
Damage-Associated Molecular Patterns (DAMPs) in Retinal Disorders

 , and
, and 
Abstract
1. Introduction
2. DAMPs in Retinal Disorders
2.1. DAMPs in Endophthalmitis
2.2. DAMPS in Uveitis
2.3. DAMPs in Glaucoma
2.4. DAMPs in Ocular Cancer
2.5. DAMPs in Ischemic Retinopathies
2.6. DAMPs in Diabetic Retinopathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
Diabetic Retinopathy | S100A8, S100A9 [5,146] | Ca2+ binding protein | Cytoplasmic | Macroglia/plasma |
| HMGB1 [148] | Nuclear binding protein | Nuclear | Vitreous | |
| Uric acid [149] | Metabolic product | Cytoplasmic | Vitreous/perum | |
| HSP27, HSP60, HSP70 [150] | Molecular chaperones | Cytoplasmic | Retinal pndothelial cells | |
| ATP [144] | Nucleotide | Cytoplasmic | Microglia | |
| Cyclophilin A [151] | Ubiquitous protein | Cytoplasmic | Plasma | |
| Aβ [152] | Peptide | Cytoplasmic | RGC | |
| Calreticulin [153] | Multifunction soluble protein | ER | Plasma | |
| Cathelicidin [154] | Antimicrobial peptide | ER | Plasma | |
| α-defensin-1, -2, -3 [155] | Antimicrobial peptide | ER | Plasma | |
| Syndecan [156] | Proteoglycan | PM | Plasma | |
| Decorin [157,158] | Proteoglycan | ECM | Plasma/aqueous | |
| Versican [159] | Proteoglycan | ECM | Plasma | |
| LMW hyaluronan [160] | Glycosaminoglycan | ECM | Vitreous | |
| HS [161] | Glycosaminoglycan | ECM | Vitreous | |
| Fibronectin [34,162] | Glycoprotein | ECM | Plasma/vitreous/aqueous/retina | |
| Laminin [163] | Glycoprotein | ECM | Basement membrane/retina | |
| Fibrinogen [164] | Glycoprotein | ECM | Plasma | |
| Tenascin-C [165] | Glycoprotein | ECM | Vitreous |
2.7. DAMPs in Age-Related Macular Degeneration
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
AMD | S100A7, S100A8, S100A9 [196] | Ca2+ binding protein | Cytoplasmic | Drusen/Retina |
| Uric acid [197] | Metabolic product | Cytoplasmic | Serum | |
| HSP40, HSP60, HSP70, HSP90 & small HSPs [198] | Molecular chaperones | Cytoplasmic | Retina | |
| ATP [193] | Nucleotide | Cytoplasmic | Vitreous | |
| Aβ [199] | Peptide | Cytoplasmic | RPE/photoreceptors | |
| HMGB1 [7] HMGB2 [200] | Nuclear binding protein | Nuclear | RPE Photoreceptor | |
| IL-1α [201] | Cytokine | Cytoplasmic | RPE | |
| dsRNA [202] | Nucleic acid | Nuclear | Drusen, RPE | |
| mtDNA [203] | Nucleic acid | Mitchondria | RPE | |
| ET-1 [204] | Ribonuclease A | ER | Plasma | |
| Perlecan [205] | Proteoglycan | Plasma membrane | Retina | |
| Syndecan-4 [205] | Proteoglycan | Plasma membrane | Retina | |
| Versican [206] | Proteoglycan | ECM | RPE | |
| Heparan sulfate [207] | Glycosaminoglycan | ECM | Bruch’s membrane | |
| Fibronectin [208] | Glycoprotein | ECM | Basal deposits | |
| Laminin [208] | Glycoprotein | ECM | Basal deposits | |
| Tenascin-C [209] | Glycoprotein | ECM | CNV membrane |
2.8. DAMPS in Proliferative Vitreoretinopathy and Rhegmatogenous Retinal Detachment
2.9. DAMPs in Inherited Retinal Disorders
3. DAMP-Driven Signal Transduction in Retinal Disorders
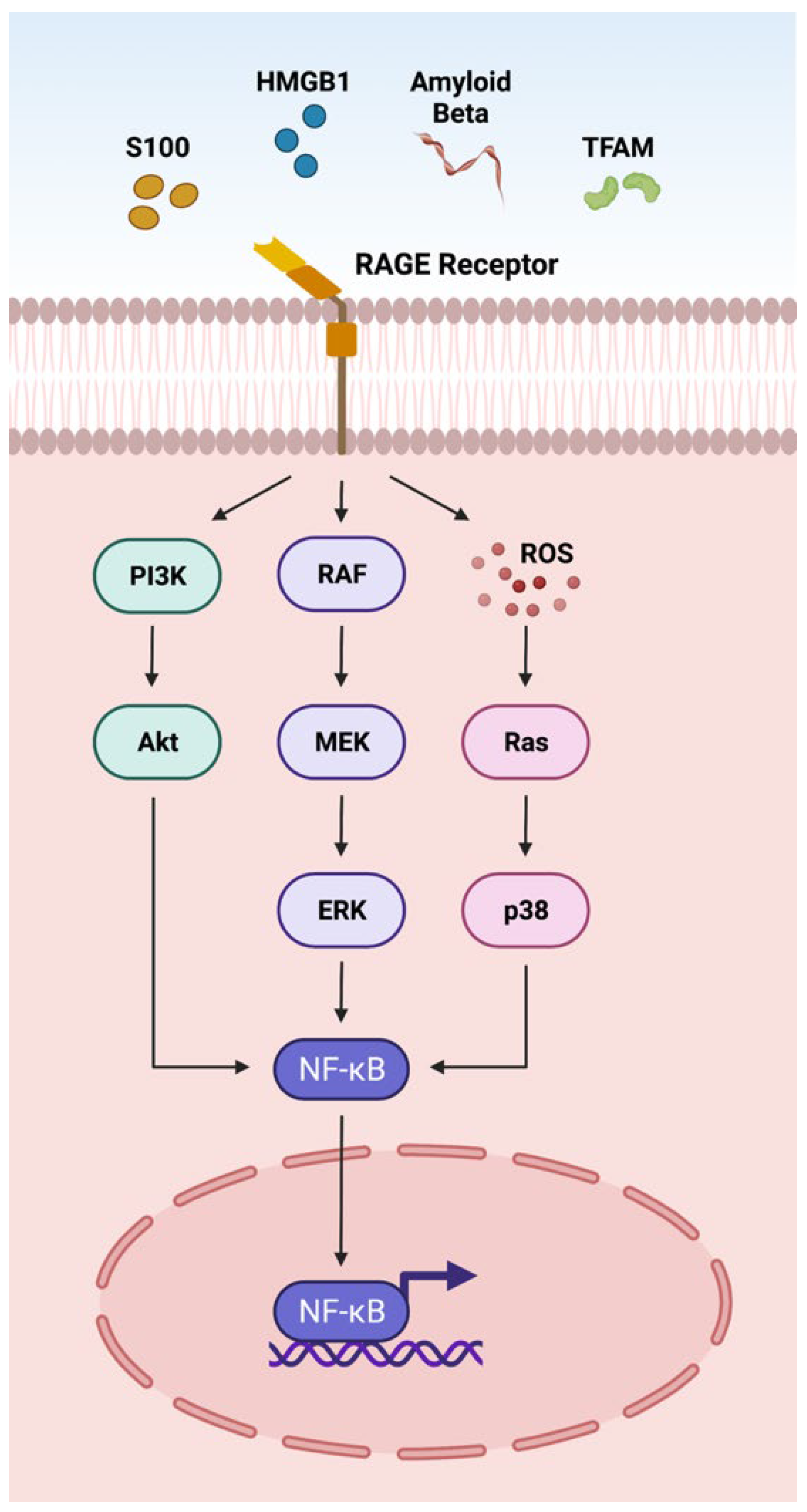
3.1. RAGE Pathway
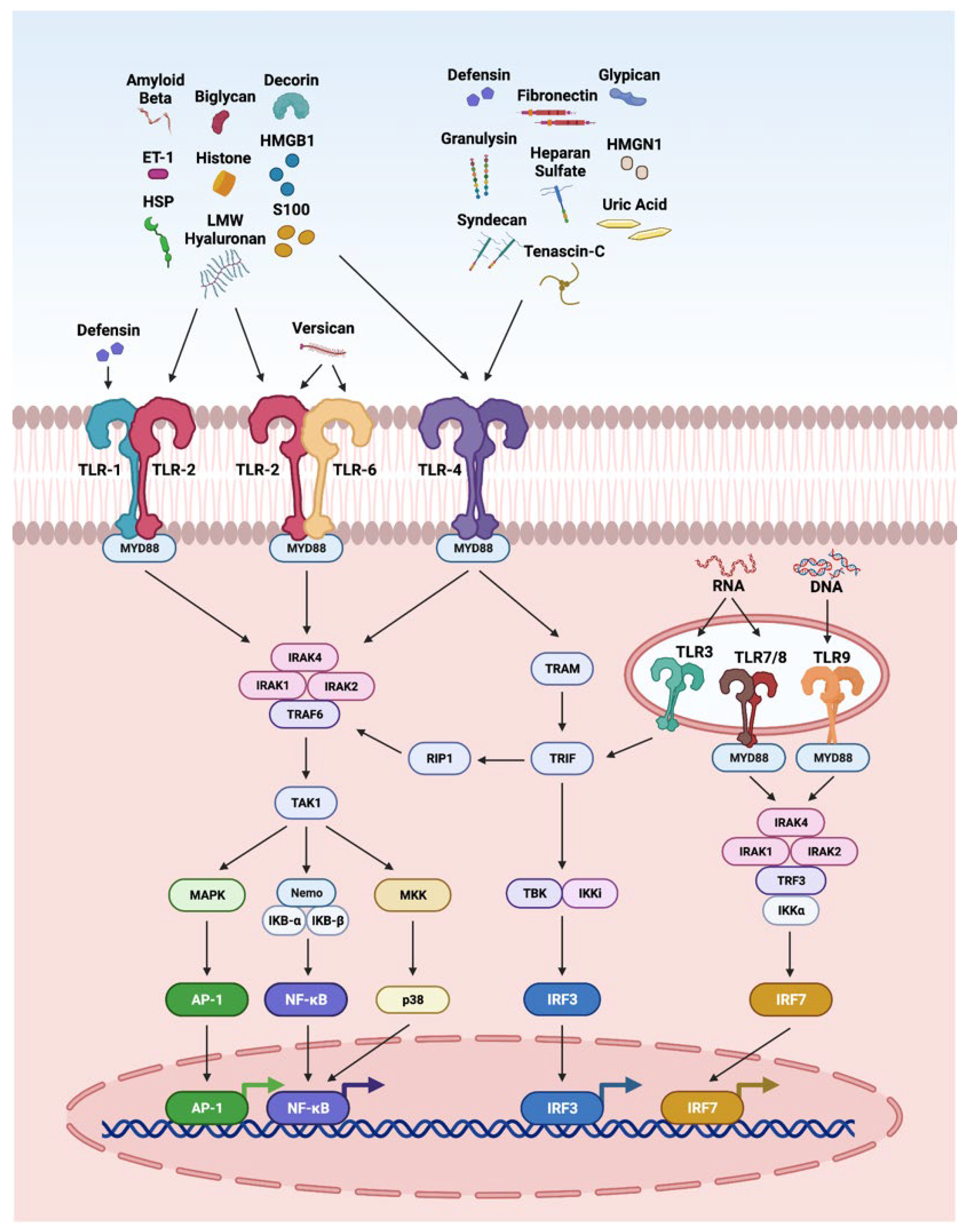
3.2. TLR Pathway
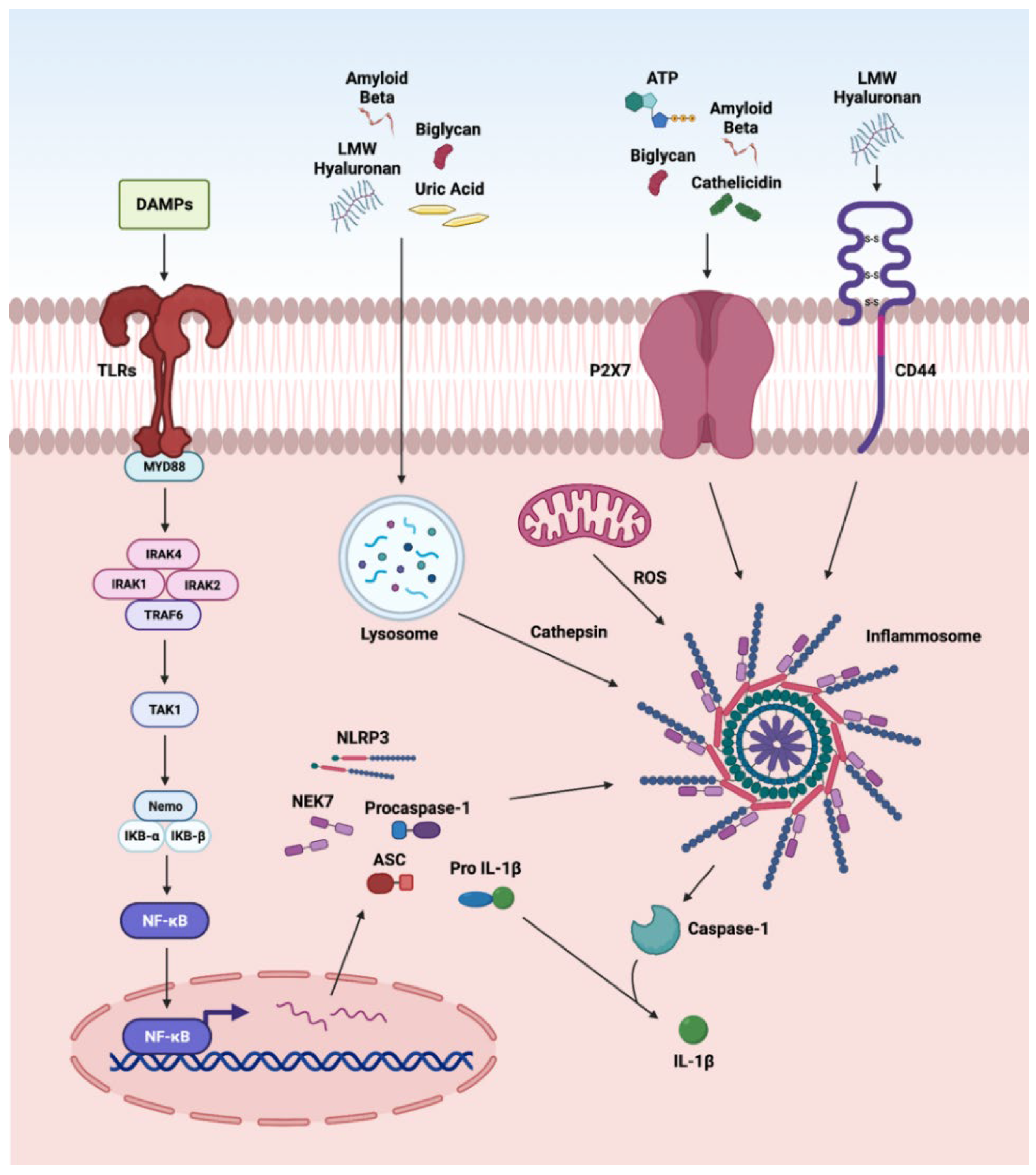
3.3. NLRP3 Inflammasome
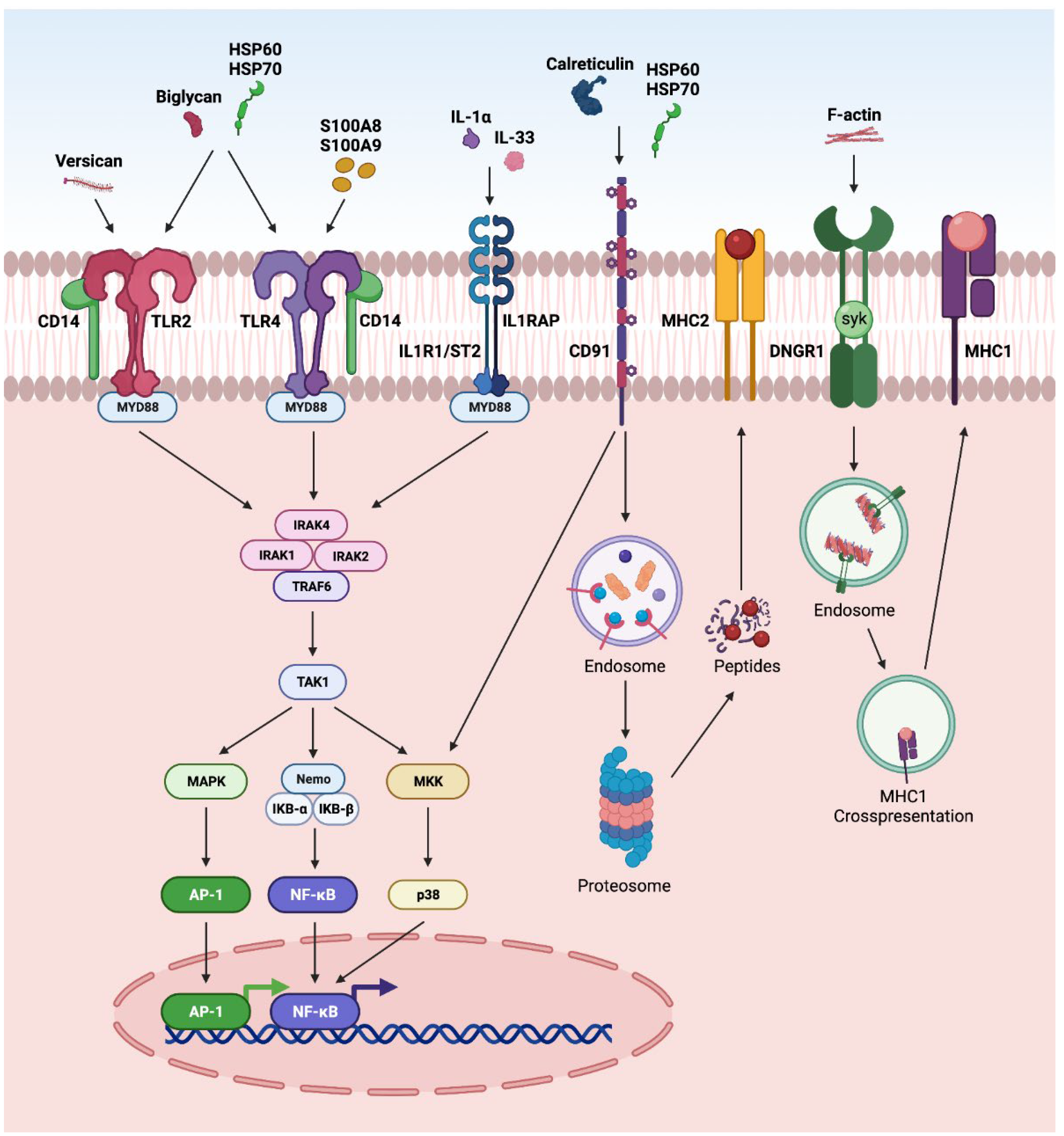
3.4. Other Pathways
4. Therapeutic Implications of DAMPs
4.1. DAMPs as Biomarkers
4.2. DAMPs as Therapeutic Targets
4.3. DAMPs as Therapeutic Agents
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Soto, I.; Howell, G.R. The complex role of neuroinflammation in glaucoma. Cold Spring Harb. Perspect. Med. 2014, 4, a017269. [Google Scholar] [CrossRef]
- Lim, R.R.; Vaidya, T.; Gadde, S.G.; Yadav, N.K.; Sethu, S.; Hainsworth, D.P.; Mohan, R.R.; Ghosh, A.; Chaurasia, S.S. Correlation between systemic S100A8 and S100A9 levels and severity of diabetic retinopathy in patients with type 2 diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 1581–1589. [Google Scholar] [CrossRef]
- Atıl, A.; Deniz, A. Could be serum uric acid a risk factor for thrombosis and/or uveitis in Behcet′s disease? Vascular 2018, 26, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhuang, G.-B.; Zhou, X.-Z. HMBG1 as a Driver of Inflammatory and Immune Processes in the Pathogenesis of Ocular Diseases. J. Ophthalmol. 2018, 2018, 5195290. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Lan, W.; Lim, R.R.; Chaurasia, S.S. S100A proteins as molecular targets in the ocular surface inflammatory diseases. Ocul. Surf. 2014, 12, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Urbak, L.; Vorum, H. Heat shock proteins in the human eye. Int. J. Proteom. 2010, 2010, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Barile, G.R.; Schmidt, A.M. RAGE and its ligands in retinal disease. Curr. Mol. Med. 2007, 7, 758–765. [Google Scholar] [CrossRef]
- Kumar, A.; Shamsuddin, N. Retinal Muller glia initiate innate response to infectious stimuli via toll-like receptor signaling. PLoS ONE 2012, 7, e29830. [Google Scholar]
- Chaurasia, S.S.; Lim, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, B.B.; Wong, T.Y.; Luu, C.D.; Agrawal, R.; Ghosh, A.; Mortellaro, A. The NLRP3 inflammasome may contribute to pathologic neovascularization in the advanced stages of diabetic retinopathy. Sci. Rep. 2018, 8, 2847. [Google Scholar] [CrossRef]
- Streilein, J.W. Ocular immune privilege: The eye takes a dim but practical view of immunity and inflammation. J. Leukoc. Biol. 2003, 74, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Stein-Streilein, J. Mechanisms of immune privilege in the posterior eye. Int. Rev. Immunol. 2013, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Akpek, E.K.; Gottsch, J.D. Immune defense at the ocular surface. Eye 2003, 17, 949–956. [Google Scholar] [CrossRef]
- Mahaling, B.; Baruah, N.; Ahamad, N.; Maisha, N.; Lavik, E.; Katti, D.S. A non-invasive nanoparticle-based sustained dual-drug delivery system as an eyedrop for endophthalmitis. Int. J. Pharm. 2021, 606, 120900. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.C.; Coburn, P.S.; Huzzatul, M.M.; LaGrow, A.L.; Livingston, E.; Callegan, M.C. Targets of immunomodulation in bacterial endophthalmitis. Prog. Retin. Eye Res. 2019, 73, 100763. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, A. Role of Staphylococcus aureus Virulence Factors in Inducing Inflammation and Vascular Permeability in a Mouse Model of Bacterial Endophthalmitis. PLoS ONE 2015, 10, e0128423. [Google Scholar] [CrossRef]
- Arimura, N.; Ki, I.Y.; Hashiguchi, T.; Sakamoto, T.; Maruyama, I. High-mobility group box 1 protein in endophthalmitis. Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 1053–1058. [Google Scholar]
- Vallejo-Garcia, J.L.; Asencio-Duran, M.; Pastora-Salvador, N.; Vinciguerra, P.; Romano, M.R. Role of Inflammation in Endophthalmitis. Mediat. Inflamm. 2012, 2012, 196094. [Google Scholar] [CrossRef] [PubMed]
- Whiston, E.A.; Sugi, N.; Kamradt, M.C.; Sack, C.; Heimer, S.R.; Engelbert, M.; Wawrousek, E.F.; Gilmore, M.S.; Ksander, B.R.; Gregory, M.S. αB-Crystallin Protects Retinal Tissue during Staphylococcus aureus-Induced Endophthalmitis. Infect. Immun. 2008, 76, 1781–1790. [Google Scholar] [CrossRef]
- Coburn, P.S.; Miller, F.C.; LaGrow, A.L.; Parkunan, S.M.; Blake Randall, C.; Staats, R.L.; Callegan, M.C. TLR4 modulates inflammatory gene targets in the retina during Bacillus cereus endophthalmitis. BMC Ophthalmol. 2018, 18, 96. [Google Scholar] [CrossRef]
- Deshmukh, D.; Chakrabarti, M.; Jayasudha, R.; Hasnat Ali, M.; Tyagi, M.; Sharma, S.; Joseph, J. Elevated cytokine levels in vitreous as biomarkers of disease severity in infectious endophthalmitis. PLoS ONE 2018, 13, e0205292. [Google Scholar] [CrossRef]
- Haynes, R.J.; McElveen, J.E.; Dua, H.S.; Tighe, P.J.; Liversidge, J. Expression of Human Beta-Defensins in Intraocular Tissues. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3026–3031. [Google Scholar]
- Singh, P.K.; Shiha, M.J.; Kumar, A. Antibacterial responses of retinal Müller glia: Production of antimicrobial peptides, oxidative burst and phagocytosis. J. Neuroinflamm. 2014, 11, 33. [Google Scholar] [CrossRef]
- Takahashi, T.; Kulkarni, N.N.; Lee, E.Y.; Zhang, L.-j.; Wong, G.C.L.; Gallo, R.L. Cathelicidin promotes inflammation by enabling binding of self-RNA to cell surface scavenger receptors. Sci. Rep. 2018, 8, 4032. [Google Scholar] [CrossRef]
- Yang, Y.-Y. Clinical value of serum amyloid A detection in the diagnosis of infective endophthalmitis. Int. Eye Sci. 2019, 12, 1738–1740. [Google Scholar]
- Chang, J.H.; McCluskey, P.J.; Wakefield, D. Recent advances in Toll-like receptors and anterior uveitis. Clin. Exp. Ophthalmol. 2012, 40, 821–828. [Google Scholar] [CrossRef] [PubMed]
- John Curnow, S.; Denniston, K.O.A.; Murray, P.I.; Wallace, G.R. Intraocular immune mechanisms in uveitis. Curr. Immunol. Rev. 2011, 7, 350–359. [Google Scholar] [CrossRef]
- Angeles-Han, S.T.; Miraldi Utz, V.; Thornton, S.; Schulert, G.; Rodriguez-Smith, J.; Kauffman, A.; Sproles, A.; Mwase, N.; Hennard, T.; Grom, A.; et al. S100 Proteins, Cytokines, and Chemokines as Tear Biomarkers in Children with Juvenile Idiopathic Arthritis-associated Uveitis. Ocul. Immunol. Inflamm. 2020, 29, 1616–1620. [Google Scholar] [CrossRef]
- Jiang, G.; Sun, D.; Yang, H.; Lu, Q.; Kaplan, H.J.; Shao, H. HMGB1 is an early and critical mediator in an animal model of uveitis induced by IRBP-specific T cells. J. Leukoc. Biol. 2014, 95, 599–607. [Google Scholar] [CrossRef]
- Sahebari, M.; Hashemzadeh, K.; Mahmoudi, M.; Saremi, Z.; Mirfeizi, Z. Diagnostic Yield of Heat Shock Protein 70 (HSP-70) and Anti-HSP-70 in Behcet-Induced Uveitis. Scand. J. Immunol. 2013, 77, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Kasper, M.; Walscheid, K.; Koch, J.M.; Müther, P.S.; Kirchhof, B.; Heiligenhaus, A.; Heinz, C. Multiplex Cytokine Analysis of Aqueous Humor in Juvenile Idiopathic Arthritis-Associated Anterior Uveitis with or without Secondary Glaucoma. Front. Immunol. 2018, 9, 708. [Google Scholar] [CrossRef]
- Probst, K.; Fijnheer, R.; Schellekens, P.; Rothova, A. Intraocular and plasma levels of cellular fibronectin in patients with uveitis and diabetes mellitus. Br. J. Ophthalmol. 2004, 88, 667. [Google Scholar] [CrossRef][Green Version]
- Ni, M.; Chan, C.-C.; Nussenblatt, R.B.; Li, S.; Mao, W. Iris Inflammatory Cells, Fibronectin, Fibrinogen, and Immunoglobulin in Various Ocular Diseases. Arch. Ophthalmol. 1988, 106, 392–395. [Google Scholar] [CrossRef]
