
Changes in Non-Deamidated versus Deamidated Epitope Targeting and Disease Prediction during the Antibody Response to Gliadin and Transglutaminase of Infants at Risk for Celiac Disease

 , , , and
, , , and 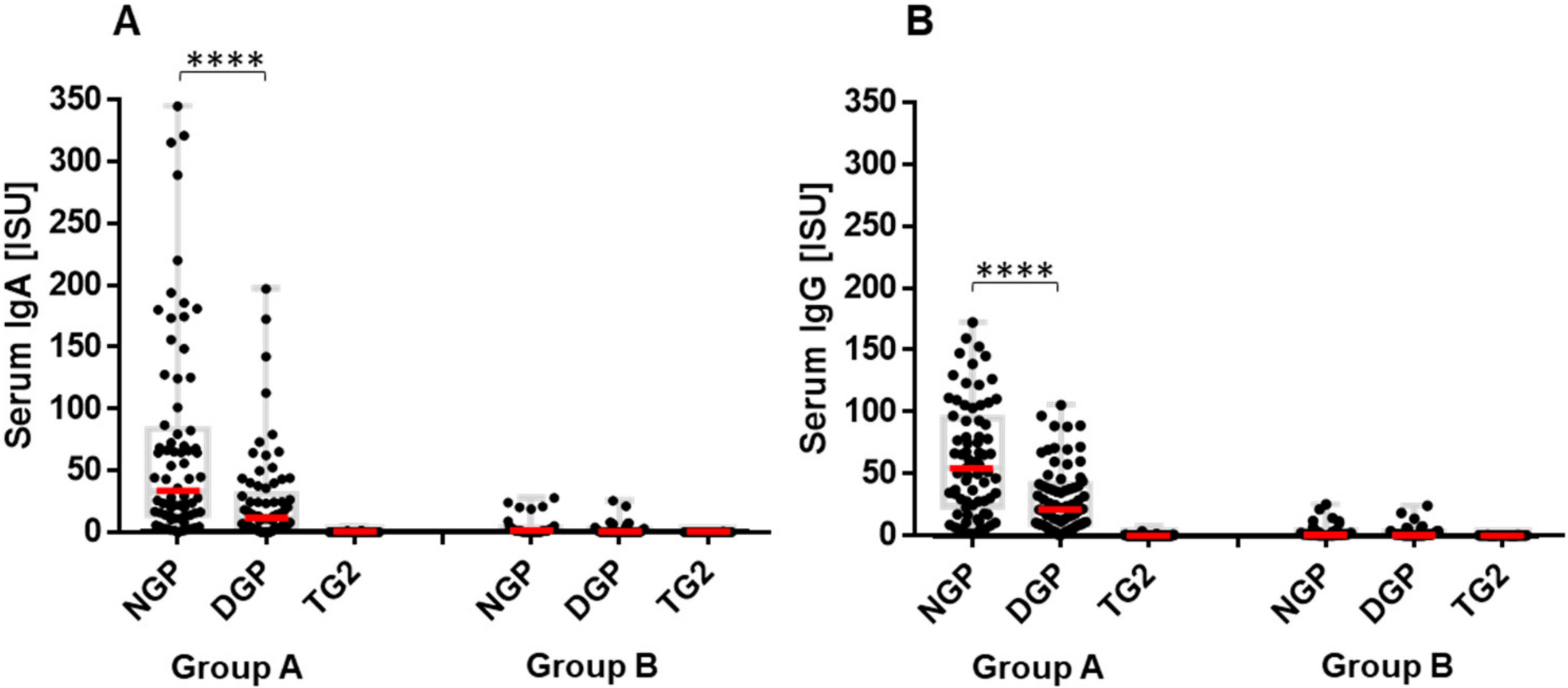{