
Fibroblast Growth Factor 23 and Osteoporosis: Evidence from Bench to Bedside

 ,
, 
Abstract
1. Introduction
2. Regulation of FGF23 Expression
3. FGF23 Effects on Mineral Homeostasis
4. Local Effects of FGF23 on Osteoblast and Bone Formation
5. Local Effects of FGF23 on Osteoclast and Bone Resorption
6. Role of FGF23 in Postmenopausal and Age-Related Osteoporosis Pathogenesis
7. The Association between FGF23 and Bone Fragility in the Elderly
8. Role of FGF23 in CKD-MBD Pathogenesis
9. The Association of FGF23 and CKD-MBD
10. Potential Clinical Application of FGF23 in Osteoporosis and CKD-MBD
11. FGF23 Measurement in Routine Clinical Practices
12. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, J.; Fu, S.; Song, Y. Relationship of Fibroblast Growth Factor 23 (FGF-23) Serum Levels With Low Bone Mass in Postmenopausal Women. J. Cell. Biochem. 2017, 118, 4454–4459. [Google Scholar] [CrossRef] [PubMed]
- Marsell, R.; Mirza, M.A.; Mallmin, H.; Karlsson, M.; Mellström, D.; Orwoll, E.; Ohlsson, C.; Jonsson, K.B.; Ljunggren, O.; Larsson, T.E. Relation between fibroblast growth factor-23, body weight and bone mineral density in elderly men. Osteoporos. Int. 2009, 20, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Rupp, T.; Butscheidt, S.; Vettorazzi, E.; Oheim, R.; Barvencik, F.; Amling, M.; Rolvien, T. High FGF23 levels are associated with impaired trabecular bone microarchitecture in patients with osteoporosis. Osteoporos. Int. 2019, 30, 1655–1662. [Google Scholar] [CrossRef]
- Bilha, S.C.; Bilha, A.; Ungureanu, M.C.; Matei, A.; Florescu, A.; Preda, C.; Covic, A.; Branisteanu, D. FGF23 Beyond the Kidney: A New Bone Mass Regulator in the General Population. Horm. Metab. Res. 2020, 52, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Kawakami, K.; Furushima, K.; Miyajima, M.; Sakaguchi, K. Central role of the proximal tubular αKlotho/FGF receptor complex in FGF23-regulated phosphate and vitamin D metabolism. Sci. Rep. 2018, 8, 6917. [Google Scholar] [CrossRef]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Manabe, N.; Miyaura, C.; Chikuda, H.; Nakamura, K.; Kuro-o, M. Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J. Clin. Investig. 1999, 104, 229–237. [Google Scholar] [CrossRef]
- Masuyama, R.; Stockmans, I.; Torrekens, S.; Van Looveren, R.; Maes, C.; Carmeliet, P.; Bouillon, R.; Carmeliet, G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Investig. 2006, 116, 3150–3159. [Google Scholar] [CrossRef]
- Yoshiko, Y.; Wang, H.; Minamizaki, T.; Ijuin, C.; Yamamoto, R.; Suemune, S.; Kozai, K.; Tanne, K.; Aubin, J.E.; Maeda, N. Mineralized tissue cells are a principal source of FGF23. Bone 2007, 40, 1565–1573. [Google Scholar] [CrossRef]