- Walscheid, K.; Heiligenhaus, A.; Holzinger, D.; Roth, J.; Heinz, C.; Tappeiner, C.; Kasper, M.; Foell, D. Elevated S100A8/A9 and S100A12 Serum Levels Reflect Intraocular Inflammation in Juvenile Idiopathic Arthritis-Associated Uveitis: Results from a Pilot Study. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7653–7660. [Google Scholar] [CrossRef]
- Wakefield, D.; Gray, P.; Chang, J.; Di Girolamo, N.; McCluskey, P. The role of PAMPs and DAMPs in the pathogenesis of acute and recurrent anterior uveitis. Br. J. Ophthalmol. 2010, 94, 271. [Google Scholar] [CrossRef]
- Poulaki, V.; Iliaki, E.; Mitsiades, N.; Mitsiades, C.S.; Paulus, Y.N.; Bula, D.V.; Gragoudas, E.S.; Miller, J.W. Inhibition of Hsp90 attenuates inflammation in endotoxin-induced uveitis. FASEB J. 2007, 21, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Hasan, A.; Wilson, A.; Stanford, M.R.; Li-Yang, Y.; Todryk, S.; Whiston, R.; Shinnick, T.; Mizushima, Y.; Van der Zee, R.; et al. Experimental mucosal induction of uveitis with the 60-kDa heat shock protein-derived peptide 336–351. Eur. J. Immunol. 1998, 28, 2444–2455. [Google Scholar] [CrossRef]
- Stanford, M.R.; Kasp, E.; Whiston, R.; Hasan, A.; Todryk, S.; Shinnick, T.; Mizushima, Y.; Dumonde, D.C.; van der Zee, R.; Lehner, T. Heat shock protein peptides reactive in patients with Behçet′s disease are uveitogenic in Lewis rats. Clin. Exp. Immunol. 1994, 97, 226–231. [Google Scholar] [CrossRef]
- Dai, M.-L.; Fan, S.; Li, Z.; Yu, X.; Lin, D.; Huang, X.-F.; Wang, Y. Correlation of serum amyloid A levels, clinical manifestations, treatment, and disease activity in patients with acute anterior uveitis. Eye 2020, 34, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, G.; Lucherini, O.M.; Vitale, A.; Talarico, R.; Lopalco, A.; Galeazzi, M.; Lapadula, G.; Cantarini, L.; Iannone, F. Putative Role of Serum Amyloid-A and Proinflammatory Cytokines as Biomarkers for Behcet′s Disease. Medicine 2015, 94, e1858. [Google Scholar] [CrossRef] [PubMed]
- Niemi, K.; Teirilä, L.; Lappalainen, J.; Rajamäki, K.; Baumann, M.H.; Öörni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Hsu, K.; Yamen, E.; Yan, W.; Fock, J.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis 2009, 207, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Barbour, M.; Allan, D.; Xu, H.; Pei, C.; Chen, M.; Niedbala, W.; Fukada, S.Y.; Besnard, A.-G.; Alves-Filho, J.C.; Tong, X.; et al. IL-33 attenuates the development of experimental autoimmune uveitis. Eur. J. Immunol. 2014, 44, 3320–3329. [Google Scholar] [CrossRef]
- Reinehr, S.; Kuehn, S.; Casola, C.; Koch, D.; Stute, G.; Grotegut, P.; Dick, H.B.; Joachim, S.C. HSP27 immunization reinforces AII amacrine cell and synapse damage induced by S100 in an autoimmune glaucoma model. Cell Tissue Res. 2018, 371, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Huang, C.; Wang, Y.; Wu, R. Expression of leukemia inhibitory factor in the rat retina following acute ocular hypertension. Mol. Med. Rep. 2015, 12, 6577–6583. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Murat, D.; Kimura, I.; Ohtake, Y.; Tsubota, K. Reduced-serum vitamin C and increased uric acid levels in normal-tension glaucoma. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 248, 243–248. [Google Scholar]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Pulukool, S.K.; Bhagavatham, S.K.S.; Kannan, V.; Sukumar, P.; Dandamudi, R.B.; Ghaisas, S.; Kunchala, H.; Saieesh, D.; Naik, A.A.; Pargaonkar, A.; et al. Elevated dimethylarginine, ATP, cytokines, metabolic remodeling involving tryptophan metabolism and potential microglial inflammation characterize primary open angle glaucoma. Sci. Rep. 2021, 11, 9766. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Gupta, V.B.; Chitranshi, N.; Gangoda, S.; Vander Wall, R.; Abbasi, M.; Golzan, M.; Dheer, Y.; Shah, T.; Avolio, A.; et al. One protein, multiple pathologies: Multifaceted involvement of amyloid β in neurodegenerative disorders of the brain and retina. Cell. Mol. Life Sci. 2016, 73, 4279–4297. [Google Scholar] [CrossRef]
- Reichelt, J.; Joachim, S.C.; Pfeiffer, N.; Grus, F.H. Analysis of autoantibodies against human retinal antigens in sera of patients with glaucoma and ocular hypertension. Curr. Eye Res. 2008, 33, 253–261. [Google Scholar] [CrossRef]
- He, W.; Xu, F.; Chen, L.; Huang, W.; Jiang, L.; Tang, F.; Yan, W.; Zhong, S.; Shen, C.; Huang, H.; et al. Association of High-Mobility Group Box-1 with Inflammationrelated Cytokines in the Aqueous Humor with Acute Primary Angle-Closure Eyes. Curr. Mol. Med. 2021, 21, 237–245. [Google Scholar] [CrossRef]
- Pantalon, A.; Obadă, O.; Constantinescu, D.; Feraru, C.; Chiseliţă, D. Inflammatory model in patients with primary open angle glaucoma and diabetes. Int. J. Ophthalmol. 2019, 12, 795–801. [Google Scholar] [PubMed]
- Izzotti, A.; Saccà, S.C.; Cartiglia, C.; De Flora, S. Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am. J. Med. 2003, 114, 638–646. [Google Scholar] [CrossRef]
- Stowell, C.; Jang, G.-F.; Zhang, L.; Crabb, J.; Reynaud, J.; Gardiner, S.K.; Willard, b.; Marsh-Armstrong, N.; Crabb, J.W.; Burgoyne, C.F. Alterations in Optic Nerve Head (ONH) Endoplasmic Reticulum (ER) Stress Response Proteins in Non-Human Primate (NHP) Early Experimental Glaucoma (EG). Investig. Ophthalmol. Vis. Sci. 2018, 59, 3514. [Google Scholar]
- Chauhan, B.C. Endothelin and its potential role in glaucoma. Can. J. Ophthalmol. 2008, 43, 356–360. [Google Scholar] [CrossRef]
- NikhalaShree, S.; Karthikkeyan, G.; George, R.; Shantha, B.; Vijaya, L.; Ratra, V.; Sulochana, K.N.; Coral, K. Lowered Decorin with Aberrant Extracellular Matrix Remodeling in Aqueous Humor and Tenon′s Tissue from Primary Glaucoma Patients. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4661–4669. [Google Scholar] [CrossRef]
- Burgoyne, C.; Stowell, C.; Jang, G.-F.; Zhang, L.; Willard, B.; Crabb, J.S.; Crabb, J.W. Non-human primate (NHP) optic nerve head (ONH) proteomic change in early experimental glaucoma (EEG). Investig. Ophthalmol. Vis. Sci. 2014, 55, 4555. [Google Scholar]
- Keller, K.E.; Bradley, J.M.; Vranka, J.A.; Acott, T.S. Segmental Versican Expression in the Trabecular Meshwork and Involvement in Outflow Facility. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5049–5057. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.B.; Carter-Dawson, L.; Harwerth, R.S. Extracellular Matrix Tissue Content of the Neuroretinal Rim differs in Healthy and Glaucomatous Eyes. Investig. Ophthalmol. Vis. Sci. 2019, 60, 662. [Google Scholar]
- Reinhard, J.; Wiemann, S.; Hildebrandt, S.; Faissner, A. Extracellular Matrix Remodeling in the Retina and Optic Nerve of a Novel Glaucoma Mouse Model. Biology 2021, 10, 169. [Google Scholar] [CrossRef]
- Tezel, G.; Edward, D.P.; Wax, M.B. Serum autoantibodies to optic nerve head glycosaminoglycans in patients with glaucoma. Arch. Ophthalmol. 1999, 117, 917–924. [Google Scholar] [CrossRef]
- Faralli, J.A.; Filla, M.S.; Peters, D.M. Role of Fibronectin in Primary Open Angle Glaucoma. Cells 2019, 8, 1518. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; McNatt, L.G.; Pang, I.-H.; Hellberg, P.E.; Fingert, J.H.; McCartney, M.D.; Clark, A.F. Increased Expression of Serum Amyloid A in Glaucoma and Its Effect on Intraocular Pressure. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1916–1923. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Casola, C.; Schiwek, J.E.; Reinehr, S.; Kuehn, S.; Grus, F.H.; Kramer, M.; Dick, H.B.; Joachim, S.C. S100 alone has the same destructive effect on retinal ganglion cells as in combination with HSP 27 in an autoimmune glaucoma model. J. Mol. Neurosci. 2015, 56, 228–236. [Google Scholar] [CrossRef]
- Joly, S.; Lange, C.; Thiersch, M.; Samardzija, M.; Grimm, C. Leukemia Inhibitory Factor Extends the Lifespan of Injured Photoreceptors In Vivo. J. Neurosci. 2008, 28, 13765–13774. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.-W.; Liu, K.-M.; Yee, R.W.; Lee, P.-F. Detection of uric acid in aqueous humor by high pressure liquid chromatography. Curr. Eye Res. 1982, 2, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shao, M.; Tang, B.; Zhang, A.; Cao, W.; Sun, X. The association between serum uric acid and glaucoma severity in primary angle closure glaucoma: A retrospective case-control study. Oncotarget 2017, 8, 2816–2824. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.; Grotegut, P.; Reinehr, S.; Joachim, S.C. Role of Heat Shock Proteins in Glaucoma. Int. J. Mol. Sci. 2019, 20, 5160. [Google Scholar] [CrossRef]
- Guo, L.; Salt, T.E.; Luong, V.; Wood, N.; Cheung, W.; Maass, A.; Ferrari, G.; Russo-Marie, F.; Sillito, A.M.; Cheetham, M.E.; et al. Targeting amyloid-beta in glaucoma treatment. Proc. Natl. Acad. Sci. USA 2007, 104, 13444–13449. [Google Scholar] [CrossRef]
- Mohd Lazaldin, M.A.; Iezhitsa, I.; Agarwal, R.; Bakar, N.S.; Agarwal, P.; Mohd Ismail, N. Neuroprotective effects of brain-derived neurotrophic factor against amyloid beta 1-40-induced retinal and optic nerve damage. Eur. J. Neurosci. 2020, 51, 2394–2411. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mao, X. Role of Retinal Amyloid-β in Neurodegenerative Diseases: Overlapping Mechanisms and Emerging Clinical Applications. Int. J. Mol. Sci. 2021, 22, 2360. [Google Scholar] [CrossRef]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-H.; Zhang, S.-H.; Nickerson, J.M.; Gao, F.-J.; Sun, Z.; Chen, X.-Y.; Zhang, S.-J.; Gao, F.; Chen, J.-Y.; Luo, Y.; et al. Cumulative mtDNA damage and mutations contribute to the progressive loss of RGCs in a rat model of glaucoma. Neurobiol. Dis. 2015, 74, 167–179. [Google Scholar] [CrossRef]
- Marola, O.J.; Syc-Mazurek, S.B.; Howell, G.R.; Libby, R.T. Endothelin 1-induced retinal ganglion cell death is largely mediated by JUN activation. Cell Death Dis. 2020, 11, 811. [Google Scholar] [CrossRef]