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
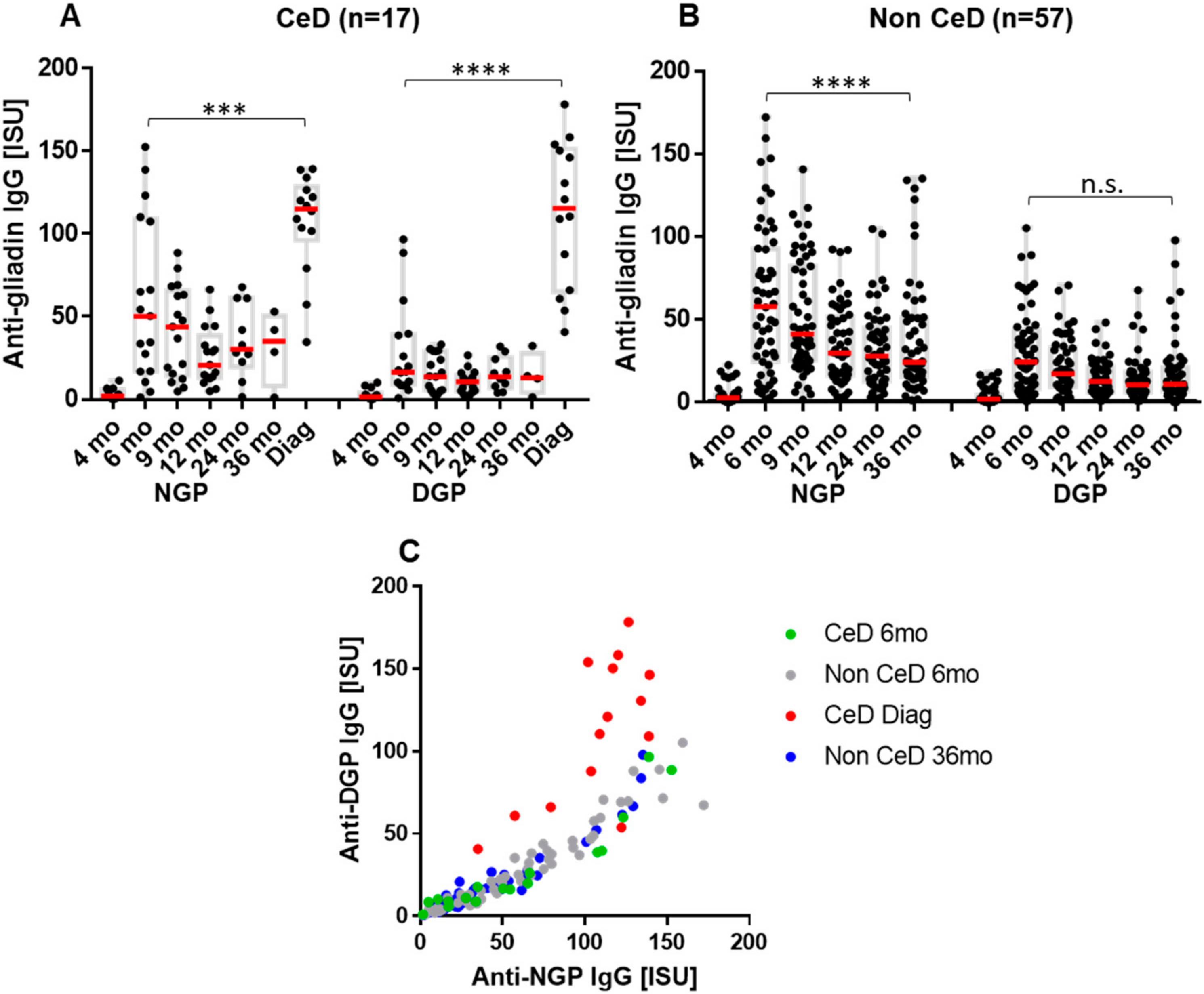
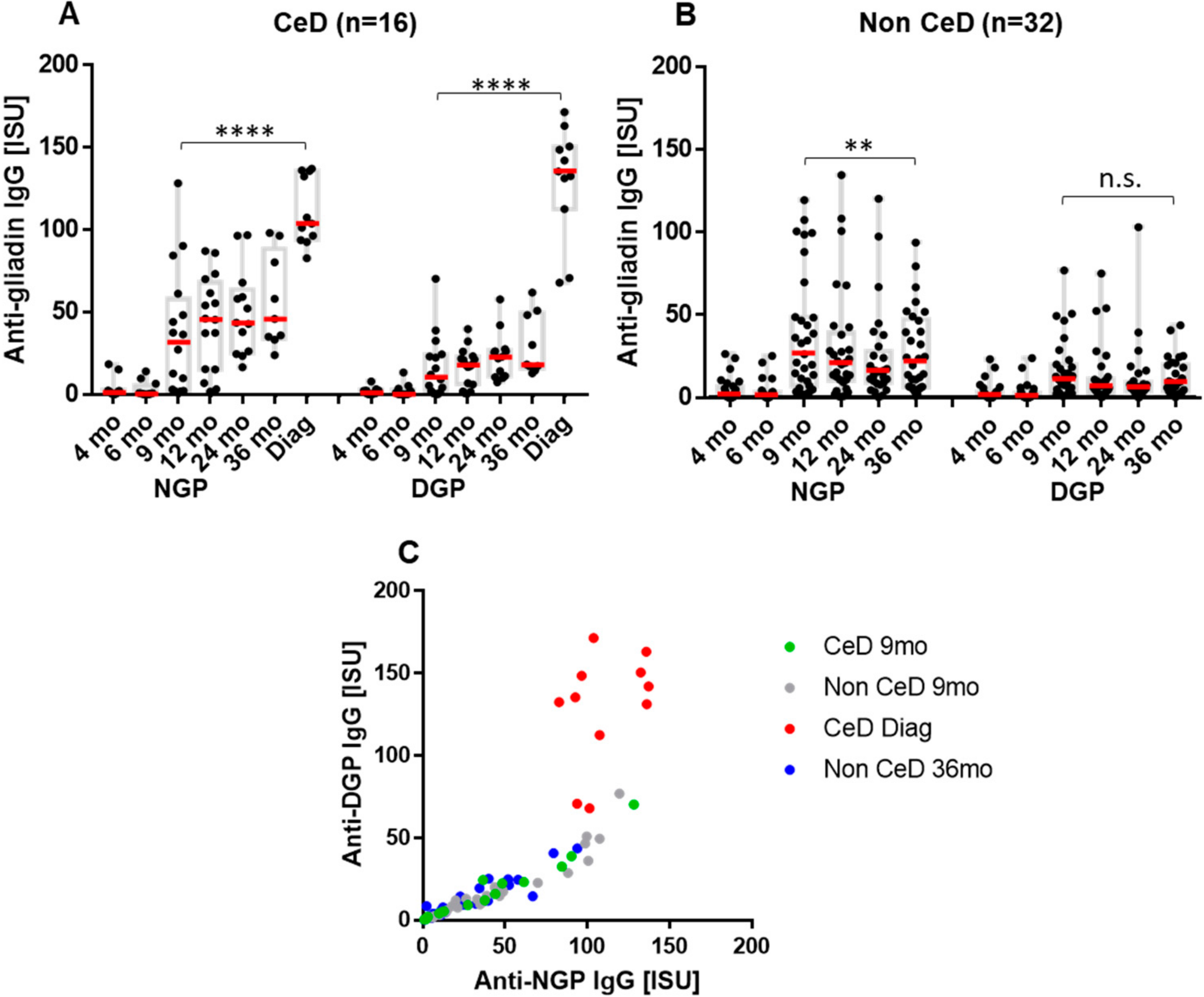
