
The Antidepressant Duloxetine Inhibits Platelet Function and Protects against Thrombosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Duloxetine Inhibits Agonist-Induced In Vitro and Ex Vivo Platelet Aggregation in a Dose-Dependent Manner
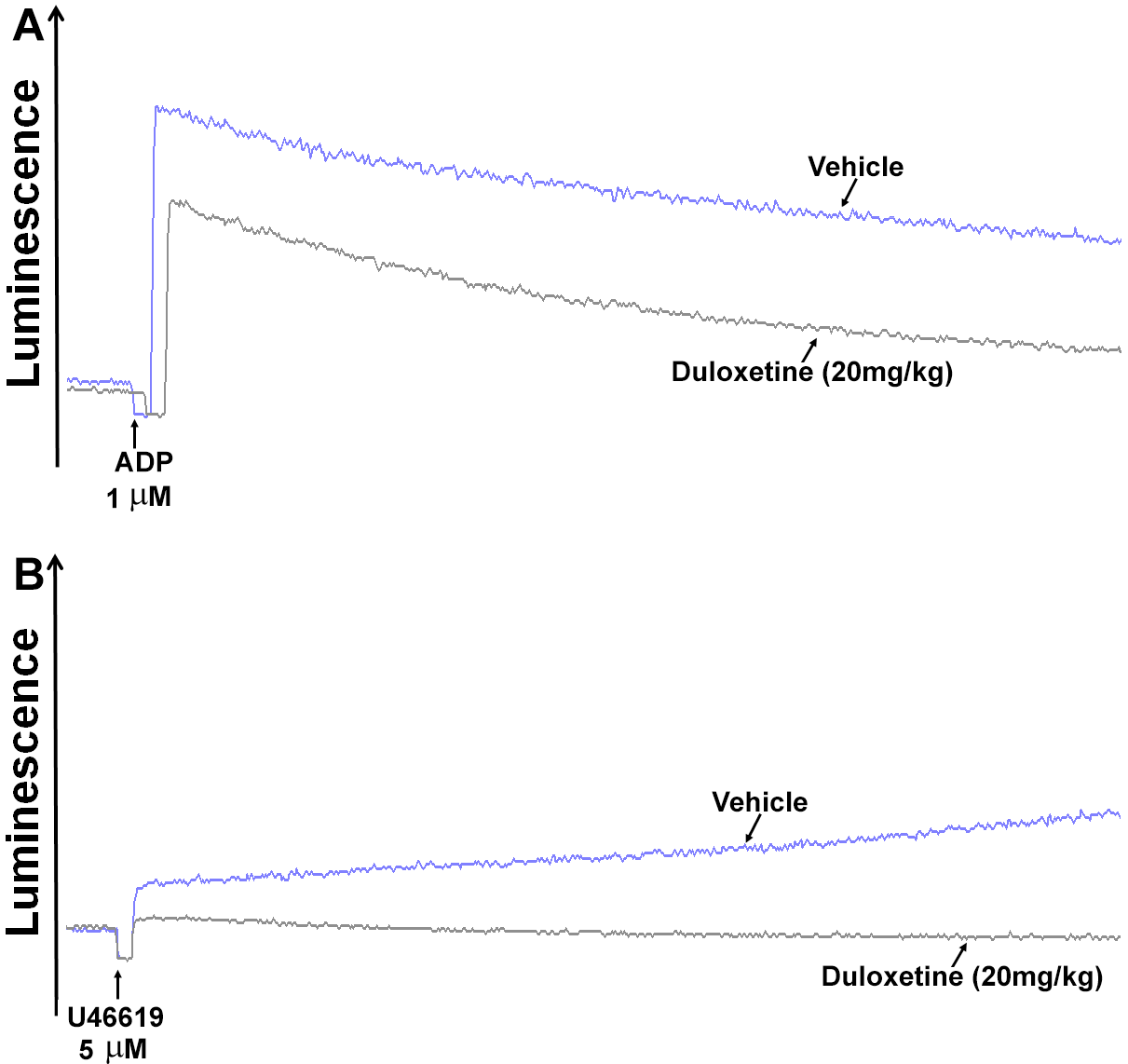
2.2. Duloxetine Inhibits P-Selectin Expression and ATP Release in Stimulated Platelets
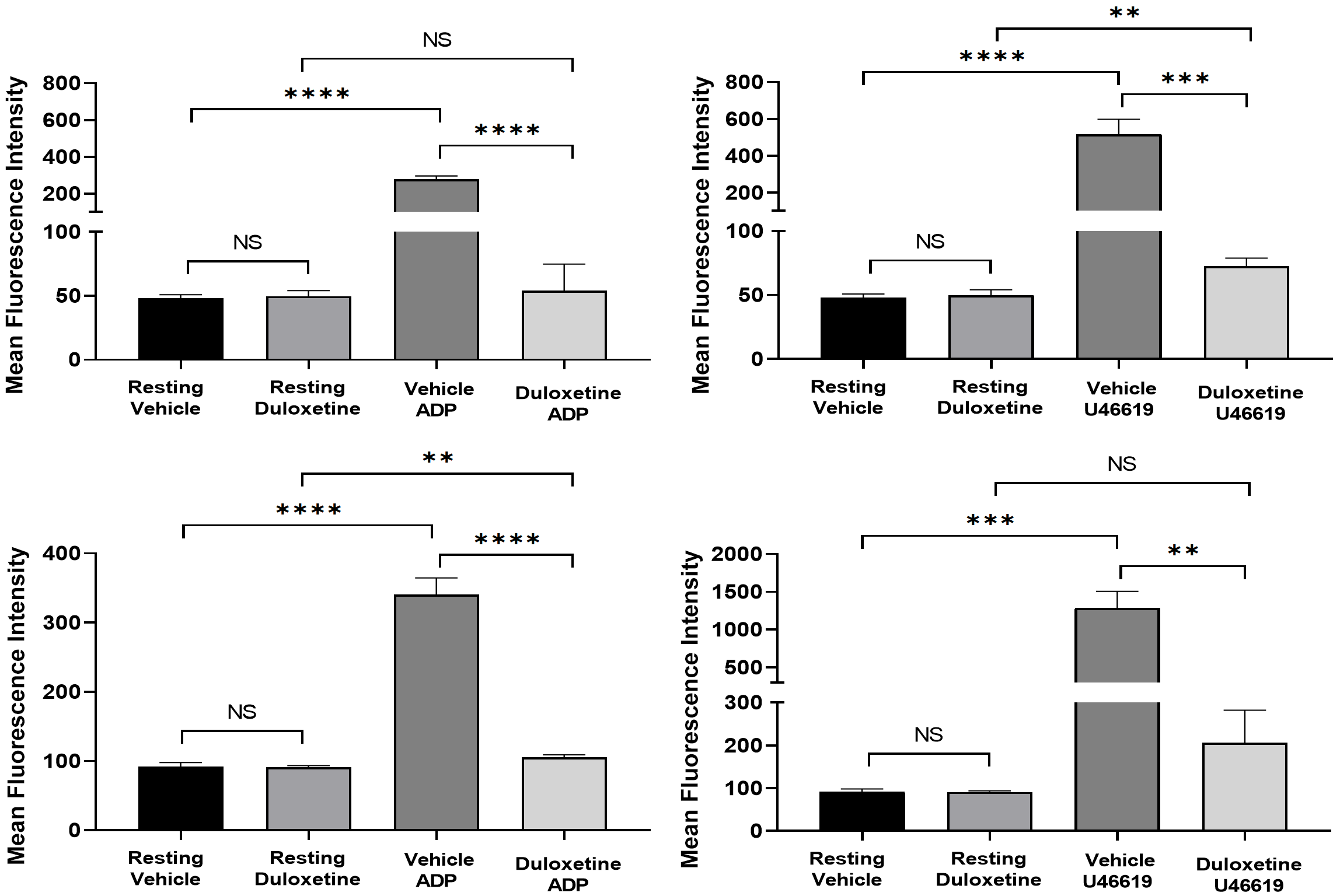