- Hill, L.J.; Mead, B.; Blanch, R.J.; Ahmed, Z.; De Cogan, F.; Morgan-Warren, P.J.; Mohamed, S.; Leadbeater, W.; Scott, R.A.H.; Berry, M.; et al. Decorin Reduces Intraocular Pressure and Retinal Ganglion Cell Loss in Rodents Through Fibrolysis of the Scarred Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3743–3757. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, S.; Reinhard, J.; Wiemann, S.; Stute, G.; Kuehn, S.; Woestmann, J.; Dick, H.B.; Faissner, A.; Joachim, S.C. Early remodelling of the extracellular matrix proteins tenascin-C and phosphacan in retina and optic nerve of an experimental autoimmune glaucoma model. J. Cell. Mol. Med. 2016, 20, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Eagle, R.C. The pathology of ocular cancer. Eye 2013, 27, 128–136. [Google Scholar] [CrossRef]
- Jovanovic, P.; Mihajlovic, M.; Djordjevic-Jocic, J.; Vlajkovic, S.; Cekic, S.; Stefanovic, V. Ocular melanoma: An overview of the current status. Int. J. Clin. Exp. Pathol. 2013, 6, 1230–1244. [Google Scholar] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Karim, M.M.; Itoh, H. Demonstration of S-100 protein immunoreactivity in normal human retina and retinoblastoma. Ophthalmologica 1997, 211, 351–353. [Google Scholar] [CrossRef]
- He, W.; Inomata, H. Dual immunologic property of S-100 protein in normal eyes and eyes with retinoblastoma: A histopathological and immunohistochemical study of 88 cases. Ophthalmologica 1993, 206, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Kan-Mitchell, J.; Rao, N.; Albert, D.M.; Van Eldik, L.J.; Taylor, C.R. S100 immunophenotypes of uveal melanomas. Investig. Ophthalmol. Vis. Sci. 1990, 31, 1492–1496. [Google Scholar]
- Keijser, S.; Missotten, G.S.; Bonfrer, J.M.; de Wolff-Rouendaal, D.; Jager, M.J.; de Keizer, R.J. Immunophenotypic markers to differentiate between benign and malignant melanocytic lesions. Br. J. Ophthalmol. 2006, 90, 213–217. [Google Scholar] [CrossRef]
- Missotten, G.S.; Korse, C.M.; van Dehn, C.; Linders, T.C.; Keunen, J.E.; Jager, M.J.; Bonfrer, J.M. S-100B Protein and Melanoma Inhibitory Activity Protein in Uveal Melanoma Screening. Tumor Biol. 2007, 28, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, M.E.; Abramson, D.H.; Senft, S.; Servodidio, C.A.; Gamache, P.H. Uric acid in the aqueous humor and tears of retinoblastoma patients. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 1998, 2, 369–371. [Google Scholar] [CrossRef]
- Venkatesan, N.; Kanwar, J.R.; Deepa, P.R.; Navaneethakrishnan, S.; Joseph, C.; Krishnakumar, S. Targeting HSP90/Survivin using a cell permeable structure based peptido-mimetic shepherdin in retinoblastoma. Chem.-Biol. Interact. 2016, 252, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Tsering, T.; Laskaris, A.; Abdouh, M.; Bustamante, P.; Parent, S.; Jin, E.; Ferrier, S.T.; Arena, G.; Burnier, J.V. Uveal Melanoma-Derived Extracellular Vesicles Display Transforming Potential and Carry Protein Cargo Involved in Metastatic Niche Preparation. Cancers 2020, 12, 2923. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; García, Á.; Thomas, B.; Piñeiro, A.; Akoulitchev, A.; Dwek, R.A.; Zitzmann, N. The characterization of the invasion phenotype of uveal melanoma tumour cells shows the presence of MUC18 and HMG-1 metastasis markers and leads to the identification of DJ-1 as a potential serum biomarker. Int. J. Cancer 2006, 119, 1014–1022. [Google Scholar] [CrossRef]
- Madic, J.; Piperno-Neumann, S.; Servois, V.; Rampanou, A.; Milder, M.; Trouiller, B.; Gentien, D.; Saada, S.; Assayag, F.; Thuleau, A.; et al. Pyrophosphorolysis-activated polymerization detects circulating tumor DNA in metastatic uveal melanoma. Clin. Cancer Res. 2012, 18, 3934–3941. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, Y.; Sun, H.; Xiao, Q.; Ye, P. Identification of Versican as an Independent Prognostic Factor in Uveal Melanoma. Int. J. Gen. Med. 2021, 14, 4639–4651. [Google Scholar] [CrossRef]
- Albakova, Z.; Siam, M.K.S.; Sacitharan, P.K.; Ziganshin, R.H.; Ryazantsev, D.Y.; Sapozhnikov, A.M. Extracellular heat shock proteins and cancer: New perspectives. Transl. Oncol. 2021, 14, 100995. [Google Scholar] [CrossRef]
- Missotten, G.S.; Journée-de Korver, J.G.; de Wolff-Rouendaal, D.; Keunen, J.E.; Schlingemann, R.O.; Jager, M.J. Heat shock protein expression in the eye and in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3059–3065. [Google Scholar] [CrossRef]
- Faingold, D.; Marshall, J.-C.; Antecka, E.; Di Cesare, S.; Odashiro, A.N.; Bakalian, S.; Fernandes, B.F.; Burnier, M.N. Immune Expression and Inhibition of Heat Shock Protein 90 in Uveal Melanoma. Clin. Cancer Res. 2008, 14, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-L.; Feng, Y.-Q.; Cheng, Y.-H. Effect on proliferation and apoptosis of retinoblastoma cell by RNA inhibiting high mobility group protein box-1 expression. Int. J. Ophthalmol. 2017, 10, 30–34. [Google Scholar] [PubMed]
- Singh, M.K.; Singh, L.; Pushker, N.; Sen, S.; Sharma, A.; Chauhan, F.A.; Kashyap, S. Correlation of High Mobility Group Box-1 Protein (HMGB1) with Clinicopathological Parameters in Primary Retinoblastoma. Pathol. Oncol. Res. 2015, 21, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; Piñeiro, A.; de la Fuente, M.; García, A.; Prabhakar, S.; Zitzmann, N.; Dwek, R.A.; Sánchez-Salorio, M.; Domínguez, F.; Capeans, C. Abnormal cell cycle regulation in primary human uveal melanoma cultures. J. Cell. Biochem. 2004, 93, 708–720. [Google Scholar] [CrossRef]
- Berry, J.L.; Xu, L.; Murphree, A.L.; Krishnan, S.; Stachelek, K.; Zolfaghari, E.; McGovern, K.; Lee, T.C.; Carlsson, A.; Kuhn, P.; et al. Potential of Aqueous Humor as a Surrogate Tumor Biopsy for Retinoblastoma. JAMA Ophthalmol. 2017, 135, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Kothari, P.; Marass, F.; Yang, J.L.; Stewart, C.M.; Stephens, D.; Patel, J.; Hasan, M.; Jing, X.; Meng, F.; Enriquez, J.; et al. Cell-free DNA profiling in retinoblastoma patients with advanced intraocular disease: An MSKCC experience. Cancer Med. 2020, 9, 6093–6101. [Google Scholar] [CrossRef]
- Jin, E.; Burnier, J.V. Liquid Biopsy in Uveal Melanoma: Are We There Yet? Ocul. Oncol. Pathol. 2021, 7, 1–16. [Google Scholar] [CrossRef]
- Bek, T. Capillary closure secondary to retinal vein occlusion. A morphological, histopathological, and immunohistochemical study. Acta Ophthalmol. Scand. 1998, 76, 643–648. [Google Scholar] [CrossRef]
- Yang, J.; Yang, N.; Luo, J.; Cheng, G.; Zhang, X.; He, T.; Xing, Y. Overexpression of S100A4 protects retinal ganglion cells against retinal ischemia-reperfusion injury in mice. Exp. Eye Res. 2020, 201, 108281. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Kanematsu, E.; Yoshiura, M.; Iwamoto, T.; Takizawa, N.; Tokuhisa, T.; Mizuno, A. Uric acid as biochemical marker for retinal and optic nerve damage after occlusion and reperfusion of common carotid and vertebral arteries in rat. Jpn. J. Ophthalmol. 1992, 36, 76–83. [Google Scholar] [PubMed]
- Li, Y.; Roth, S.; Laser, M.; Ma, J.-x.; Crosson, C.E. Retinal preconditioning and the induction of heat-shock protein 27. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1299–1304. [Google Scholar] [CrossRef]
- Kalesnykas, G.; Tuulos, T.; Uusitalo, H.; Jolkkonen, J. Neurodegeneration and cellular stress in the retina and optic nerve in rat cerebral ischemia and hypoperfusion models. Neuroscience 2008, 155, 937–947. [Google Scholar] [CrossRef]
- Strunnikova, N.; Baffi, J.; Gonzalez, A.; Silk, W.; Cousins, S.W.; Csaky, K.G. Regulated heat shock protein 27 expression in human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2130–2138. [Google Scholar]
- Arckens, L.; Van der Gucht, E.; Van den Bergh, G.; Massie, A.; Leysen, I.; Vandenbussche, E.; Eysel, U.T.; Huybrechts, R.; Vandesande, F. Differential display implicates cyclophilin A in adult cortical plasticity. Eur. J. Neurosci. 2003, 18, 61–75. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Hernandez, E.; Grant, J.; Santos, A.R.C.; Yang, H.; Ivanov, D. The high-mobility group box-1 nuclear factor mediates retinal injury after ischemia reperfusion. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7187–7194. [Google Scholar] [CrossRef]
- Li, X.; Ye, Z.; Pei, S.; Zheng, D.; Zhu, L. Neuroprotective effect of minocycline on rat retinal ischemia-reperfusion injury. Mol. Vis. 2021, 27, 438. [Google Scholar] [PubMed]
- Ozacmak, H.S.; Ozacmak, V.H.; Barut, F.; Araslı, M.; Ucan, B.H. Pretreatment with mineralocorticoid receptor blocker reduces intestinal injury induced by ischemia and reperfusion: Involvement of inhibition of inflammatory response, oxidative stress, nuclear factor κB, and inducible nitric oxide synthase. J. Surg. Res. 2014, 191, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Shim, M.S.; Kim, K.Y.; Weinreb, R.N.; Wheeler, L.A.; Ju, W.K. Inhibition of cyclophilin D by cyclosporin A promotes retinal ganglion cell survival by preventing mitochondrial alteration in ischemic injury. Cell Death Dis. 2014, 5, e1105. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kim, K.-Y.; Noh, Y.H.; Chai, S.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.-K. Brimonidine blocks glutamate excitotoxicity-induced oxidative stress and preserves mitochondrial transcription factor a in ischemic retinal injury. PLoS ONE 2012, 7, e47098. [Google Scholar] [CrossRef]
- Reinhard, J.; Renner, M.; Wiemann, S.; Shakoor, D.A.; Stute, G.; Dick, H.B.; Faissner, A.; Joachim, S.C. Ischemic injury leads to extracellular matrix alterations in retina and optic nerve. Sci. Rep. 2017, 7, 43470. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Kataoka, K.; Kachi, S.; Komeima, K.; Terasaki, H. Regulation of pathologic retinal angiogenesis in mice and inhibition of VEGF-VEGFR2 binding by soluble heparan sulfate. PLoS ONE 2010, 5, e13493. [Google Scholar] [CrossRef]
- Cheng, G.; He, T.; Xing, Y. Silencing of S100A4, a metastasis-associated protein, inhibits retinal neovascularization via the downregulation of BDNF in oxygen-induced ischaemic retinopathy. Eye 2016, 30, 877–887. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, G.; Tian, K.; Zhang, L.; Yang, N.; Xing, Y.; He, T. S100A4 gene silencing in oxygen-induced ischemic retinopathy inhibits retinal neovascularization via down-regulation of CREB expression. Graefe′s Arch. Clin. Exp. Ophthalmol. 2016, 254, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Inatani, M.; Tanihara, H.; Honjo, M.; Hangai, M.; Kresse, H. Expression of Proteoglycan Decorin in Neural Retina. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1783–1791. [Google Scholar]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.Y.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Windisch, B.K.; LeVatte, T.L.; Archibald, M.L.; Chauhan, B.C. Induction of heat shock proteins 27 and 72 in retinal ganglion cells after acute pressure-induced ischaemia. Clin. Exp. Ophthalmol. 2009, 37, 299–307. [Google Scholar] [CrossRef]