2.1. Infants at Risk of CeD Responded with Anti-Gliadin Antibody Production to Gluten Intake
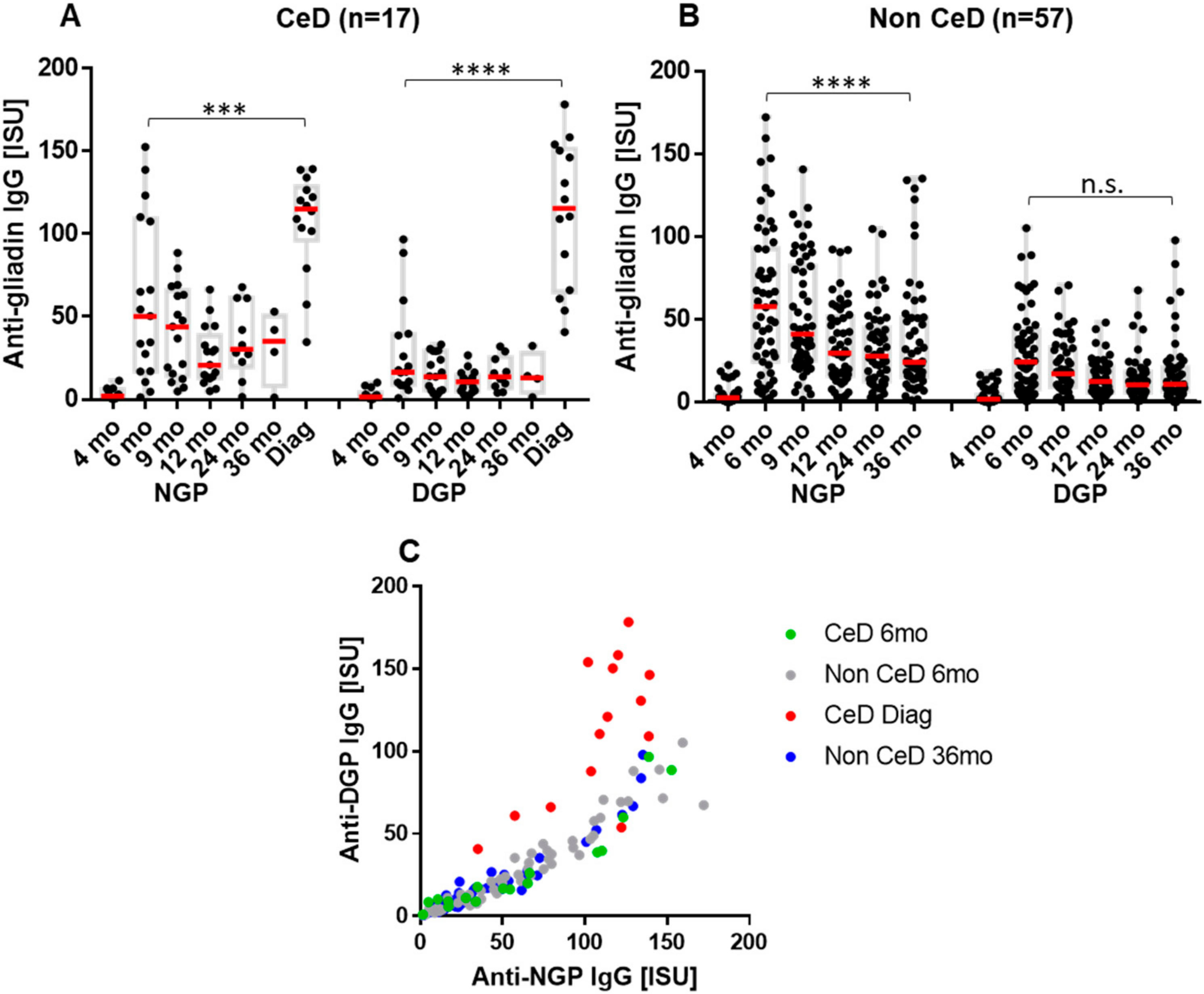