- Mirams, M.; Robinson, B.G.; Mason, R.S.; Nelson, A.E. Bone as a source of FGF23: Regulation by phosphate? Bone 2004, 35, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Olauson, H.; Larsson, T.E.; Lindgren, U. FGF23 affects the lineage fate determination of mesenchymal stem cells. Calcif. Tissue Int. 2013, 93, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Bon, N.; Frangi, G.; Sourice, S.; Guicheux, J.; Beck-Cormier, S.; Beck, L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol. Metab. 2018, 11, 197–204. [Google Scholar] [CrossRef]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef] [PubMed]
- Krajisnik, T.; Björklund, P.; Marsell, R.; Ljunggren, O.; Akerström, G.; Jonsson, K.B.; Westin, G.; Larsson, T.E. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J. Endocrinol. 2007, 195, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Olauson, H.; Lindberg, K.; Amin, R.; Sato, T.; Jia, T.; Goetz, R.; Mohammadi, M.; Andersson, G.; Lanske, B.; Larsson, T.E. Parathyroid-Specific Deletion of Klotho Unravels a Novel Calcineurin-Dependent FGF23 Signaling Pathway That Regulates PTH Secretion. PLoS Genet. 2013, 9, e1003975. [Google Scholar] [CrossRef] [PubMed]
- Kantham, L.; Quinn, S.J.; Egbuna, O.I.; Baxi, K.; Butters, R.; Pang, J.L.; Pollak, M.R.; Goltzman, D.; Brown, E.M. The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E915–E923. [Google Scholar] [CrossRef]
- Ferrè, S.; Hoenderop, J.G.J.; Bindels, R.J.M. Sensing mechanisms involved in Ca2+ and Mg2+ homeostasis. Kidney Int. 2012, 82, 1157–1166. [Google Scholar] [CrossRef]
- Murali, S.K.; Roschger, P.; Zeitz, U.; Klaushofer, K.; Andrukhova, O.; Erben, R.G. FGF23 Regulates Bone Mineralization in a 1,25(OH)2D3 and Klotho-Independent Manner. J. Bone Mineral. Res. 2016, 31, 129–142. [Google Scholar] [CrossRef]
- Mackenzie, N.C.W.; Zhu, D.; Milne, E.M.; van’t Hof, R.; Martin, A.; Quarles, D.L.; Millán, J.L.; Farquharson, C.; MacRae, V.E. Altered Bone Development and an Increase in FGF-23 Expression in Enpp1−/− Mice. PLoS ONE 2012, 7, e32177. [Google Scholar] [CrossRef]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G.; Bisaz, S.; Donath, A.; Morgan, D.B.; Fleisch, H. Inorganic pyrophosphate in plasma in normal persons and in patients with hypophosphatasia, osteogenesis imperfecta, and other disorders of bone. J. Clin. Investig. 1971, 50, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Lieben, L.; Masuyama, R.; Torrekens, S.; Van Looveren, R.; Schrooten, J.; Baatsen, P.; Lafage-Proust, M.H.; Dresselaers, T.; Feng, J.Q.; Bonewald, L.F.; et al. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J. Clin. Investig. 2012, 122, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Jiang, Y.; Zhao, X.; Sato, T.; Densmore, M.; Schüler, C.; Erben, R.G.; McKee, M.D.; Lanske, B. Increased osteopontin contributes to inhibition of bone mineralization in FGF23-deficient mice. J. Bone Miner. Res. Off. J. Am. Soc. Bone Mineral. Res. 2014, 29, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Harmey, D.; Millán, J.L.; McKee, M.D.; Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005, 19, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Azari, F.; Sørensen, E.S.; Kaartinen, M.T.; McKee, M.D. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J. Biol. Chem. 2007, 282, 15872–15883. [Google Scholar] [CrossRef]