- Whitlock, N.A.; Agarwal, N.; Ma, J.-X.; Crosson, C.E. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Oshitari, T.; Negishi, H.; Dezawa, M.; Mizota, A.; Adachi-Usami, E. Protection of retinal ganglion cells from ischemia-reperfusion injury by electrically applied Hsp27. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3283–3286. [Google Scholar]
- Lewden, O.; Garcher, C.; Assem, M.; Morales, C.; Rochette, L.; Bron, A. Changes of the inducible heat shock protein 70 mRNA level in rat retina after ischemia and reperfusion. Ophthalmic Res. 1998, 30, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Liu, J.; Zheng, Z.; Vexler, Z.S.; Lee, J.E.; Giffard, R.G. Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann. N. Y. Acad. Sci. 2005, 1053, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.M.; Lam, T.T.; Caprioli, J. Hyperthermic pre-conditioning protects retinal neurons from N-methyl-D-aspartate (NMDA)-induced apoptosis in rat. Brain Res. 2003, 970, 119–130. [Google Scholar] [CrossRef]
- Joachim, S.C.; Jehle, T.; Boehm, N.; Gramlich, O.W.; Lagreze, W.A.; Pfeiffer, N.; Grus, F.H. Effect of ischemia duration on autoantibody response in rats undergoing retinal ischemia-reperfusion. Ophthalmic Res. 2012, 48, 67–74. [Google Scholar] [CrossRef]
- Yoneda, S.; Hara, H.; Hirata, A.; Fukushima, M.; Inomata, Y.; Tanihara, H. Vitreous fluid levels of β-amyloid (1–42) and tau in patients with retinal diseases. Jpn. J. Ophthalmol. 2005, 49, 106–108. [Google Scholar] [CrossRef]
- Yu, P.; Zhou, W.; Liu, L.; Tang, Y.-B.; Song, Y.; Lu, J.-J.; Hou, L.-N.; Chen, H.-Z.; Cui, Y.-Y. L-satropane Prevents Retinal Neuron Damage by Attenuating Cell Apoptosis and aβ Production via Activation of M1 Muscarinic Acetylcholine Receptor. Curr. Eye Res. 2017, 42, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Ash, J.; McLeod, D.S.; Lutty, G.A. Transgenic expression of leukemia inhibitory factor (LIF) blocks normal vascular development but not pathological neovascularization in the eye. Mol. Vis. 2005, 11, 298–308. [Google Scholar]
- Rivera-Pérez, J.; Martínez-Rosas, M.; Conde-Castañón, C.A.; Toscano-Garibay, J.D.; Ruiz-Pérez, N.J.; Flores, P.L.; Mera Jimenez, E.; Flores-Estrada, J. Epigallocatechin 3-Gallate has a neuroprotective effect in retinas of rabbits with ischemia/reperfusion through the activation of Nrf2/HO-1. Int. J. Mol. Sci. 2020, 21, 3716. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Sun, Y.; Ren, X.; Lin, Q.; Hu, X.; Huang, X.; Su, S.-B.; Liu, Y.; Liu, X. Angiogenesis mediated by toll-like receptor 4 in ischemic neural tissue. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 330–338. [Google Scholar] [CrossRef][Green Version]
- Yang, S.; Hirooka, K.; Liu, Y.; Fujita, T.; Fukuda, K.; Nakamutra, T.; Itano, T.; Zhang, J.; Nishibori, M.; Shiraga, F. Deleterious role of anti-high mobility group box 1 monoclonal antibody in retinal ischemia-reperfusion injury. Curr. Eye Res. 2011, 36, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jiang, Y.; Steinle, J.J. Inhibition of HMGB1 protects the retina from ischemia-reperfusion, as well as reduces insulin resistance proteins. PLoS ONE 2017, 12, e0178236. [Google Scholar] [CrossRef] [PubMed]
- Hangai, M.; Yoshimura, N.; Yoshida, M.; Yabuuchi, K.; Honda, Y. Interleukin-1 gene expression in transient retinal ischemia in the rat. Investig. Ophthalmol. Vis. Sci. 1995, 36, 571–578. [Google Scholar]
- Hokari, M.; Kuroda, S.; Kinugawa, S.; Ide, T.; Tsutsui, H.; Iwasaki, Y. Overexpression of mitochondrial transcription factor A (TFAM) ameliorates delayed neuronal death due to transient forebrain ischemia in mice. Neuropathology 2010, 30, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhao, T.; Zhang, Y.; Ma, Y.; Jiang, Y. Differences in aqueous concentrations of cytokines in macular edema secondary to branch and central retinal vein occlusion. PLoS ONE 2013, 8, e68149. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, S.; Yousf, A.; Joachim, S.C.; Peters, C.; Mueller-Buehl, A.M.; Wagner, N.; Reinhard, J. Knock-out of tenascin-C ameliorates ischemia-induced rod-photoreceptor degeneration and retinal dysfunction. Front. Neurosci. 2021, 15, 15. [Google Scholar] [CrossRef]
- Mahaling, B.; Srinivasarao, D.A.; Raghu, G.; Kasam, R.K.; Bhanuprakash Reddy, G.; Katti, D.S. A non-invasive nanoparticle mediated delivery of triamcinolone acetonide ameliorates diabetic retinopathy in rats. Nanoscale 2018, 10, 16485–16498. [Google Scholar] [CrossRef] [PubMed]
- Steinle, J.J. Role of HMGB1 signaling in the inflammatory process in diabetic retinopathy. Cell. Signal. 2020, 73, 109687. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.-d.; Wang, Y.-z.; Zou, C.; She, X.-p.; Zheng, Z. The role of uric acid in the pathogenesis of diabetic retinopathy based on Notch pathway. Biochem. Biophys. Res. Commun. 2018, 503, 921–929. [Google Scholar] [CrossRef]
- Joussen, A.M.; Huang, S.; Poulaki, V.; Camphausen, K.; Beecken, W.-D.; Kirchhof, B.; Adamis, A.P. In Vivo Retinal Gene Expression in Early Diabetes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3047–3057. [Google Scholar]
- Brucklacher, R.M.; Patel, K.M.; VanGuilder, H.D.; Bixler, G.V.; Barber, A.J.; Antonetti, D.A.; Lin, C.-M.; LaNoue, K.F.; Gardner, T.W.; Bronson, S.K.; et al. Whole genome assessment of the retinal response to diabetes reveals a progressive neurovascular inflammatory response. BMC Med. Genom. 2008, 1, 26. [Google Scholar] [CrossRef]
- Zeiner, J.; Loukovaara, S.; Losenkova, K.; Zuccarini, M.; Korhonen, A.M.; Lehti, K.; Kauppinen, A.; Kaarniranta, K.; Müller, C.E.; Jalkanen, S.; et al. Soluble and membrane-bound adenylate kinase and nucleotidases augment ATP-mediated inflammation in diabetic retinopathy eyes with vitreous hemorrhage. J. Mol. Med. 2019, 97, 341–354. [Google Scholar] [CrossRef]
- Singh, L.P.; Devi, T.S.; Yumnamcha, T. The Role of Txnip in Mitophagy Dysregulation and Inflammasome Activation in Diabetic Retinopathy: A New Perspective. JOJ Ophthalmol. 2017, 4, 10. [Google Scholar] [CrossRef]
- Van Hove, I.; De Groef, L.; Boeckx, B.; Modave, E.; Hu, T.-T.; Beets, K.; Etienne, I.; Van Bergen, T.; Lambrechts, D.; Moons, L.; et al. Single-cell transcriptome analysis of the Akimba mouse retina reveals cell-type-specific insights into the pathobiology of diabetic retinopathy. Diabetologia 2020, 63, 2235–2248. [Google Scholar] [CrossRef]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 Proteins as an Important Regulator of Macrophage Inflammation. Front. Immunol. 2018, 8, 1908. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Alam, K.; Garcia-Ramirez, M.; Ahmad, A.; Siddiquei, M.M.; Mohammad, G.; Mousa, A.; De Hertogh, G.; Opdenakker, G.; Simó, R. Association of HMGB1 with oxidative stress markers and regulators in PDR. Mol. Vis. 2017, 23, 853–871. [Google Scholar]
- Krizova, L.; Kalousova, M.; Kubena, A.; Benakova, H.; Zima, T.; Kovarik, Z.; Kalvoda, J.; Kalvodova, B. Increased Uric Acid and Glucose Concentrations in Vitreous and Serum of Patients with Diabetic Macular Oedema. Ophthalmic Res. 2011, 46, 73–79. [Google Scholar] [CrossRef]
- Bellini, S.; Barutta, F.; Mastrocola, R.; Imperatore, L.; Bruno, G.; Gruden, G. Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective. Int. J. Mol. Sci. 2017, 18, 2709. [Google Scholar] [CrossRef]
- Ramachandran, S.; Venugopal, A.; Kutty, V.A.D.G.V.; Chitrasree, V.; Mullassari, A.; Pratapchandran, N.S.; Santosh, K.R.; Pillai, M.R.; Santosh, K.R.; Pillai, M.R.; et al. Plasma level of cyclophilin A is increased in patients with type 2 diabetes mellitus and suggests presence of vascular disease. Cardiovasc. Diabetol. 2014, 13, 38. [Google Scholar] [CrossRef]
- Nagai, N.; Ito, Y.; Tanino, T. Effect of High Glucose Levels on Amyloid β Production in Retinas of Spontaneous Diabetes Mellitus Otsuka Long-Evans Tokushima Fatty Rats. Biol. Pharm. Bull. 2015, 38, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Shen, S.; Wang, J.; He, Z.; Poon, A.; Li, J.; Qu, J.; Zhang, S. Comparative Proteomic Analysis of the Mitochondria-associated ER Membrane (MAM) in a Long-term Type 2 Diabetic Rodent Model. Sci. Rep. 2017, 7, 2062. [Google Scholar] [CrossRef]
- Uruska, A.; Michalska, A.; Ostrowska, J.; Skonieczna, P.; Lipski, D.; Uruski, P.; Pakuła, M.; Tykarski, A.; Zozulinska-Ziolkiewicz, D. Is cathelicidin a novel marker of diabetic microangiopathy in patients with type 1 diabetes? Clin. Biochem. 2017, 50, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Saraheimo, M.; Forsblom, C.; Pettersson-Fernholm, K.; Flyvbjerg, A.; Groop, P.H.; Frystyk, J. Increased levels of alpha-defensin (-1, -2 and -3) in type 1 diabetic patients with nephropathy. Nephrol. Dial. Transplant. 2008, 23, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Kolseth, I.B.M.; Reine, T.M.; Parker, K.; Sudworth, A.; Witczak, B.J.; Jenssen, T.G.; Kolset, S.O. Increased levels of inflammatory mediators and proinflammatory monocytes in patients with type I diabetes mellitus and nephropathy. J. Diabetes Its Complicat. 2017, 31, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Low, S.W.Y.; Vaidya, T.; Gadde, S.G.K.; Mochi, T.B.; Kumar, D.; Kassem, I.S.; Costakos, D.M.; Ahmad, B.; Sethu, S.; Ghosh, A.; et al. Decorin Concentrations in Aqueous Humor of Patients with Diabetic Retinopathy. Life 2021, 11, 1421. [Google Scholar] [CrossRef]
- Schaefer, L.; Raslik, I.; Gröne, H.-J.; Schönherr, E.; Macakova, K.; Ugorcakova, J.; Budny, S.; Schaefer, R.M.; Kresse, H. Small proteoglycans in human diabetic nephropathy: Discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican, and fibromodulin. FASEB J. 2001, 15, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Osterbauer, K.J. Is There a Role for Myeloid Cell-Derived Versican in Diabetes-Accelerated Atherosclerosis? Master’s Thesis, University of Washington, Washington, DC, USA, 2018. [Google Scholar]