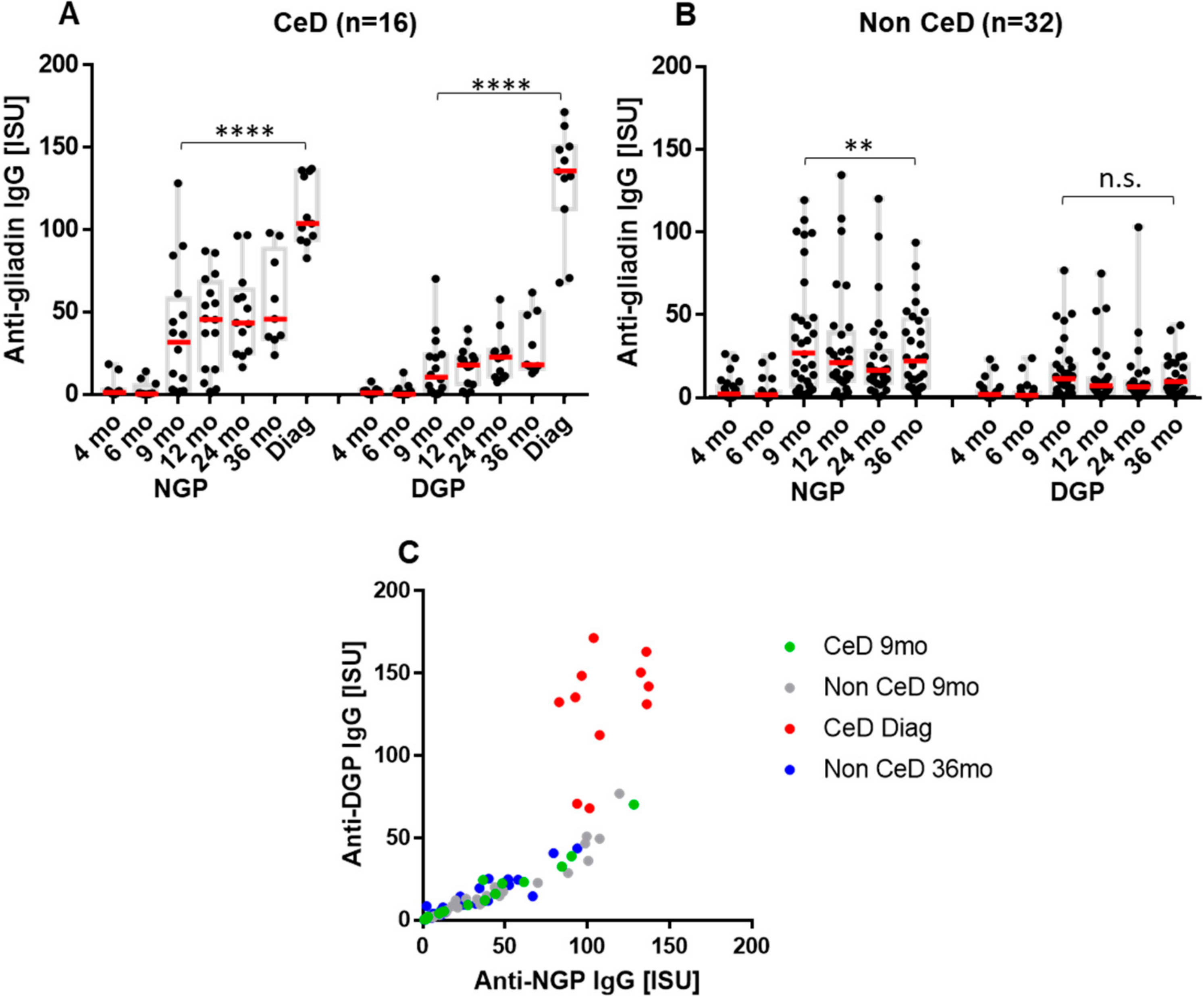
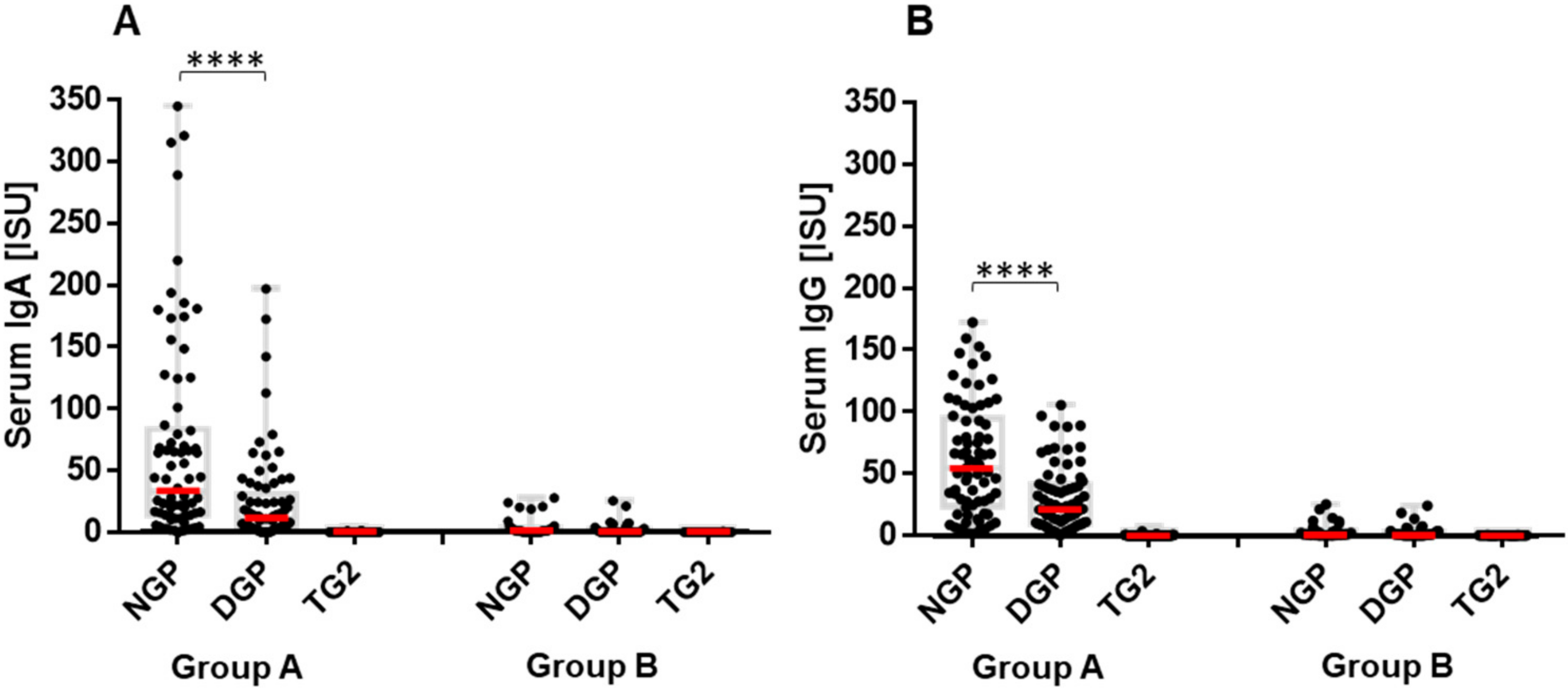
2.2. The Early Antibody Response Is Directed