2.3. Duloxetine Inhibits Agonist-Induced Glycoprotein IIb-IIIa Activation and Phosphatidylserine Expression in Stimulated Platelets
2.4. Duloxetine Impairs Clot Retraction
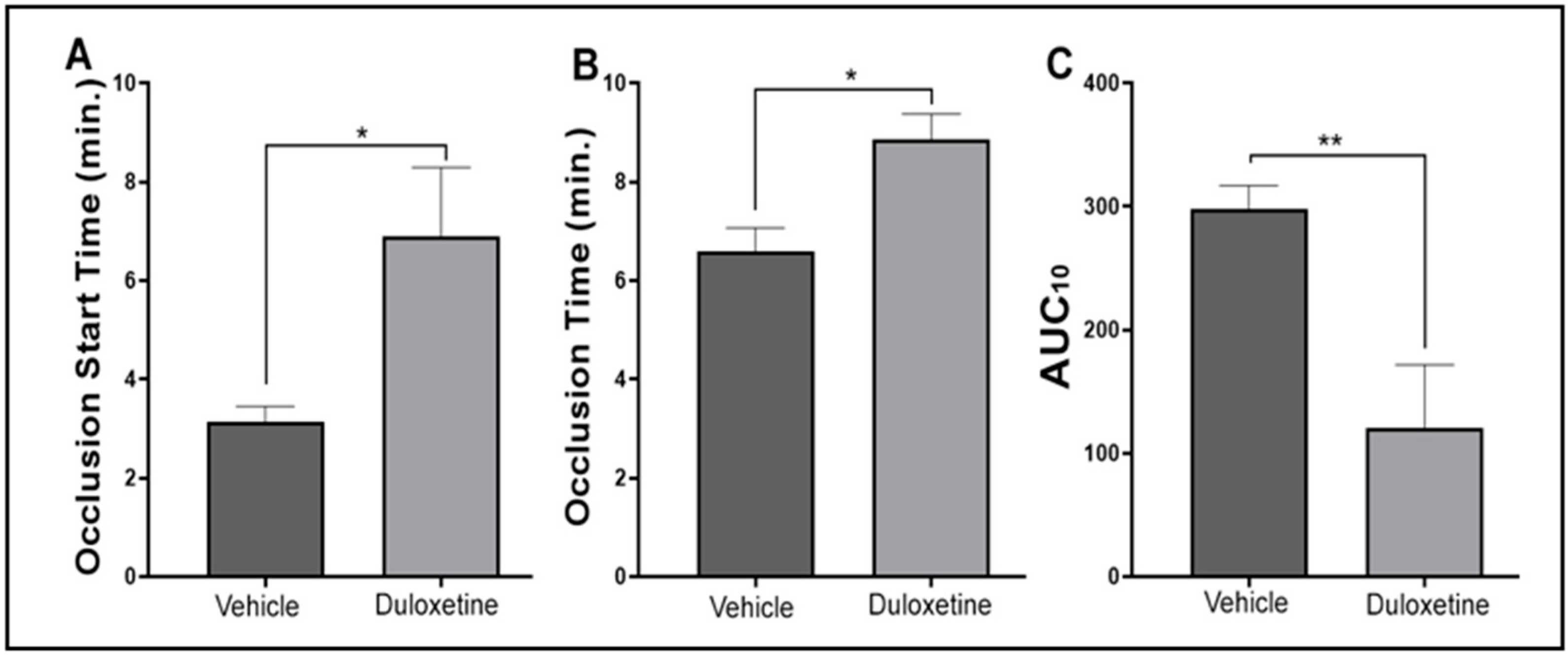
2.5. Duloxetine Prolongs Occlusion Time and Tail Bleeding Time
2.6. Duloxetine Modulates Hemostasis Response in Human Platelets
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Human Blood Samples
4.3. Animals
4.4. Human and Mouse Platelet-Rich Plasma Preparation
4.5. Washed Platelets Preparation
4.6. In Vitro Platelet Aggregation
4.7. Ex Vivo Platelet Aggregation and ATP Release
4.8. Flow Cytometric Analysis
4.9. Fibrin Clot Retraction Assay
4.10. Tail Bleeding Time
4.11. Human Hemostasis Assessment
4.12. In Vivo Ferric Chloride Carotid Artery Injury–Induced Thrombosis Model
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 20 December 2021).
- Major Depression. Available online: https://www.nimh.nih.gov/health/statistics/major-depression.shtml (accessed on 20 December 2021).