- Leskova, W.; Pickett, H.; Eshaq, R.S.; Shrestha, B.; Pattillo, C.B.; Harris, N.R. Effect of diabetes and hyaluronidase on the retinal endothelial glycocalyx in mice. Exp. Eye Res. 2019, 179, 125–131. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Ushida, H.; Tomida, D.; Kachi, S.; Kondo, M.; Terasaki, H. Age-dependent alteration of intraocular soluble heparan sulfate levels and its implications for proliferative diabetic retinopathy. Mol. Vis. 2013, 19, 1125–1131. [Google Scholar]
- Singh, L.P.; Devi, T.S. TXNIP Regulates Aberrant ECM Expression and Impaired Innate Immune Responses in Early Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3579. [Google Scholar]
- Oshitari, T.; Polewski, P.; Chadda, M.; Li, A.-F.; Sato, T.; Roy, S. Effect of Combined Antisense Oligonucleotides Against High-Glucose–and Diabetes-Induced Overexpression of Extracellular Matrix Components and Increased Vascular Permeability. Diabetes 2006, 55, 86. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Le, D.S.; Miles, R.; Savage, P.J.; Cornell, E.; Tracy, R.P.; Knowler, W.C.; Krakoff, J. The association of plasma fibrinogen concentration with diabetic microvascular complications in young adults with early-onset of type 2 diabetes. Diabetes Res. Clin. Pract. 2008, 82, 317–323. [Google Scholar] [CrossRef]
- Kubo, Y.; Ishikawa, K.; Mori, K.; Kobayashi, Y.; Nakama, T.; Arima, M.; Nakao, S.; Hisatomi, T.; Haruta, M.; Sonoda, K.-H.; et al. Periostin and tenascin-C interaction promotes angiogenesis in ischemic proliferative retinopathy. Sci. Rep. 2020, 10, 9299. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Mei, Z. Uric Acid Promotes Diabetic Retinopathy Progression through Transforming Growth Factor β2 (TGF-β2) Signaling and Beneficial Effects of Fenofibrate. 2021; preprint. [Google Scholar] [CrossRef]
- Seclì, L.; Fusella, F.; Avalle, L.; Brancaccio, M. The dark-side of the outside: How extracellular heat shock proteins promote cancer. Cell. Mol. Life Sci. 2021, 78, 4069–4083. [Google Scholar] [CrossRef]
- Qu, X.; Wang, C.; Zhang, J.; Qie, G.; Zhou, J. The Roles of CD147 and/or Cyclophilin A in Kidney Diseases. Mediat. Inflamm. 2014, 2014, 728673. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-J.; Wang, P.-W.; Yang, I.H.; Wu, C.-L.; Chuang, J.-H. Amyloid-beta mediates the receptor of advanced glycation end product-induced pro-inflammatory response via toll-like receptor 4 signaling pathway in retinal ganglion cell line RGC-5. Int. J. Biochem. Cell Biol. 2015, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.C.; Lee, T.-C.; Juo, S.-H.H.; Yang, D.-I. Hyperglycemia Increases the Production of Amyloid Beta-Peptide Leading to Decreased Endothelial Tight Junction. CNS Neurosci. Ther. 2016, 22, 291–297. [Google Scholar] [CrossRef]
- Scuderi, S.; D’amico, A.G.; Federico, C.; Saccone, S.; Magro, G.; Bucolo, C.; Drago, F.; D’Agata, V. Different Retinal Expression Patterns of IL-1α, IL-1β, and Their Receptors in a Rat Model of Type 1 STZ-Induced Diabetes. J. Mol. Neurosci. 2015, 56, 431–439. [Google Scholar] [CrossRef]
- Xu, D.; Mu, R.; Wei, X. The Roles of IL-1 Family Cytokines in the Pathogenesis of Systemic Sclerosis. Front. Immunol. 2019, 10, 2025. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sato, T.; Tanaka, A.; Muraoka, T.; Taguchi, M.; Sakurai, Y.; Karasawa, Y.; Ito, M. Elevated Levels of Cytokines Associated with Th2 and Th17 Cells in Vitreous Fluid of Proliferative Diabetic Retinopathy Patients. PLoS ONE 2015, 10, e0137358. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sato, T.; Sakurai, Y.; Taguchi, M.; Harimoto, K.; Karasawa, Y.; Ito, M. Association between aqueous humor and vitreous fluid levels of Th17 cell-related cytokines in patients with proliferative diabetic retinopathy. PLoS ONE 2017, 12, e0178230. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Yin, H.; Yuan, B.; Liu, T.; Luo, L.; Huang, P.; Dai, L.; Zeng, K. IL-33 improves wound healing through enhanced M2 macrophage polarization in diabetic mice. Mol. Immunol. 2017, 90, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Pallero, M.A.; Owusu, B.Y.; Borovjagin, A.V.; Lei, W.; Sanders, P.W.; Murphy-Ullrich, J.E. Calreticulin is important for the development of renal fibrosis and dysfunction in diabetic nephropathy. Matrix Biol. Plus 2020, 8, 100034. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Joussen, A.M.; Klein, C.; Andre, P.; Wagner, D.D.; Hinkes, M.T.; Kirchhof, B.; Adamis, A.P.; Bernfield, M. Role of Syndecan-1 in Leukocyte–Endothelial Interactions in the Ocular Vasculature. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1135–1141. [Google Scholar]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived from the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Myren, M.; Kirby, D.J.; Noonan, M.L.; Maeda, A.; Owens, R.T.; Ricard-Blum, S.; Kram, V.; Kilts, T.M.; Young, M.F. Biglycan potentially regulates angiogenesis during fracture repair by altering expression and function of endostatin. Matrix Biol. 2016, 52–54, 141–150. [Google Scholar] [CrossRef]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef]
- Moreth, K.; Iozzo, R.V.; Schaefer, L. Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 2012, 11, 2084–2091. [Google Scholar] [CrossRef]
- Wang, S.; Du, S.; Wu, Q.; Hu, J.; Li, T. Decorin Prevents Retinal Pigment Epithelial Barrier Breakdown Under Diabetic Conditions by Suppressing p38 MAPK Activation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2971–2979. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican-A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Ievdokimova, N. Hyaluronic acid, receptor CD44, and their role in diabetic complications. Ukr. Biokhim. Zh. 2008, 80, 5–44. [Google Scholar]
- Tavianatou, A.G.; Caon, I.; Franchi, M.; Piperigkou, Z.; Galesso, D.; Karamanos, N.K. Hyaluronan: Molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J. 2019, 286, 2883–2908. [Google Scholar] [CrossRef] [PubMed]
- Kanters, S.D.J.M.; Banga, J.-D.; Algra, A.; Frijns, R.C.J.M.; Beutler, J.J.; Fijnheer, R. Plasma Levels of Cellular Fibronectin in Diabetes. Diabetes Care 2001, 24, 323. [Google Scholar] [CrossRef]
- Milner, R.; Crocker, S.J.; Hung, S.; Wang, X.; Frausto, R.F.; del Zoppo, G.J. Fibronectin-and vitronectin-induced microglial activation and matrix metalloproteinase-9 expression is mediated by integrins α5β1 and αvβ5. J. Immunol. 2007, 178, 8158–8167. [Google Scholar] [CrossRef] [PubMed]
- Sarker, B.; Cardona, S.M.; Vanegas, D.; Church, K.; Mendiola, A.S.; Cardona, A. Fibrinogen in Microglia-Mediated Inflammation in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 418. [Google Scholar]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef]
- Castellon, R.; Caballero, S.; Hamdi, H.K.; Atilano, S.R.; Aoki, A.M.; Tarnuzzer, R.W.; Kenney, M.C.; Grant, M.B.; Ljubimov, A.V. Effects of Tenascin-C on Normal and Diabetic Retinal Endothelial Cells in Culture. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2758–2766. [Google Scholar]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Zuliani-Alvarez, L.; Marzeda, A.M.; Deligne, C.; Schwenzer, A.; McCann, F.E.; Marsden, B.D.; Piccinini, A.M.; Midwood, K.S. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat. Commun. 2017, 8, 1595. [Google Scholar] [CrossRef] [PubMed]
- Notomi, S.; Hisatomi, T.; Kanemaru, T.; Takeda, A.; Ikeda, Y.; Enaida, H.; Kroemer, G.; Ishibashi, T. Critical involvement of extracellular ATP acting on P2RX7 purinergic receptors in photoreceptor cell death. Am. J. Pathol. 2011, 179, 2798–2809. [Google Scholar] [CrossRef] [PubMed]
- Notomi, S.; Hisatomi, T.; Murakami, Y.; Terasaki, H.; Sonoda, S.; Asato, R.; Takeda, A.; Ikeda, Y.; Enaida, H.; Sakamoto, T.; et al. Dynamic increase in extracellular ATP accelerates photoreceptor cell apoptosis via ligation of P2RX7 in subretinal hemorrhage. PLoS ONE 2013, 8, e53338. [Google Scholar] [CrossRef] [PubMed]
- Ardeljan, C.P.; Ardeljan, D.; Abu-Asab, M.; Chan, C.C. Inflammation and Cell Death in Age-Related Macular Degeneration: An Immunopathological and Ultrastructural Model. J. Clin. Med. 2014, 3, 1542–1560. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W. The proteomics of drusen. Cold Spring Harb. Perspect. Med. 2014, 4, a017194. [Google Scholar] [CrossRef]
- Subramani, S.; Khor, S.E.; Livingstone, B.I.; Kulkarni, U.V. Serum uric acid levels and its association with age-related macular degeneration (ARMD). Med. J. Malays. 2010, 65, 36–40. [Google Scholar]
- Kaarniranta, K.; Salminen, A.; Eskelinen, E.L.; Kopitz, J. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2009, 8, 128–139. [Google Scholar] [CrossRef]
- Dinet, V.; Bruban, J.; Chalour, N.; Maoui, A.; An, N.; Jonet, L.; Buret, A.; Behar-Cohen, F.; Klein, C.; Tréton, J.; et al. Distinct effects of inflammation on gliosis, osmohomeostasis, and vascular integrity during amyloid beta-induced retinal degeneration. Aging Cell 2012, 11, 683–693. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Z.; Zhao, X.; Xie, H.; Zhang, C.; Sun, X.; Zhang, J. HMGB2 causes photoreceptor death via down-regulating Nrf2/HO-1 and up-regulating NF-κB/NLRP3 signaling pathways in light-induced retinal degeneration model. Free Radic. Biol. Med. 2022, 181, 14–28. [Google Scholar] [CrossRef]
- Wooff, Y.; Man, S.M.; Aggio-Bruce, R.; Natoli, R.; Fernando, N. IL-1 Family Members Mediate Cell Death, Inflammation and Angiogenesis in Retinal Degenerative Diseases. Front. Immunol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef]
- Lin, H.; Xu, H.; Liang, F.Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3521–3529. [Google Scholar] [CrossRef] [PubMed]
- Totan, Y.; Koca, C.; Erdurmuş, M.; Keskin, U.; Yiğitoğlu, R. Endothelin-1 and Nitric Oxide Levels in Exudative Age-Related Macular Degeneration. J. Ophthalmic Vis. Res. 2015, 10, 151–154. [Google Scholar] [PubMed]
- Regatieri, C.; Dreyfuss, J.L.; Melo, G.; Lavinsky, D.; Hossaka, S.; Rodrigues, E.B.; Farah, M.E.; Maia, M.; Nader, H. Quantitative evaluation of experimental choroidal neovascularization by confocal scanning laser ophthalmoscopy: Fluorescein angiogram parallels heparan sulfate proteoglycan expression. Braz. J. Med. Biol. Res. 2010, 43, 627–633. [Google Scholar] [CrossRef]