- Allard, L.; Demoncheaux, N.; Machuca-Gayet, I.; Georgess, D.; Coury-Lucas, F.; Jurdic, P.; Bacchetta, J. Biphasic Effects of Vitamin D and FGF23 on Human Osteoclast Biology. Calcif. Tissue Int. 2015, 97, 69–79. [Google Scholar] [CrossRef]
- Lane, N.E.; Parimi, N.; Corr, M.; Yao, W.; Cauley, J.A.; Nielson, C.M.; Ix, J.H.; Kado, D.; Orwoll, E. Association of serum fibroblast growth factor 23 (FGF23) and incident fractures in older men: The Osteoporotic Fractures in Men (MrOS) study. J. Bone Miner. Res. 2013, 28, 2325–2332. [Google Scholar] [CrossRef]
- Mirza, M.A.; Karlsson, M.K.; Mellström, D.; Orwoll, E.; Ohlsson, C.; Ljunggren, O.; Larsson, T.E. Serum fibroblast growth factor-23 (FGF-23) and fracture risk in elderly men. J. Bone Miner. Res. 2011, 26, 857–864. [Google Scholar] [CrossRef]
- Lewerin, C.; Ljunggren, Ö.; Nilsson-Ehle, H.; Karlsson, M.K.; Herlitz, H.; Lorentzon, M.; Ohlsson, C.; Mellström, D. Low serum iron is associated with high serum intact FGF23 in elderly men: The Swedish MrOS study. Bone 2017, 98, 1–8. [Google Scholar] [CrossRef]
- Yamamoto, S.; Koyama, D.; Igarashi, R.; Maki, T.; Mizuno, H.; Furukawa, Y.; Kuro, O.M. Serum Endocrine Fibroblast Growth Factors as Potential Biomarkers for Chronic Kidney Disease and Various Metabolic Dysfunctions in Aged Patients. Intern. Med. 2020, 59, 345–355. [Google Scholar] [CrossRef]
- Moe, S.M.; Drüeke, T.; Lameire, N.; Eknoyan, G. Chronic kidney disease-mineral-bone disorder: A new paradigm. Adv. Chronic Kidney Dis. 2007, 14, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M. Fibroblast Growth Factor 23 and Disordered Vitamin D Metabolism in Chronic Kidney Disease: Updating the “Trade-off” Hypothesis. Clin. J. Am. Soc. Nephrol. 2010, 5, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Kanda, E.; Yoshida, M.; Sasaki, S. Applicability of fibroblast growth factor 23 for evaluation of risk of vertebral fracture and chronic kidney disease-mineral bone disease in elderly chronic kidney disease patients. BMC Nephrol. 2012, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Desbiens, L.C.; Sidibé, A.; Ung, R.V.; Fortier, C.; Munger, M.; Wang, Y.P.; Bisson, S.K.; Marquis, K.; Agharazii, M.; Mac-Way, F. FGF23-klotho axis, bone fractures, and arterial stiffness in dialysis: A case-control study. Osteoporos. Int. 2018, 29, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Tanaka, H.; Tominaga, Y.; Fukagawa, M.; Kurokawa, K.; Seino, Y. Decreased 1,25-dihydroxyvitamin D3 receptor density is associated with a more severe form of parathyroid hyperplasia in chronic uremic patients. J. Clin. Investig. 1993, 92, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Sugimoto, T.; Tsukamoto, T.; Chihara, K.; Kobayashi, A.; Kitazawa, S.; Maeda, S.; Kitazawa, R. Association of decreased calcium-sensing receptor expression with proliferation of parathyroid cells in secondary hyperparathyroidism. Kidney Int. 2000, 58, 1980–1986. [Google Scholar] [CrossRef]