- Dahlstroem, A.; Fuxe, K. Evidence for the Existence of Monoamine-Containing Neurons in the Central Nervous System. I. Demonstration of Monoamines in the Cell Bodies of Brain Stem Neurons. Acta Physiol. Scand. 1964, 62 (Suppl. 232), 1–80. [Google Scholar]
- Vikenes, K.; Farstad, M.; Nordrehaug, J.E. Serotonin is associated with coronary artery disease and cardiac events. Circulation 1999, 100, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Mercado, C.P.; Kilic, F. Molecular mechanisms of SERT in platelets: Regulation of plasma serotonin levels. Mol. Interv. 2010, 10, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M. Platelet-derived serotonin, the endothelium, and cardiovascular disease. J. Cardiovasc. Pharmacol. 1991, 17 (Suppl. 5), S6–S12. [Google Scholar] [CrossRef] [PubMed]
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The pathogenesis of coronary artery disease and the acute coronary syndromes (2). N. Engl. J. Med. 1992, 326, 310–318. [Google Scholar] [PubMed]
- Walther, D.J.; Peter, J.U.; Winter, S.; Holtje, M.; Paulmann, N.; Grohmann, M.; Vowinckel, J.; Alamo-Bethencourt, V.; Wilhelm, C.S.; Ahnert-Hilger, G.; et al. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 2003, 115, 851–862. [Google Scholar] [CrossRef]
- Milne, W.L.; Cohn, S.H. Role of serotonin in blood coagulation. Am. J. Physiol. 1957, 189, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.L.; Woulfe, D.; Kilic, F. Responses of Plasma Catecholamine, Serotonin, and the Platelet Serotonin Transporter to Cigarette Smoking. Front. Neurosci. 2019, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A.D.; Vittinghoff, E.; Maselli, J.; Pekow, P.S.; Young, J.Q.; Lindenauer, P.K. Perioperative use of selective serotonin reuptake inhibitors and risks for adverse outcomes of surgery. JAMA Intern. Med. 2013, 173, 1075–1081. [Google Scholar] [CrossRef]
- Ferreiro, J.L.; Angiolillo, D.J. New directions in antiplatelet therapy. Circ. Cardiovasc. Interv. 2012, 5, 433–445. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G.; Lasagna, L. Cost of innovation in the pharmaceutical industry. J. Health Econ. 1991, 10, 107–142. [Google Scholar] [CrossRef]
- Onuțu, A.H.D.D.; Petrișor, C. Serotonin Reuptake Inhibitors and Their Role in Chronic Pain Management. In Serotonin; IntechOpen: London, UK, 2018. [Google Scholar]
- Tawfik, M.K.; Helmy, S.A.; Badran, D.I.; Zaitone, S.A. Neuroprotective effect of duloxetine in a mouse model of diabetic neuropathy: Role of glia suppressing mechanisms. Life Sci. 2018, 205, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Santana-Coelho, D.; Souza-Monteiro, J.R.; Paraense, R.S.O.; Busanello, G.L.; Arrifano, G.P.F.; Mendonca, J.R.; Silveira-Junior, M.E.P.; Royes, L.F.F.; Crespo-Lopez, M.E. Antidepressant drugs in convulsive seizures: Pre-clinical evaluation of duloxetine in mice. Neurochem. Int. 2016, 99, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Hakenjos, J.M.; MacKenzie, K.R.; Barzi, M.; Chavan, H.; Nyshadham, P.; Wang, J.; Jung, S.Y.; Guner, J.Z.; Chen, S.; et al. Metabolism of a Selective Serotonin and Norepinephrine Reuptake Inhibitor Duloxetine in Liver Microsomes and Mice. Drug Metab. Dispos. 2022, 50, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Yoshioka, T.; Yamada, D.; Hamano, T.; Ohashi, M.; Matsumoto, M.; Iio, K.; Ikeda, M.; Kamei, M.; Otsuki, T.; et al. Cold-restraint stress-induced ultrasonic vocalization as a novel tool to measure anxiety in mice. Biol. Pharm. Bull. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Salat, K.; Furgala-Wojas, A.; Salat, R. The Microglial Activation Inhibitor Minocycline, Used Alone and in Combination with Duloxetine, Attenuates Pain Caused by Oxaliplatin in Mice. Molecules 2021, 26, 3577. [Google Scholar] [CrossRef]