- Lin, M.K.; Yang, J.; Hsu, C.W.; Gore, A.; Bassuk, A.G.; Brown, L.M.; Colligan, R.; Sengillo, J.D.; Mahajan, V.B.; Tsang, S.H. HTRA1, an age-related macular degeneration protease, processes extracellular matrix proteins EFEMP1 and TSP1. Aging Cell 2018, 17, e12710. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J.; Shukla, D. Role of heparan sulfate in ocular diseases. Exp. Eye Res. 2013, 110, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Budoff, G.; Prenner, J.L.; Schwarzbauer, J.E. Minireview: Fibronectin in retinal disease. Exp. Biol. Med. 2017, 242, 1–7. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yoshida, S.; Zhou, Y.; Nakama, T.; Ishikawa, K.; Kubo, Y.; Arima, M.; Nakao, S.; Hisatomi, T.; Ikeda, Y.; et al. Tenascin-C secreted by transdifferentiated retinal pigment epithelial cells promotes choroidal neovascularization via integrin αV. Lab. Investig. 2016, 96, 1178–1188. [Google Scholar] [CrossRef]
- Li, P.; Li, Q.; Biswas, N.; Xin, H.; Diemer, T.; Liu, L.; Perez Gutierrez, L.; Paternostro, G.; Piermarocchi, C.; Domanskyi, S.; et al. LIF, a mitogen for choroidal endothelial cells, protects the choriocapillaris: Implications for prevention of geographic atrophy. EMBO Mol. Med. 2021, 14, e14511. [Google Scholar] [CrossRef]
- Singh, J.A.; Cleveland, J.D. Gout and the risk of age-related macular degeneration in the elderly. PLoS ONE 2018, 13, e0199562. [Google Scholar] [CrossRef]
- Aguilà, M.; Cheetham, M.E. Hsp90 as a Potential Therapeutic Target in Retinal Disease. Adv. Exp. Med. Biol. 2016, 854, 161–167. [Google Scholar]
- Kumar, R.; Soni, R.; Heindl, S.E.; Wiltshire, D.A.; Khan, S. Unravelling the Role of HSP70 as the Unexplored Molecular Target in Age-Related Macular Degeneration. Cureus 2020, 12, e8960. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Takeda, A.; Yoshimura, T.; Oshima, Y.; Sonoda, K.H.; Ishibashi, T. IL-10 is significantly involved in HSP70-regulation of experimental subretinal fibrosis. PLoS ONE 2013, 8, e80288. [Google Scholar]
- Hoh Kam, J.; Lenassi, E.; Jeffery, G. Viewing ageing eyes: Diverse sites of amyloid Beta accumulation in the ageing mouse retina and the up-regulation of macrophages. PLoS ONE 2010, 5, e13127. [Google Scholar] [CrossRef]
- Liu, R.T.; Wang, A.; To, E.; Gao, J.; Cao, S.; Cui, J.Z.; Matsubara, J.A. Vinpocetine inhibits amyloid-beta induced activation of NF-κB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp. Eye Res. 2014, 127, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Cai, B.; Li, Y.; Su, W.; Zhao, X.; Gong, B.; Li, Z.; Zhang, X.; Wu, Y.; Chen, C.; et al. HMGB1 and Caveolin-1 related to RPE cell senescence in age-related macular degeneration. Aging 2019, 11, 4323–4337. [Google Scholar] [CrossRef] [PubMed]
- Nassar, K.; Grisanti, S.; Elfar, E.; Lüke, J.; Lüke, M.; Grisanti, S. Serum cytokines as biomarkers for age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 699–704. [Google Scholar] [CrossRef]
- Sakurada, Y.; Nakamura, Y.; Yoneyama, S.; Mabuchi, F.; Gotoh, T.; Tateno, Y.; Sugiyama, A.; Kubota, T.; Iijima, H. Aqueous humor cytokine levels in patients with polypoidal choroidal vasculopathy and neovascular age-related macular degeneration. Ophthalmic Res. 2015, 53, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Giani, A.; Kataoka, K.; Morizane, Y.; Kayama, M.; Thanos, A.; Nakatake, S.; Notomi, S.; et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014, 21, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Simtchouk, S.; Schindler, S.M.; Villanueva, E.B.; Gill, N.E.; Walker, D.G.; Wolthers, K.R.; Klegeris, A. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol. Cell. Neurosci. 2014, 60, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Wang, J.H.; Barathi, V.A.; Prea, S.M.; He, Z.; Lee, J.H.; Bender, J.; King, A.E.; Logan, G.J.; Alexander, I.E.; et al. AAV-mediated gene delivery of the calreticulin anti-angiogenic domain inhibits ocular neovascularization. Angiogenesis 2018, 21, 95–109. [Google Scholar] [CrossRef]
- Wang, X.; Ma, W.; Han, S.; Meng, Z.; Zhao, L.; Yin, Y.; Wang, Y.; Li, J. TGF-β participates choroid neovascularization through Smad2/3-VEGF/TNF-α signaling in mice with Laser-induced wet age-related macular degeneration. Sci. Rep. 2017, 7, 9672. [Google Scholar] [CrossRef]
- Low, S.W.Y.; Connor, T.B.; Kassem, I.S.; Costakos, D.M.; Chaurasia, S.S. Small Leucine-Rich Proteoglycans (SLRPs) in the Retina. Int. J. Mol. Sci. 2021, 22, 7293. [Google Scholar] [CrossRef] [PubMed]
- Austin, B.A.; Liu, B.; Li, Z.; Nussenblatt, R.B. Biologically Active Fibronectin Fragments Stimulate Release of MCP-1 and Catabolic Cytokines from Murine Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2896–2902. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.; Mitchell, P.; Leeder, S.R.; Wang, J.J. Plasma fibrinogen levels, other cardiovascular risk factors, and age-related maculopathy: The Blue Mountains Eye Study. Arch. Ophthalmol. 1998, 116, 583–587. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yoshida, S.; Zhou, Y.; Nakama, T.; Ishikawa, K.; Arima, M.; Nakao, S.; Sassa, Y.; Takeda, A.; Hisatomi, T.; et al. Tenascin-C promotes angiogenesis in fibrovascular membranes in eyes with proliferative diabetic retinopathy. Mol. Vis. 2016, 22, 436–445. [Google Scholar]
- Pachydaki, S.I.; Tari, S.R.; Lee, S.E.; Ma, W.; Tseng, J.J.; Sosunov, A.A.; Cataldergirmen, G.; Scarmeas, N.; Caspersen, C.; Chang, S.; et al. Upregulation of RAGE and its ligands in proliferative retinal disease. Exp. Eye Res. 2006, 82, 807–815. [Google Scholar] [CrossRef]
- Quintyn, J.C.; Pereira, F.; Hellot, M.F.; Brasseur, G.; Coquerel, A. Concentration of neuron-specific enolase and S100 protein in the subretinal fluid of rhegmatogenous retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. 2005, 243, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Ki-i, Y.; Hashiguchi, T.; Kawahara, K.-i.; Biswas, K.K.; Nakamura, M.; Sonoda, Y.; Yamakiri, K.; Okubo, A.; Sakamoto, T.; et al. Intraocular expression and release of high-mobility group box 1 protein in retinal detachment. Lab. Investig. 2009, 89, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Moreno, K.M.; Gentile, A.M.; Sabbadini, R.A.; Stoller, G.L. Sphingosine-1-phosphate (S1P) is a novel fibrotic mediator in the eye. Exp. Eye Res. 2008, 87, 367–375. [Google Scholar] [CrossRef]
- Kayama, M.; Nakazawa, T.; Thanos, A.; Morizane, Y.; Murakami, Y.; Theodoropoulou, S.; Abe, T.; Vavvas, D.; Miller, J.W. Heat shock protein 70 (HSP70) is critical for the photoreceptor stress response after retinal detachment via modulating anti-apoptotic Akt kinase. Am. J. Pathol. 2011, 178, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, T.; Hisatomi, T.; Notomi, S.; Nakatake, S.; Murakami, Y.; Sengoku, A.; Ikeda, Y.; Yoshida, S.; Ishibashi, T.; Sonoda, K.-H. Vitreous and subretinal fluid ATP concentrations in rhegmatogenous retinal detachment. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5376. [Google Scholar]
- Kawano, H.; Ito, T.; Yamada, S.; Hashiguchi, T.; Maruyama, I.; Hisatomi, T.; Nakamura, M.; Sakamoto, T. Toxic effects of extracellular histones and their neutralization by vitreous in retinal detachment. Lab. Investig. 2014, 94, 569–585. [Google Scholar] [CrossRef]
- Ricker, L.J.A.G.; Kijlstra, A.; Kessels, A.G.H.; de Jager, W.; Liem, A.T.A.; Hendrikse, F.; La Heij, E.C. Interleukin and growth factor levels in subretinal fluid in rhegmatogenous retinal detachment: A case-control study. PLoS ONE 2011, 6, e19141. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Hirakata, T.; Asada, Y.; Iwamoto, S.; Nakae, S.; Matsuda, A. The role of interleukin-33 in retinal tissue fibrosis after laser injury. Investig. Ophthalmol. Vis. Sci. 2018, 59, 344. [Google Scholar]
- Wang, J.B.; Tian, C.W.; Guo, C.M.; Du, H.J.; Liu, H.L.; Zhang, Y.J.; Hui, Y.N. Increased levels of soluble syndecan-1 in the subretinal fluid and the vitreous of eyes with rhegmatogenous retinal detachment. Curr. Eye Res. 2008, 33, 101–107. [Google Scholar] [CrossRef]
- Farjo, R.; Peterson, W.M.; Naash, M.I. Expression profiling after retinal detachment and reattachment: A possible role for aquaporin-0. Investig. Ophthalmol. Vis. Sci. 2008, 49, 511–521. [Google Scholar] [CrossRef]
- Begum, G.; O′Neill, J.; Chaudhary, R.; Blachford, K.; Snead, D.R.J.; Berry, M.; Scott, R.A.H.; Logan, A.; Blanch, R.J. Altered Decorin Biology in Proliferative Vitreoretinopathy: A Mechanistic and Cohort Study. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4929–4936. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, M.; Esser, P.; Wiedemann, P.; Heimann, K. Tenascin and decorin in epiretinal membranes of proliferative vitreoretinopathy and proliferative diabetic retinopathy. Ger. J. Ophthalmol. 1993, 2, 28–31. [Google Scholar]
- Mitamura, Y.; Takeuchi, S.; Ohtsuka, K.; Matsuda, A.; Hiraiwa, N.; Kusakabe, M. Tenascin-C Levels in the Vitreous of Patients with Proliferative Diabetic Retinopathy. Diabetes Care 2002, 25, 1899. [Google Scholar] [CrossRef][Green Version]
- Theocharis, I. Fibrinogen and rhegmatogenous retinal detachment: A pilot prospective study. Clin. Ophthalmol. 2010, 4, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.R.; Dubovy, S.R.; Relhan, N.; Flynn, H.W., Jr. Clinicopathologic Correlation of a Subretinal Proliferative Vitreoretinopathy Band in a Patient with Chronic Recurrent Retinal Detachment. Case Rep. Ophthalmol. 2018, 9, 279–282. [Google Scholar] [CrossRef]
- El-Asrar, A.M.A.; Missotten, L.; Geboes, K. Expression of high-mobility groups box-1/receptor for advanced glycation end products/osteopontin/early growth response-1 pathway in proliferative vitreoretinal epiretinal membranes. Mol. Vis. 2011, 17, 508–518. [Google Scholar]
- Abu El-Asrar, A.M.; Imtiaz Nawaz, M.; Kangave, D.; Siddiquei, M.M.; Geboes, K. Osteopontin and Other Regulators of Angiogenesis and Fibrogenesis in the Vitreous from Patients with Proliferative Vitreoretinal Disorders. Mediat. Inflamm. 2012, 2012, 493043. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Lin, C.-W.; Hsieh, M.-C.; Wu, H.-J.; Wu, W.-S.; Wu, W.-C.; Kao, Y.-H. High mobility group B1 up-regulates angiogenic and fibrogenic factors in human retinal pigment epithelial ARPE-19 cells. Cell. Signal. 2017, 40, 248–257. [Google Scholar] [CrossRef]
- Tosi, G.M.; Regoli, M.; Altera, A.; Galvagni, F.; Arcuri, C.; Bacci, T.; Elia, I.; Realini, G.; Orlandini, M.; Bertelli, E. Heat Shock Protein 90 Involvement in the Development of Idiopathic Epiretinal Membranes. Investig. Ophthalmol. Vis. Sci. 2020, 61, 34. [Google Scholar] [CrossRef]