- Tokumoto, M.; Tsuruya, K.; Fukuda, K.; Kanai, H.; Kuroki, S.; Hirakata, H. Reduced p21, p27 and vitamin D receptor in the nodular hyperplasia in patients with advanced secondary hyperparathyroidism. Kidney Int. 2002, 62, 1196–1207. [Google Scholar] [CrossRef][Green Version]
- Andrukhova, O.; Schüler, C.; Bergow, C.; Petric, A.; Erben, R.G. Augmented Fibroblast Growth Factor-23 Secretion in Bone Locally Contributes to Impaired Bone Mineralization in Chronic Kidney Disease in Mice. Front. Endocrinol. 2018, 9, 311. [Google Scholar] [CrossRef]
- Sun, N.; Guo, Y.; Liu, W.; Densmore, M.; Shalhoub, V.; Erben, R.G.; Ye, L.; Lanske, B.; Yuan, Q. FGF23 neutralization improves bone quality and osseointegration of titanium implants in chronic kidney disease mice. Sci. Rep. 2015, 5, 8304. [Google Scholar] [CrossRef]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Coskun, Y.; Paydas, S.; Balal, M.; Soyupak, S.; Kara, E. Bone Disease and Serum Fibroblast Growth Factor-23 Levels in Renal Transplant Recipients. Transplant. Proc. 2016, 48, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Yavropoulou, M.P.; Vaios, V.; Pikilidou, M.; Chryssogonidis, I.; Sachinidou, M.; Tournis, S.; Makris, K.; Kotsa, K.; Daniilidis, M.; Haritanti, A.; et al. Bone Quality Assessment as Measured by Trabecular Bone Score in Patients with End-Stage Renal Disease on Dialysis. J. Clin. Densitom. 2017, 20, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Xiao, D.M.; Fan, W.F.; Ye, X.W.; Niu, J.Y.; Gu, Y. Effect of serum fibroblast growth factor-23, matrix Gla protein and Fetuin-A in predicting osteoporosis in maintenance hemodialysis patients. Ther. Apher Dial 2014, 18, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Slouma, M.; Sahli, H.; Bahlous, A.; Laadhar, L.; Smaoui, W.; Rekik, S.; Gharsallah, I.; Sallami, M.; Moussa, F.B.; Elleuch, M.; et al. Mineral bone disorder and osteoporosis in hemodialysis patients. Adv. Rheumatol. 2020, 60, 15. [Google Scholar] [CrossRef]
- Desjardins, L.; Liabeuf, S.; Renard, C.; Lenglet, A.; Lemke, H.D.; Choukroun, G.; Drueke, T.B.; Massy, Z.A. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos. Int. 2012, 23, 2017–2025. [Google Scholar] [CrossRef]
- Smith, E.R.; Cai, M.M.; McMahon, L.P.; Holt, S.G. Biological Variability of Plasma Intact and C-Terminal FGF23 Measurements. J. Clin. Endocrinol. Metab. 2012, 97, 3357–3365. [Google Scholar] [CrossRef]
- Dirks, N.F.; Smith, E.R.; van Schoor, N.M.; Vervloet, M.G.; Ackermans, M.T.; de Jonge, R.; Heijboer, A.C. Pre-analytical stability of FGF23 with the contemporary immunoassays. Clin. Chim. Acta 2019, 493, 104–106. [Google Scholar] [CrossRef]