- Meejuru, G.F.; Somavarapu, A.; Danduga, R.; Nissankara Roa, L.S.; Kola, P.K. Protective effects of duloxetine against chronic immobilisation stress-induced anxiety, depression, cognitive impairment and neurodegeneration in mice. J. Pharm. Pharmacol. 2021, 73, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Storrie, B.; Whiteheart, S.W. Editorial: Platelet Secretion. Platelets 2017, 28, 107. [Google Scholar] [CrossRef][Green Version]
- Mumford, A.D.; Iii, A.L.F.; Gachet, C.; Gresele, P.; Noris, P.; Harrison, P.; Mezzano, D. A review of platelet secretion assays for the diagnosis of inherited platelet secretion disorders. Thromb. Haemost. 2015, 114, 14–25. [Google Scholar] [CrossRef]
- Bennett, J.S. Structure and function of the platelet integrin IIb 3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef] [PubMed]
- Schoenwaelder, S.M.; Yuan, Y.; Josefsson, E.C.; White, M.J.; Yao, Y.; Mason, K.D.; O’Reilly, L.A.; Henley, K.J.; Ono, A.; Hsiao, S.; et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 2009, 114, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Schnetzer, G.W. Platelets and thrombogenesis—Current concepts. Am. Hear. J. 1972, 83, 552–564. [Google Scholar] [CrossRef][Green Version]
- Nagareddy, P.; Smyth, S.S. Inflammation and thrombosis in cardiovascular disease. Curr. Opin. Hematol. 2013, 20, 457–463. [Google Scholar] [CrossRef]
- Hernandez, K.R.; Karim, Z.A.; Qasim, H.; Druey, K.M.; Alshbool, F.Z.; Khasawneh, F.T. Regulator of G-Protein Signaling 16 Is a Negative Modulator of Platelet Function and Thrombosis. J. Am. Hear. Assoc. 2019, 8, e011273. [Google Scholar] [CrossRef] [PubMed]
- Kaikita, K.; Hosokawa, K.; Dahlen, J.R.; Tsujita, K. Total Thrombus-Formation Analysis System (T-TAS): Clinical Application of Quantitative Analysis of Thrombus Formation in Cardiovascular Disease. Thromb. Haemost. 2019, 119, 1554–1562. [Google Scholar] [CrossRef]
- Hosokawa, K.; Ohnishi-Wada, T.; Nagasato, T.; Sameshima-Kaneko, H.; Oyamada, C.; Dahlen, J. New methodological approaches for assessing thrombus formation in cardiovascular disease. Kardiol. Pol. 2020, 78, 667–673. [Google Scholar] [CrossRef]
- Ernst, M.; Brähler, E.; Otten, D.; Werner, A.M.; Tibubos, A.N.; Reiner, I.; Wicke, F.; Wiltink, J.; Michal, M.; Nagler, M.; et al. Inflammation predicts new onset of depression in men, but not in women within a prospective, representative community cohort. Sci. Rep. 2021, 11, 2271. [Google Scholar] [CrossRef]
- Huffman, J.C.; Celano, C.M.; Beach, S.R.; Motiwala, S.R.; Januzzi, J.L. Depression and cardiac disease: Epidemiology, mechanisms, and diagnosis. Cardiovasc. Psychiatry Neurol. 2013, 2013, 695925. [Google Scholar] [CrossRef]
- Cohen, F.J. Macro trends in pharmaceutical innovation. Nat. Rev. Drug Discov. 2005, 4, 78–84. [Google Scholar] [CrossRef]
- Aronson, J.K. Old drugs—New uses. Br. J. Clin. Pharmacol. 2007, 64, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Sullivan, D.J., Jr. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef] [PubMed]
- McPhie, D.C. Old drugs, new uses: Solving a Hatch-Waxman patent predicament. Food Drug Law J. 2004, 59, 155–168. [Google Scholar] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.J.; Murray, W.J.; Khasawneh, F.T. Repurposing an old drug for a new use: Glybenclamide exerts antiplatelet activity by interacting with the thromboxane A(2) receptor. Acta Pharmacol. Sin. 2010, 31, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.J.; Khasawneh, F.T. Glybenclamide: An antidiabetic with in vivo antithrombotic activity. Eur. J. Pharmacol. 2010, 649, 249–254. [Google Scholar] [CrossRef]
- Murad, J.P.; Espinosa, E.V.; Ting, H.J.; Khasawneh, F.T. Characterization of the in vivo antiplatelet activity of the antihypertensive agent losartan. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 308–314. [Google Scholar] [CrossRef]