- Aplin, F.P.; Vessey, K.A.; Luu, C.D.; Guymer, R.H.; Shepherd, R.K.; Fletcher, E.L. Retinal Changes in an ATP-Induced Model of Retinal Degeneration. Front. Neuroanat. 2016, 10, 46. [Google Scholar] [CrossRef]
- Sugita, J.; Asada, Y.; Kawano, H.; Ebihara, N.; Murakami, A.; Nakae, S.; Matsuda, A. The role of interleukin-33 expression in retinal tissue. Investig. Ophthalmol. Vis. Sci. 2014, 55, 708. [Google Scholar]
- Augustine, J.; Pavlou, S.; Ali, I.; Harkin, K.; Ozaki, E.; Campbell, M.; Stitt, A.W.; Xu, H.; Chen, M. IL-33 deficiency causes persistent inflammation and severe neurodegeneration in retinal detachment. J. Neuroinflamm. 2019, 16, 251. [Google Scholar] [CrossRef]
- Kaprinis, K.; Symeonidis, C.; Papakonstantinou, E.; Tsinopoulos, I.; Dimitrakos, S.A. Decreased hyaluronan concentration during primary rhegmatogenous retinal detachment. Eur. J. Ophthalmol. 2016, 26, 633–638. [Google Scholar] [CrossRef]
- Mitamura, Y.; Takeuchi, S.; Ohtsuka, K.; Matsuda, A.; Yamamoto, T.; Yamamoto, S.; Hiraiwa, N.; Kusakabe, M. Tenascin-C levels in the vitreous of patients with proliferative vitreoretinopathy. Ophthalmologica 2003, 217, 422–425. [Google Scholar] [CrossRef]
- Yeo, J.H.; Sadeghi, J.; Campochiaro, P.A.; Green, W.R.; Glaser, B.M. Intravitreous Fibronectin and Platelet-Derived Growth Factor: New Model for Traction Retinal Detachment. Arch. Ophthalmol. 1986, 104, 417–421. [Google Scholar] [CrossRef]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal inflammation, cell death and inherited retinal dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef]
- Roesch, K.; Stadler, M.B.; Cepko, C.L. Gene expression changes within Müller glial cells in retinitis pigmentosa. Mol. Vis. 2012, 18, 1197. [Google Scholar]
- Chusova, G.; Ostapenko, I.; Shabanova, M.; Eliseeva, R. Changes in the blood uric acid levels in patients with retinitis pigmentosa and in rats with hereditary retinal degeneration. Biull.’ Eksp. Biol. Med. 1982, 94, 21–23. [Google Scholar]
- Gawęcki, M. Laser treatment in retinitis pigmentosa-a review. Lasers Med. Sci. 2020, 35, 1663–1670. [Google Scholar] [CrossRef]
- Löffler, K.U.; Edward, D.P.; Tso, M.O. Immunoreactivity against tau, amyloid precursor protein, and beta-amyloid in the human retina. Investig. Ophthalmol. Vis. Sci. 1995, 36, 24–31. [Google Scholar]
- Murakami, Y.; Ikeda, Y.; Nakatake, S.; Tachibana, T.; Fujiwara, K.; Yoshida, N.; Notomi, S.; Nakao, S.; Hisatomi, T.; Miller, J. Necrotic enlargement of cone photoreceptor cells and the release of high-mobility group box-1 in retinitis pigmentosa. Cell Death Discov. 2015, 1, 1–7. [Google Scholar] [CrossRef]
- Landers, R.A.; Rayborn, M.E.; Myers, K.M.; Hollyfield, J.G. Increased retinal synthesis of heparan sulfate proteoglycan and HNK-1 glycoproteins following photoreceptor degeneration. J. Neurochem. 1994, 63, 737–750. [Google Scholar] [CrossRef]
- Maïza, A.; Chantepie, S.; Vera, C.; Fifre, A.; Huynh, M.B.; Stettler, O.; Ouidja, M.O.; Papy-Garcia, D. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 2018, 592, 3806–3818. [Google Scholar] [CrossRef]
- Yun, J.; Jiang, G.; Wang, Y.; Xiao, T.; Zhao, Y.; Sun, D.; Kaplan, H.J.; Shao, H. The HMGB1-CXCL12 Complex Promotes Inflammatory Cell Infiltration in Uveitogenic T Cell-Induced Chronic Experimental Autoimmune Uveitis. Front. Immunol. 2017, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.J.; Sun, D.; Shao, H. Damage-associated Molecular Patterns in Clinical and Animal Models of Uveitis. Ocul. Immunol. Inflamm. 2021, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Minhas, G.; Sharma, J.; Khan, N. Cellular stress response and immune signaling in retinal ischemia–reperfusion injury. Front. Immunol. 2016, 7, 444. [Google Scholar] [CrossRef] [PubMed]
- Mulfaul, K.; Rhatigan, M.; Doyle, S. Toll-Like Receptors and Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 19–28. [Google Scholar]
- Sánchez-Cruz, A.; Méndez, A.C.; Lizasoain, I.; de la Villa, P.; de la Rosa, E.J.; Hernández-Sánchez, C. Tlr2 Gene Deletion Delays Retinal Degeneration in Two Genetically Distinct Mouse Models of Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 7815. [Google Scholar] [CrossRef]
- Hooper, M.J.; Wang, J.; Browning, R.; Ash, J.D. Damage-associated molecular pattern recognition is required for induction of retinal neuroprotective pathways in a sex-dependent manner. Sci. Rep. 2018, 8, 9115. [Google Scholar] [CrossRef]
- Allocca, M.; Corrigan, J.J.; Mazumder, A.; Fake, K.R.; Samson, L.D. Inflammation, necrosis, and the kinase RIP3 are key mediators of AAG-dependent alkylation-induced retinal degeneration. Sci. Signal. 2019, 12, 568. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, S.; Zhang, J.; Guan, X.L.; Sun, B.G.; Sun, L. A virulent Bacillus cereus strain from deep-sea cold seep induces pyroptosis in a manner that involves NLRP3 inflammasome, JNK pathway, and lysosomal rupture. Virulence 2021, 12, 1362–1376. [Google Scholar] [CrossRef]
- Willermain, F.; Rosenbaum, J.T.; Bodaghi, B.; Rosenzweig, H.L.; Childers, S.; Behrend, T.; Wildner, G.; Dick, A.D. Interplay between innate and adaptive immunity in the development of non-infectious uveitis. Prog. Retin. Eye Res. 2012, 31, 182–194. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, M.; Bai, Y.; Huang, L.; Yu, W.; Bian, Z.; Zhao, M.; Li, X. Retinal ischemia/reperfusion injury is mediated by Toll-like receptor 4 activation of NLRP3 inflammasomes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5466–5475. [Google Scholar] [CrossRef]
- Lim, R.R.; Wieser, M.E.; Ganga, R.R.; Barathi, V.A.; Lakshminarayanan, R.; Mohan, R.R.; Hainsworth, D.P.; Chaurasia, S.S. NOD-like Receptors in the Eye: Uncovering Its Role in Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 21, 899. [Google Scholar] [CrossRef]
- Kataoka, K.; Matsumoto, H.; Kaneko, H.; Notomi, S.; Takeuchi, K.; Sweigard, J.; Atik, A.; Murakami, Y.; Connor, K.; Terasaki, H. Macrophage-and RIP3-dependent inflammasome activation exacerbates retinal detachment-induced photoreceptor cell death. Cell Death Dis. 2015, 6, e1731. [Google Scholar] [CrossRef]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jiang, Y.; Steinle, J.J. Epac1 and Glycyrrhizin Both Inhibit HMGB1 Levels to Reduce Diabetes-Induced Neuronal and Vascular Damage in the Mouse Retina. J. Clin. Med. 2019, 8, 772. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Huang, J.; Xie, M.; Yu, Y.; Zhu, S.; Kang, R.; Cao, L.; Tang, D.; Duan, X. MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy 2014, 10, 442–452. [Google Scholar] [CrossRef]
- Liu, M.; Wang, S.M.; Jiang, Z.X.; Lauren, H.; Tao, L.M. Effects of miR-22 on viability, migration, invasion and apoptosis in retinoblastoma Y79 cells by targeting high-mobility group box 1. Int. J. Ophthalmol. 2018, 11, 1600–1607. [Google Scholar] [PubMed]
- Land, W.G. Use of DAMPs and SAMPs as Therapeutic Targets or Therapeutics: A Note of Caution. Mol. Diagn. Ther. 2020, 24, 251–262. [Google Scholar] [CrossRef]
- Sakamoto, K.; Endo, K.; Suzuki, T.; Fujimura, K.; Kurauchi, Y.; Mori, A.; Nakahara, T.; Ishii, K. P2X7 receptor antagonists protect against N-methyl-d-aspartic acid-induced neuronal injury in the rat retina. Eur. J. Pharmacol. 2015, 756, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Srethapakdi, M.; Liu, F.; Tavorath, R.; Rosen, N. Inhibition of Hsp90 function by ansamycins causes retinoblastoma gene product-dependent G1 arrest. Cancer Res. 2000, 60, 3940–3946. [Google Scholar] [PubMed]
- Lee, D.; Kim, K.Y.; Shim, M.S.; Kim, S.Y.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 ameliorates oxidative stress and prevents mitochondrial alteration in ischemic retinal injury. Apoptosis 2014, 19, 603–614. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, J.; Bu, S. Tasquinimod efficacy and S100A9 expression in glucose-treated HREC cells. Int. Ophthalmol. 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.C.; Kao, Y.H.; Hu, P.S.; Chen, J.H. Geldanamycin, a HSP90 inhibitor, attenuates the hypoxia-induced vascular endothelial growth factor expression in retinal pigment epithelium cells in vitro. Exp. Eye Res. 2007, 85, 721–731. [Google Scholar] [CrossRef]
- Van Harten, R.M.; van Woudenbergh, E.; van Dijk, A.; Haagsman, H.P. Cathelicidins: Immunomodulatory Antimicrobials. Vaccines 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, S.; Szurman, P.; Warga, M.; Kaczmarek, R.; Ziemssen, F.; Tatar, O.; Bartz-Schmidt, K.U. Decorin Modulates Wound Healing in Experimental Glaucoma Filtration Surgery: A Pilot Study. Investig. Ophthalmol. Vis. Sci. 2005, 46, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Nassar, K.; Lüke, J.; Lüke, M.; Kamal, M.; Abd El-Nabi, E.; Soliman, M.; Rohrbach, M.; Grisanti, S. The novel use of decorin in prevention of the development of proliferative vitreoretinopathy (PVR). Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Q.; Wang, R.Q.; Wang, R.Z.; Wang, J. Protection of leukemia inhibitory factor against high-glucose-induced human retinal endothelial cell dysfunction. Arch. Physiol. Biochem. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, S.; Copland, D.A.; Liu, J.; Wu, J.; Gardner, P.J.; Ozaki, E.; Doyle, S.L.; Campbell, M.; Dick, A.D. Interleukin-33 regulates tissue remodelling and inhibits angiogenesis in the eye. J. Pathol. 2017, 241, 45–56. [Google Scholar] [CrossRef]
- He, S.; Li, X.; Chan, N.; Hinton, D.R. Review: Epigenetic mechanisms in ocular disease. Mol. Vis. 2013, 19, 665–674. [Google Scholar]
- Nanini, H.F.; Bernardazzi, C.; Castro, F.; de Souza, H.S.P. Damage-associated molecular patterns in inflammatory bowel disease: From biomarkers to therapeutic targets. World J. Gastroenterol. 2018, 24, 4622–4634. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xie, Y.; Sun, X.; Zeh, H.J., 3rd; Kang, R.; Lotze, M.T.; Tang, D. DAMPs, ageing, and cancer: The ‘DAMP Hypothesis’. Ageing Res. Rev. 2015, 24 Pt A, 3–16. [Google Scholar] [CrossRef]
- Jimenez-Mateos, E.M.; Smith, J.; Nicke, A.; Engel, T. Regulation of P2X7 receptor expression and function in the brain. Brain Res. Bull. 2019, 151, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L.; Tang, D.; Lotze, M.T. Life after death: Targeting high mobility group box 1 in emergent cancer therapies. Am. J. Cancer Res. 2013, 3, 1–20. [Google Scholar] [PubMed]




| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
Endophthalmitis | S100A7, S100A9 [18] | Ca2+ binding protein | Cytoplasmic | Retina |