- Vervloet, M.G.; van Ittersum, F.J.; Buttler, R.M.; Heijboer, A.C.; Blankenstein, M.A.; ter Wee, P.M. Effects of Dietary Phosphate and Calcium Intake on Fibroblast Growth Factor-23. Clin. J. Am. Soc. Nephrol. 2011, 6, 383–389. [Google Scholar] [CrossRef]
- Burnett, S.M.; Gunawardene, S.C.; Bringhurst, F.R.; Jüppner, H.; Lee, H.; Finkelstein, J.S. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J. Bone Min. Res. 2006, 21, 1187–1196. [Google Scholar] [CrossRef]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
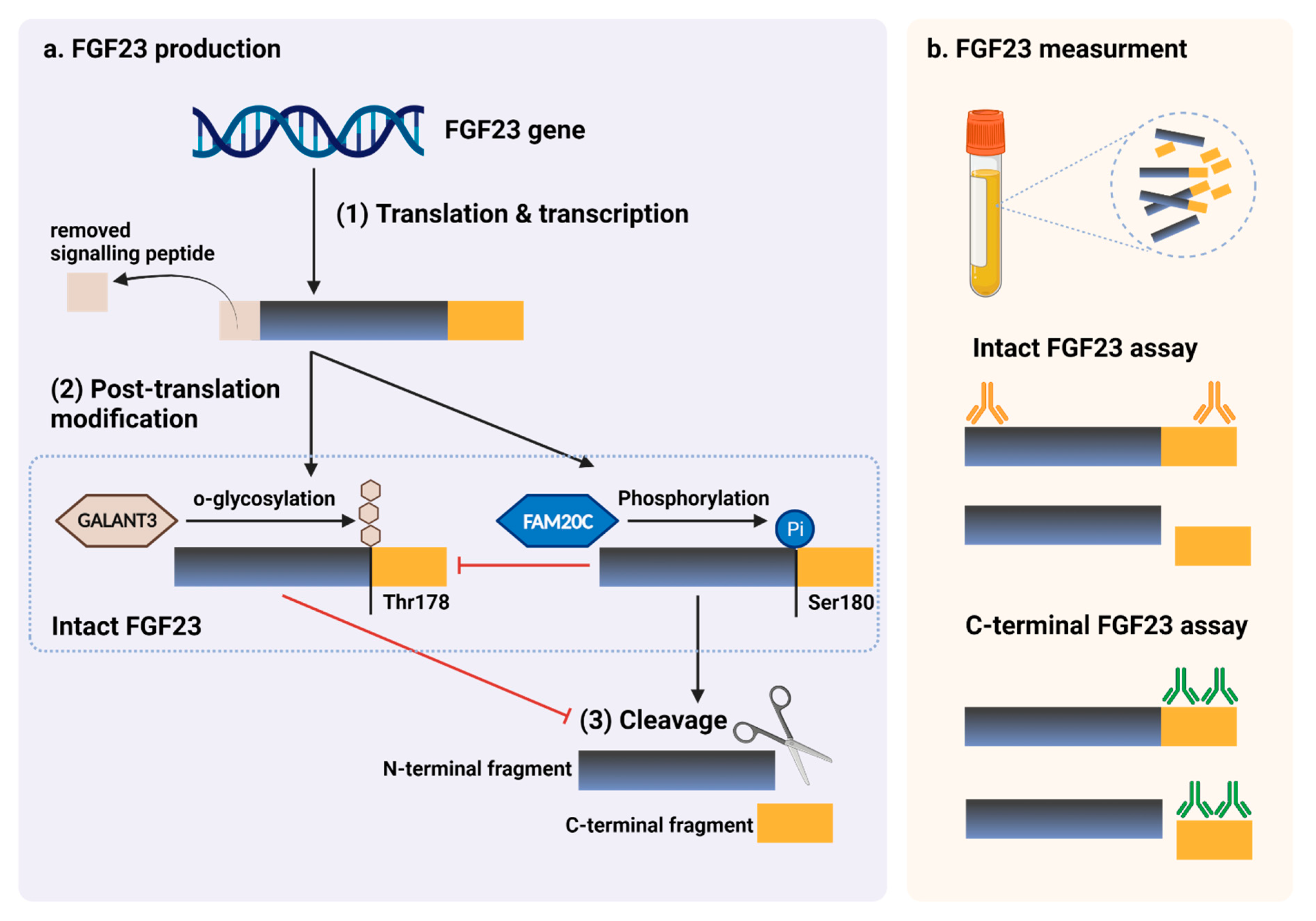{kind=link}
| Study Design | Key Findings | Interpretation | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient Group | Age (Year) | Study Base | Serum FGF23 (pg./mL) | OB Activity/Proliferation | OC Activity/Proliferation | BMD/ Bone Architecture | Fragility Fracture | ||
| Men (n = 2782) | 75.4 ± 3.1 | cross- sectional (Sweden) | 49 ± 40.8 E | N/A | N/A | ↔ BMD a (age, smoking, height, weight) | N/A | FGF23 weakly associated with BMD in elderly men, but there was not clinical and statistical significance after adjustment with potential confounders. | [2] |
| ↑ BMD a (Pi, Ca, PTH, eGFR, 25(OH)2D3) | |||||||||
| Men (n = 3014) | 75.4 ± 3.2 | prospective cohort | ref. > 57.4 E (median) | N/A | N/A | N/A | ↑ a (BMI, FN BMD, eFGR, Pi, PTH, 25(OH)2D3, other fracture risks) | High circulating FGF23 was an independent risk factor of overall fragility fracture in elderly men. | [29] |
| (Sweden) | |||||||||
| Men (n = 1680) eGFR > 60 | 73.7 ± 5.8 | prospective case-cohort (USA) | 22.4–111.1 I (quartile 4) | N/A | N/A | ↔ BMD | ↔ a (FN BMD, Pi, PTH, 25(OH)2D3, other fracture risks) | There was no significant association between BMD or osteoporosis fracture with FGF23 in elderly men who had eFGR > 60. | [28] |
| Men (n = 997) | 75.3 ± 3.2 | cross- sectional | 42.2 (20.6) I | N/A | N/A | ↑ LS BMD a (age, BMI, cysC, Pi, PTH, 25(OH)2D3, Apo-B/A1 ratio, Iron study) | N/A | Increasing serum FGF23 level in elderly men was weakly associated with lumbar BMD. | [30] |
| (Sweden) | |||||||||
| pre- menopause (n = 60) | 43.8 ± 5.3 | cross- sectional (China) | 44.5 ± 9.2 E | ref. | ref. | ref. | N/A | In early post-menopause, estrogen deprivation caused BMD reduction from excessive bone resorption and decreased bone formation. Increased FGF23 might be a compensational response to this process. In post-menopausal women with low bone mass, increasing FGF23 showed a strong negative correlation with BMD. | [1] |
| early menopause (n = 60) | 48.6 ± 4.7 | 76.7 ± 11.6 E | ↓ OC ↑↑P1NP | ↑ CTX-1 | ↓ PF BMD ↓ LS BMD | N/A | |||
| late menopause (n = 60) | 53.4 ± 3.2 | 29.2 ± 8.6 E | ↓ OC ↔P1NP | ↑ CTX-1 | ↓↓ PF BMD ↓↓ LS BMD | N/A | |||
| post menopause with low bone mass (subgroups) | |||||||||
| PF t-score −1 to −2 | 73.5 ± 9.6 E | N/A | N/A | ↓↓ PF BMD b | N/A | ||||