- Carneiro, A.M.; Cook, E.H.; Murphy, D.L.; Blakely, R.D. Interactions between integrin alphaIIbbeta3 and the serotonin transporter regulate serotonin transport and platelet aggregation in mice and humans. J. Clin. Investig. 2008, 118, 1544–1552. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Ghirardello, S.; Lecchi, A.; Artoni, A.; Panigada, M.; Aliberti, S.; Scalambrino, E.; La Marca, S.; Boscarino, M.; Gramegna, A.; Properzi, P.; et al. Assessment of Platelet Thrombus Formation under Flow Conditions in Adult Patients with COVID-19: An Observational Study. Thromb. Haemost. 2021, 121, 1087–1096. [Google Scholar]
- Karim, Z.A.; Zhang, J.; Banerjee, M.; Chicka, M.C.; Al Hawas, R.; Hamilton, T.R.; Roche, P.A.; Whiteheart, S.W. IkappaB kinase phosphorylation of SNAP-23 controls platelet secretion. Blood 2013, 121, 4567–4574. [Google Scholar] [CrossRef] [PubMed]
- Alshbool, F.Z.; Karim, Z.A.; Vemana, H.P.; Conlon, C.; Lin, O.A.; Khasawneh, F.T. The regulator of G-protein signaling 18 regulates platelet aggregation, hemostasis and thrombosis. Biochem. Biophys. Res. Commun. 2015, 462, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Hensch, N.R.; Karim, Z.A.; Druey, K.M.; Tansey, M.G.; Khasawneh, F.T. RGS10 Negatively Regulates Platelet Activation and Thrombogenesis. PLoS ONE 2016, 11, e0165984. [Google Scholar] [CrossRef]
- Karim, Z.A.; Alshbool, F.Z.; Vemana, H.P.; Conlon, C.; Druey, K.M.; Khasawneh, F.T. CXCL12 regulates platelet activation via the regulator of G-protein signaling 16. Biochim. Biophys. Acta 2016, 1863, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Park, J.H.; Ahn, J.H.; Hong, S.; Cho, J.H.; Won, M.H.; Lee, C.H. The anti-inflammatory activity of duloxetine, a serotonin/norepinephrine reuptake inhibitor, prevents kainic acid-induced hippocampal neuronal death in mice. J. Neurol. Sci. 2015, 358, 390–397. [Google Scholar] [CrossRef]
- Lin, O.A.; Karim, Z.A.; Vemana, H.P.; Espinosa, E.V.; Khasawneh, F.T. The antidepressant 5-HT2A receptor antagonists pizotifen and cyproheptadine inhibit serotonin-enhanced platelet function. PLoS ONE 2014, 9, e87026. [Google Scholar] [CrossRef] [PubMed]
- Vemana, H.P.; Karim, Z.A.; Conlon, C.; Khasawneh, F.T. A critical role for the transient receptor potential channel type 6 in human platelet activation. PLoS ONE 2015, 10, e0125764. [Google Scholar] [CrossRef] [PubMed]
- Murad, J.P.; Espinosa, E.V.; Ting, H.J.; McClure, D.; Khasawneh, F.T. A novel antibody targeting the ligand binding domain of the thromboxane A(2) receptor exhibits antithrombotic properties in vivo. Biochem. Biophys. Res. Commun. 2012, 421, 456–461. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozano, P.A.; Alarabi, A.B.; Garcia, S.E.; Boakye, E.T.; Kingbong, H.T.; Naddour, E.; Villalobos-García, D.; Badejo, P.; El-Halawany, M.S.; Khasawneh, F.T.; et al. The Antidepressant Duloxetine Inhibits Platelet Function and Protects against Thrombosis. Int. J. Mol. Sci. 2022, 23, 2587. https://doi.org/10.3390/ijms23052587
Lozano PA, Alarabi AB, Garcia SE, Boakye ET, Kingbong HT, Naddour E, Villalobos-García D, Badejo P, El-Halawany MS, Khasawneh FT, et al. The Antidepressant Duloxetine Inhibits Platelet Function and Protects against Thrombosis. International Journal of Molecular Sciences. 2022; 23(5):2587. https://doi.org/10.3390/ijms23052587
Chicago/Turabian StyleLozano, Patricia A., Ahmed B. Alarabi, Sarah E. Garcia, Erica T. Boakye, Hendreta T. Kingbong, Elie Naddour, Daniel Villalobos-García, Precious Badejo, Medhat S. El-Halawany, Fadi T. Khasawneh, and et al. 2022. "The Antidepressant Duloxetine Inhibits Platelet Function and Protects against Thrombosis" International Journal of Molecular Sciences 23, no. 5: 2587. https://doi.org/10.3390/ijms23052587
APA StyleLozano, P. A., Alarabi, A. B., Garcia, S. E., Boakye, E. T., Kingbong, H. T., Naddour, E., Villalobos-García, D., Badejo, P., El-Halawany, M. S., Khasawneh, F. T., & Alshbool, F. Z. (2022). The Antidepressant Duloxetine Inhibits Platelet Function and Protects against Thrombosis. International Journal of Molecular Sciences, 23(5), 2587. https://doi.org/10.3390/ijms23052587