| HMGB1 [20] | Nuclear binding protein | Nuclear | Vitreous | |
| αβ-crystallin [21] | Molecular chaperones | Cytoplasmic | Retina | |
| LIF [22] | Cytokines | Cytoplasmic | Retina | |
| IL-1α [23] | Cytokines | Cytoplasmic | Vitreous | |
| β−defensin-1, -2 [24,25] | Antimicrobial protein | ER | RPE/CBE/Müller glia | |
| Cathelicidin LL37 [26] | Antimicrobial protein | ER | Müller glia | |
| SAA [27] | Acute-phase protein | Plasma | Serum |
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
Uveitis | S100A8, S100A9, S100A12 [30] | Ca2+ binding protein | Cytoplasmic | Serum/aqueous/tears |
| HMGB1 [31] | Nuclear binding protein | Nuclear | Retina | |
| HSP70 [32] | Molecular chaperones | Cytoplasmic | Serum | |
| SAA [33] | Acute-phase protein | Plasma | Aqueous | |
| Fibronectin [34] | Glycoprotein | ECM | Iris | |
| Fibrinogen [35] | Glycoprotein | ECM | Iris |
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
Glaucoma | S100B [46] | Ca2+ binding protein | Cytoplasmic | Astrocyte/Müller glia |
| LIF [47] | Cytokine | Cytoplasmic | Retina | |
| Uric acid [48] | Metabolic product | Cytoplasmic | Serum | |
| HSP60, HSP70 [49] | Molecular chaperones | Cytoplasmic | Retina | |
| ATP [50] | Nucleotide | Cytoplasmic | Aqueous/vitreous | |
| Aβ [51] | Peptide | Cytoplasmic | Aqueous/optic nerve | |
| Histone-H4 [52] | Nuclear binding protein | Nuclear | Serum | |
| HMGB1 [53] | Nuclear binding protein | Nuclear | Aqueous | |
| IL-1α [54] | Cytokine | Cytoplasmic | Aqueous | |
| mtDNA [55] | Nucleic acid | Mitchondria | Ganglion cell | |
| Calreticulin [56] | Multifunction soluble protein | ER | Nerve fiber layer | |
| ET-1 [57] | Ribonuclease A | ER | Astrocyte | |
| Decorin [58] | Proteoglycan | ECM | Aqueous | |
| Biglycan [59] | Proteoglycan | ECM | Optic nerve | |
| Versican [60] | Proteoglycan | ECM | Trabecular meshwork | |
| Aggrecan [61] | Proteoglycan | ECM | Optic nerve | |
| Phosphocan [62] | Proteoglycan | ECM | Retina/optic nerve | |
| HS [63] | Glycosaminoglycan | ECM | Retina/trabecular meshwork | |
| Fibronectin [62,64] | Glycoprotein | ECM | Retina/optic nerve | |
| Laminin [62] | Glycoprotein | ECM | Retina/optic nerve/astrocytes | |
| Tenascin-C [62] | Glycoprotein | ECM | Trabecular meshwork | |
| SAA [65] | Acute-phase protein | Plasma | Trabecular meshwork and plasma |
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
Ocular cancer | S100 [83] | Ca2+ binding protein | Cytoplasmic | Astrocytes/ganglion cell/Müller glia |
| S100A1 [86] | Ca2+ binding protein | Cytoplasmic | Uveal melanoma | |
| S100B [87] | Ca2+ binding protein | Cytoplasmic | Serum | |
| Uric acid [88] | Metabolic product | Cytoplasmic | Aqueous | |
| HSP70, HSP90 [89,90] | Molecular chaperones | Cytoplasmic | Retina/extracellular vesicles | |
| HMGB1 [91] | Nuclear binding protein | Nuclear | Retinoblastoma | |
| cfcDNA [92] | Nucleic acid | Nuclear | Plasma | |
| Versican [93] | Proteoglycan | ECM | Uveal melanoma |
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
Ischemic retinopathy | S100 [103], S100A4 [104] | Ca2+ binding proteins | Cytoplasmic | Ganglion cell |
| Uric acid [105] | Metabolic product | Cytoplasmic | Retina | |
| HSP27, HSP60, HSP70, αβ-crystallin [106,107,108] | Molecular chaperones | Cytoplasmic | Retina (RGC, RPE, INL) | |
| Cyclophilin A [109] | Ubiquitous protein | Cytoplasmic | Neuron | |
| LIF [47] | Peptide | Cytoplasmic | Retina | |
| HMGB1 [110] | Nuclear binding protein | Nuclear | Vitreous/retina | |
| IL-1α [111,112] | Cytokine | Cytoplasmic | Retina/plasma | |
| TFAM [113,114] | Transcription factor | Mitchondria | Retina (OPL, INL, IPL, GCL) | |
| Decorin [115] | Proteoglycan | ECM | Retina (INL) | |
| Fibronectin [115] | Glycoprotein | ECM | Retina | |
| Laminin [115] | Glycoprotein | ECM | Optic nerve | |
| Tenascin-C [115] | Glycoprotein | ECM | Optic nerve | |
| HS [116] | Glycosaminoglycan | ECM | Optic nerve | |
| Chondritin sulfate [115] | Glycosaminoglycan | ECM | Optic nerve | |
| Aggrecan [115] | Proteoglycan | ECM | Optic nerve |
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
PVR/RRD | LIF [130] | Peptide | Cytoplasmic | Pre-retinal membrane |
| S100 [228,229] | Ca2+ binding protein | Cytoplasmic | Epiretinal membrane/subretinal fluid | |
| HMGB1 [230] | Nuclear binding protein | Nuclear | Vitreous | |
| HSP47, HSP70 [231,232] | Molecular chaperones | Cytoplasmic | RPE/inner segments | |
| ATP [233] | Nucleotide | Cytoplasmic | Vitreous/subretinal fluid | |
| Histone-H3 [234] | Nuclear binding protein | Nuclear | Vitreous/ detached retina | |
| IL-1α [235] | Cytokine | Cytoplasmic | Subretinal fluid | |
| IL-33 [236] | Cytokine | Cytoplasmic | Müller glia | |
| Syndecan-1 [237] | Proteoglycan | Plasma membrane | Vitreous/subretinal fluid | |
| Biglycan [238] | Proteoglycan | ECM | Retina | |
| Decorin [239,240] | Proteoglycan | ECM | Vitreous/epiretinal membrane | |
| Tenascin-C [240,241] | Glycoprotein | ECM | Vitreous/epiretinal membrane | |
| Fibrinogen [242] | Glycoprotein | ECM | Plasma |
| Disease | DAMPs | Type | Origin | Localization |
|---|---|---|---|---|
IRD  | S100A1, S100A16 [256] | Ca2+ binding protein | Cytoplasmic | Müller glia |
| LIF [67] | Peptide | Cytoplasmic | Müller glia | |
| Uric acid [257] | Metabolic product | Cytoplasmic | Serum | |
| HSP70 [258] | Molecular chaperones | Cytoplasmic | Photoreceptors | |
| Aβ [259] | Peptide | Cytoplasmic | GCL/sub-RPE deposits | |
| HMGB1 [260] | Nuclear binding protein | Nuclear | Vitreous | |
| HS [261,262] | Glycosaminoglycan | ECM | Photoreceptors | |
| Chondritin sulfate [261,262] | Glycosaminoglycan | ECM | Photoreceptors |
| Disorders | DAMPs | Plasma/ Serum | Tear | Vitreous | Aqueous | Refs |
|---|---|---|---|---|---|---|
| Endophthalmitis | HMGB1 | ✕ | ✕ | ✓ | ✕ | [20] |
| IL-1α | ✕ | ✕ | ✓ | ✕ | [23] | |
| SAA | ✓ | ✕ | ✕ | ✕ | [27] | |
| Uveitis | S100 | ✕ | ✓ | ✕ | ✕ | [30] |
| HSP | ✓ | ✕ | ✕ | ✕ | [32] | |
| SAA | ✕ | ✕ | ✕ | ✓ | [33] | |
| Glaucoma | Uric acid | ✓ | ✕ | ✕ | ✕ | [48] |
| ATP | ✕ | ✕ | ✓ | ✓ | [50] | |
| Aβ | ✕ | ✕ | ✕ | ✓ | [51] | |
| Histone | ✓ | ✕ | ✕ | ✕ | [52] | |
| HMGB1 | ✕ | ✕ | ✕ | ✓ | [53] | |
| IL-1α | ✕ | ✕ | ✕ | ✓ | [54] | |
| Decorin | ✕ | ✕ | ✕ | ✓ | [58] | |
| SAA | ✓ | ✕ | ✕ | ✕ | [65] | |
| Ocular cancer | Uric acid | ✕ | ✕ | ✕ | ✓ | [88] |
| DNA | ✓ | ✕ | ✕ | ✕ | [92] | |
| Ischemic Retinopathies | HMGB1 | ✕ | ✕ | ✓ | ✕ | [110] |
| IL-1α | ✓ | ✕ | ✕ | ✕ | [112] | |
| Diabetic Retinopathy | S100A8, S100A9 | ✓ | ✕ | ✕ | ✕ | [5] |
| HMGB1 | ✕ | ✕ | ✓ | ✕ | [148] | |
| Uric acid | ✓ | ✕ | ✓ | ✕ | [149] | |
| Cyclophilin A | ✓ | ✕ | ✕ | ✕ | [151] | |
| Cathelicidin | ✓ | ✕ | ✕ | ✕ | [154] | |
| Defensins | ✓ | ✕ | ✕ | ✕ | [155] | |
| Syndecan | ✓ | ✕ | ✕ | ✕ | [156] | |
| Decorin | ✓ | ✕ | ✕ | ✓ | [157,158] | |
| Versican | ✓ | ✕ | ✕ | ✕ | [159] | |
| LMW hyaluronan | ✕ | ✓ | ✓ | ✕ | [160] | |
| HS | ✕ | ✕ | ✓ | ✕ | [160] | |
| Fibronectin | ✓ | ✕ | ✓ | ✓ | [34] | |
| Fibrinogen | ✓ | ✕ | ✕ | ✕ | [164] | |
| Tenascin-C | ✕ | ✕ | ✓ | ✕ | [165] | |
| AMD | Uric acid | ✓ | ✕ | ✕ | ✕ | [197] |
| ATP | ✕ | ✕ | ✓ | ✕ | [193] | |
| ET-1 | ✓ | ✕ | ✕ | ✕ | [204] | |
| PVR/RRD | HMGB1 | ✕ | ✕ | ✓ | ✕ | [19] |
| ATP | ✕ | ✕ | ✓ | ✕ | [233] | |
| Histone | ✕ | ✕ | ✓ | ✕ | [229,234] | |
| Syndecan | ✕ | ✕ | ✓ | ✕ | [237] | |
| Decorin | ✕ | ✕ | ✓ | ✕ | [239] | |
| Tenascin-C | ✕ | ✕ | ✓ | ✕ | [241] | |
| Fibrinogen | ✓ | ✕ | ✕ | ✕ | [242] | |
| IRD | Uric acid | ✓ | ✕ | ✕ | ✕ | [257] |
| HMGB1 | ✕ | ✕ | ✓ | ✕ | [260] |
| Retinal Disorders | Drug | DAMPs | Action | Refs |
|---|---|---|---|---|
| Uveitis | HMGB1 Ab Glycyrrhizin | HMGB1 | Inhibition of IRBP-specific T cell proliferation | [31] |
| Glaucoma | Brilliant Blue G and N-methyl-d-aspartic acid | ATP | Antagonists of the P2X7R | [280] |
| Ocular cancer | Ansamycins | HSP90 | G1 Arrest | [281] |
| miR34A and miR-22 | HMGB1 | Act on autophagy, migration, and invasion of RB cells | [277,278] | |
| Ischemic retinopathies | Cyclosporin A | TFAM | Preserves TFAM | [113] |
| Coenzyme Q10 | TFAM | Preserves TFAM | [282] | |
| Brimonidine | TFAM | Preserves TFAM | [114] | |
| Diabetic retinopathy | Tasquinimod Glycyrrhizin | S100 HMGB1 | Inhibits angiogenesis via TSP-1, VEGF, ICAM-1 and ERK1/2 Acts on HMGB1 via SIRT1 and provides neurovascular protection | [276,283] |
| AMD | Geldanamycin | HSP90 | Inhibits VEGF and HIFα | [284] |
| PVR/RRD | Anti-histone Antibodies | Histone | Reduced retinal damage | [234] |
| Geranylgeranylacetone | HSP70 | Activation of Akt pathway | [232] |
| Retinal Disorders | DAMPs | Therapeutic Property | Signaling Pathway | Refs |
|---|---|---|---|---|
| Endophthalmitis | Cathelicidin | Anti-microbial | TLRs | [26] |
| Uveitis | IL-33 | Anti-inflammatory | Macrophage M2 polarization | [45] |
| Glaucoma | Decorin | Anti-fibrotic | Inhibits TGF-β | [286] |
| Diabetic retinopathy | LIF HS | Anti-angiogenic Anti-angiogenic | HIF-1α and VEGF Inhibiting VEGF-VEGFR2 binding | [116,288] |
| AMD | LIF | Anti-angiogenic | STAT3 pathway | [210] |
| HSP70 | Anti-inflammatory | TLR2/TLR4 | [213,214] | |
| IL-33 | Anti-angiogenic | Not known | [289] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahaling, B.; Low, S.W.Y.; Beck, M.; Kumar, D.; Ahmed, S.; Connor, T.B.; Ahmad, B.; Chaurasia, S.S. Damage-Associated Molecular Patterns (DAMPs) in Retinal Disorders. Int. J. Mol. Sci. 2022, 23, 2591. https://doi.org/10.3390/ijms23052591
Mahaling B, Low SWY, Beck M, Kumar D, Ahmed S, Connor TB, Ahmad B, Chaurasia SS. Damage-Associated Molecular Patterns (DAMPs) in Retinal Disorders. International Journal of Molecular Sciences. 2022; 23(5):2591. https://doi.org/10.3390/ijms23052591
Chicago/Turabian StyleMahaling, Binapani, Shermaine W. Y. Low, Molly Beck, Devesh Kumar, Simrah Ahmed, Thomas B. Connor, Baseer Ahmad, and Shyam S. Chaurasia. 2022. "Damage-Associated Molecular Patterns (DAMPs) in Retinal Disorders" International Journal of Molecular Sciences 23, no. 5: 2591. https://doi.org/10.3390/ijms23052591
APA StyleMahaling, B., Low, S. W. Y., Beck, M., Kumar, D., Ahmed, S., Connor, T. B., Ahmad, B., & Chaurasia, S. S. (2022). Damage-Associated Molecular Patterns (DAMPs) in Retinal Disorders. International Journal of Molecular Sciences, 23(5), 2591. https://doi.org/10.3390/ijms23052591