| PF t-score <−2 | 82.5 ± 8.4 E | ||||||||
| LS t-score −1 to −2 | 75.5 ± 9.7 E | N/A | N/A | ↓↓ LS BMD b | N/A | ||||
| LS t-score <−2 | 82.9 ± 9.1 E | ||||||||
| osteoporosis patient (n = 82) | 64.0 ± 12.7 | cross- sectional (Germany) | 98 ± 133 C | ↔ BALP | N/A | ↓ BV/TV a (age, BMI, Pi, PTH, 25(OH)2D3, BAP) | N/A | High FGF23 was associated with reduced trabecular bone micro-architecture in osteoporosis. | [3] |
| ↓↓ Tb.N a (age, BMI, Pi, PTH, 25(OH)2D3, BAP) | |||||||||
| ↓ Tb.Th a (age, BMI, Pi, PTH, 25(OH)2D3, BAP) | |||||||||
| All genders (n = 73) | 76.2 ± 8.0 | cross- sectional (Japan) | 37 (12.7) E | ↓ BALP b | ↓ P1NP b | ↔BMD b | N/A | FGF23 did not show a clinically significant association with BMD and bone remodeling when adjusted for confounders. | [31] |
| ↔ TRAP5b a (eGFR, 25(OH)2D3) | |||||||||
| post menopause (n = 55) | 61 ± 1.1 | cross- sectional (Romania) | 81.2 ± 3.6 C | N/A | ↔ CTX-1 b | ↓ FN BMD a (PTH, 25(OH)2D3, Leptin) | N/A | Serum FGF23 level was independently associated with decreasing BMD in the femoral neck in post- menopausal women. | [4] |
| Study Design | Key Findings | Interpretation | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient Group | Age (Year) | Study-Based | Serum FGF23 (pg./mL) | OB Activity/Proliferation | OC Activity/Proliferation | BMD | Fragility Fracture | ||
| post menopause eGFR 45.7 ± 24.1 (n = 105) | 73.2 (7.7) | cross-sectional (Japan) | 49 (37) E ref. > 56.8 E | N/A | ↔NTX a (age, BMI, eFGR, Ca, Pi, PTH, 1,25(OH)2D) | N/A | ↑ vertebral a (age, eGFR) | The higher level of FGF23 was associated with vertebral fracture in the elderly with CKD. There was no significant difference between FGF23 and osteogenic biomarkers. | [34] |
| men | 73.7 | prospective | 22.4–111.1 I | N/A | N/A | N/A | ↑↑ non-vertebral a (FN BMD, Pi, PTH, 25(OH)2D3, other fracture risks) | FGF23 elevation had increased the risk of non- vertebral fractures in the subgroup of elderly men with CKD (eFGR < 60). | [28] |
| eGFR < 60 | ± 5.8 | case-cohort | (quartile 4) | ||||||
| subgroup, (n = 313) | (USA) | ||||||||
| CKD 2–5 (n = 142) | 67 ± 12 | prospective cohort | 52.55 ± 55.19 E | N/A | N/A | ↔ b | N/A | Serum FGF23 level was not associated with BMD in CKD patients. | [46] |
| (France) | |||||||||
| ESRD with MHD | 60.6 ± 11.3 | cross- sectional | N/A | N/A | N/A | ↔ b | N/A | No association between FGF23 and BMD in ESRD patients was found. FGF23 in ESRD patients with osteoporosis was significantly higher than patients without osteoporosis or osteopenia. | [44] |
| (n = 64) | (China) | ||||||||
| Subgroup | |||||||||
| normal (n = 10) | 55.4 ± 5.0 | 218.7 ± 28.6 E | ref. | N/A | t-score > −1 | N/A | |||
| osteopenia (n = 24) | 64.4 ± 3.9 | 235.6 ± 54.4 E | ↑BALP | N/A | t-score −1 to −2.5 | N/A | |||
| osteoporosis (n = 30) | 67.4 ± 3.8 | 296.2 ± 48.6 E | ↑↑BALP | N/A | t-score < −2.5 | N/A | |||
