174.4 |
| S1 | 18.8 ± 0.22 | 2.9 ± 0.17 | 234 ± 7 | 178.7 ± 4.25 |
| S2 | 16.5 ± 0.02 | 2.2 ± 0.06 | 240 ± 8 | 173.0 ± 2.74 |
| S3 | 16.9 ± 0.04 | 2.2 ± 0.04 | 241 ± 7 | 172.7 ± 3.13 |
| S4 | 20.6 ± 0.24 | 2.8 ± 0.20 | 233 ± 7 | 178.9 ± 3.77 |
| S5 | 15.6 ± 0.20 | 2.4 ± 0.19 | 234 ± 8 | 176.4 ± 4.50 |
| M1 | 14.1 ± 0.07 | 2.3 ± 0.18 | 239 ± 7 | 175.8 ± 4.28 |
| M2 | 17.3 ± 0.13 | 3.1 ± 0.08 | 231 ± 7 | 184.7 ± 2.88 |
| M3 | 11.7 ± 0.11 | 3.3 ± 0.15 | 232 ± 7 | 186.3 ± 2.60 |
| M4 | 15.6 ±0.18 | 3.0 ± 0.15 | 233 ± 8 | 183.2 ± 3.13 |
| M5 | 14.1 ±0.06 | 2.3 ± 0.18 | 238 ± 7 | 175.3 ± 4.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luís, J.P.; Mata, A.I.; Simões, C.J.V.; Brito, R.M.M. Conformational Dynamics of the Soluble and Membrane-Bound Forms of Interleukin-1 Receptor Type-1: Insights into Linker Flexibility and Domain Orientation. Int. J. Mol. Sci. 2022, 23, 2599. https://doi.org/10.3390/ijms23052599
Luís JP, Mata AI, Simões CJV, Brito RMM. Conformational Dynamics of the Soluble and Membrane-Bound Forms of Interleukin-1 Receptor Type-1: Insights into Linker Flexibility and Domain Orientation. International Journal of Molecular Sciences. 2022; 23(5):2599. https://doi.org/10.3390/ijms23052599
Chicago/Turabian StyleLuís, João P., Ana I. Mata, Carlos J. V. Simões, and Rui M. M. Brito. 2022. "Conformational Dynamics of the Soluble and Membrane-Bound Forms of Interleukin-1 Receptor Type-1: Insights into Linker Flexibility and Domain Orientation" International Journal of Molecular Sciences 23, no. 5: 2599. https://doi.org/10.3390/ijms23052599
APA StyleLuís, J. P., Mata, A. I., Simões, C. J. V., & Brito, R. M. M. (2022). Conformational Dynamics of the Soluble and Membrane-Bound Forms of Interleukin-1 Receptor Type-1: Insights into Linker Flexibility and Domain Orientation. International Journal of Molecular Sciences, 23(5), 2599. https://doi.org/10.3390/ijms23052599