| KT (n = 106) eFGR | 40.1 ± 11.1 | cross- sectional | 25.29 ± 30.81 E | N/A | N/A | ↔ b | N/A | No relationship was found between FGF23 and BMD in KT and CKD patients. | [42] |
| 72.6 ± 27.1 | (Turkey) | ||||||||
| CKD (n = 30) eGFR 65.2 ± 54.6 | 39.2 ± 11.3 | 28.86 ± 26.5 E | N/A | N/A | ↔ b | N/A | |||
| post menopause (n = 102) | prospective case-control (Greece) | N/A | N/A | N/A | ↔ b | N/A | Elevated FGF23 was not associated with BMD in ESRD with HD. However, BMD was significantly decreased when compared with health matched controls. | [43] | |
| Subgroups | |||||||||
| ESRD with MHD (n = 50) | 62 ± 9.6 | 1027.8 ± 556.7 C | N/A | N/A | ↓ | N/A | |||
| healthy control (n = 52) | 59 ± 9.5 | 100.3 ± 54.7 C | N/A | N/A | ref. | N/A | |||
| ESRD with MHD (n = 130) | 72 (14) | Prospective nested case-control (Canada) | 787 (1430) C | ↔ OC b ↔P1NP b | ↔ TRAP5b b ↔ Sclerostin b | N/A | ↑ all fracture a (PTH, ferritin, smoking, P1NP) | C-terminal FGF23 was associated with fracture incidence in ESRD with regular HD patients. There was no significant correlation between FGF23 and bone cell biomarkers. | [35] |
| ESRD with MHD | 53 ± 14.6 | cross- sectional | 221.9 ± 248.9 E | ↔BALP b | ↔CTX-1 b | ↔ b | ↔ | FGF23 was significantly increased in ESRD patients with lumbar spine osteo- porosis, but no correlation between BMD and FGF23 was observed. The biomarkers related to bone formation and resorption did not show any difference between normal bone, osteopenia and osteoporosis. | [45] |
| (n = 90) | (Tunisia) | ||||||||
| Subgroups | |||||||||
| LS osteoporosis (n = 8) | 65.8 ± 10.1 | ↑ 428.1 ± 275.6 E | ↔ BALP | ↔ CTX-1 | ↔ b | N/A | |||
| vs. normal/ osteopenia (ref.) | |||||||||
| TH osteoporosis (n = 18) | 63.9 ± 11.4 | ↔ 250.3 ± 250.3 E | ↔ BALP | ↔ CTX-1 | ↔ b | N/A | |||
| vs. normal/ osteopenia (ref.) | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirikul, W.; Siri-Angkul, N.; Chattipakorn, N.; Chattipakorn, S.C. Fibroblast Growth Factor 23 and Osteoporosis: Evidence from Bench to Bedside. Int. J. Mol. Sci. 2022, 23, 2500. https://doi.org/10.3390/ijms23052500
Sirikul W, Siri-Angkul N, Chattipakorn N, Chattipakorn SC. Fibroblast Growth Factor 23 and Osteoporosis: Evidence from Bench to Bedside. International Journal of Molecular Sciences. 2022; 23(5):2500. https://doi.org/10.3390/ijms23052500
Chicago/Turabian StyleSirikul, Wachiranun, Natthaphat Siri-Angkul, Nipon Chattipakorn, and Siriporn C. Chattipakorn. 2022. "Fibroblast Growth Factor 23 and Osteoporosis: Evidence from Bench to Bedside" International Journal of Molecular Sciences 23, no. 5: 2500. https://doi.org/10.3390/ijms23052500
APA StyleSirikul, W., Siri-Angkul, N., Chattipakorn, N., & Chattipakorn, S. C. (2022). Fibroblast Growth Factor 23 and Osteoporosis: Evidence from Bench to Bedside. International Journal of Molecular Sciences, 23(5), 2500. https://doi.org/10.3390/ijms23052500