Preferentially to NGP, while at the Time of CeD Development, Antibody Response Increases for DGP
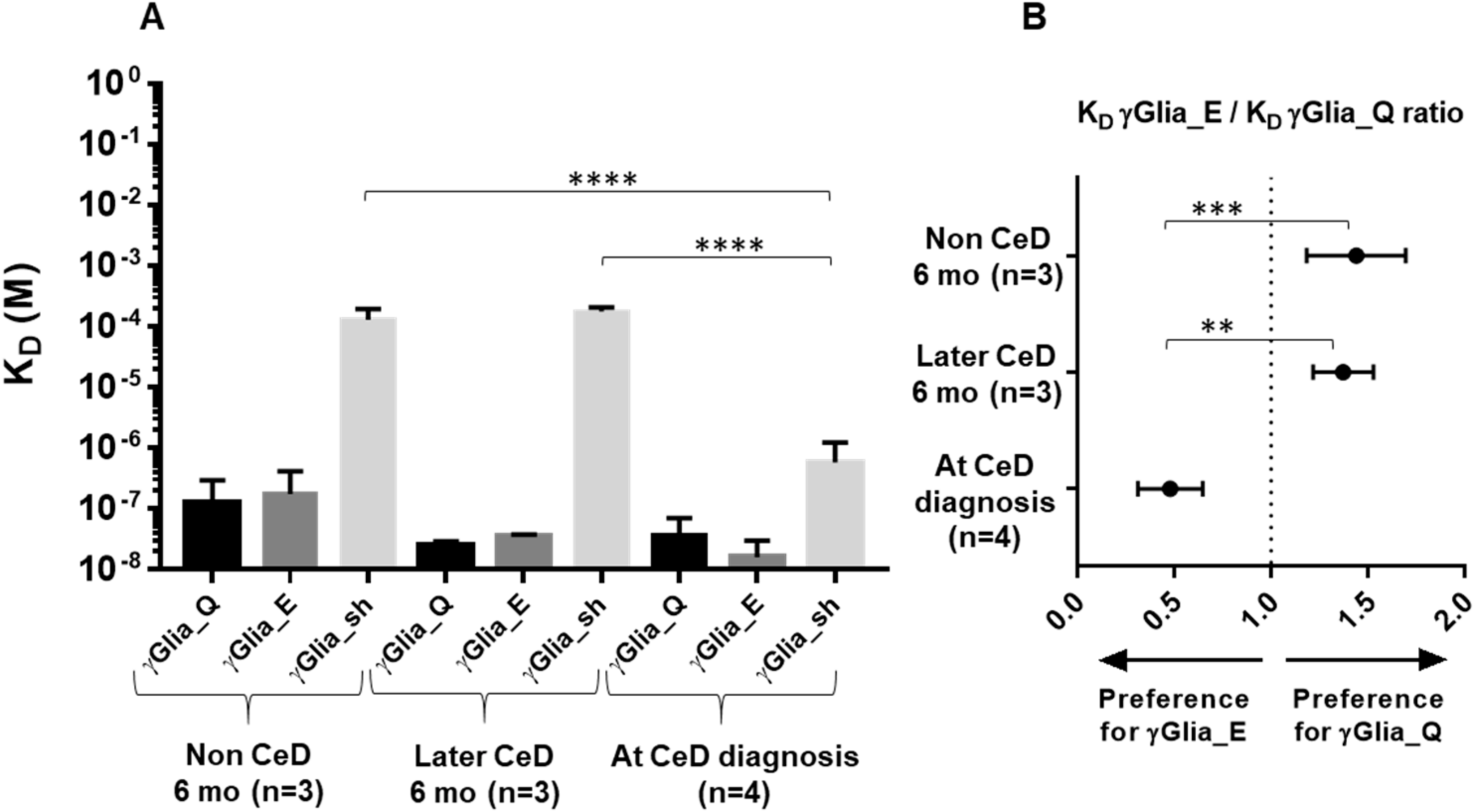
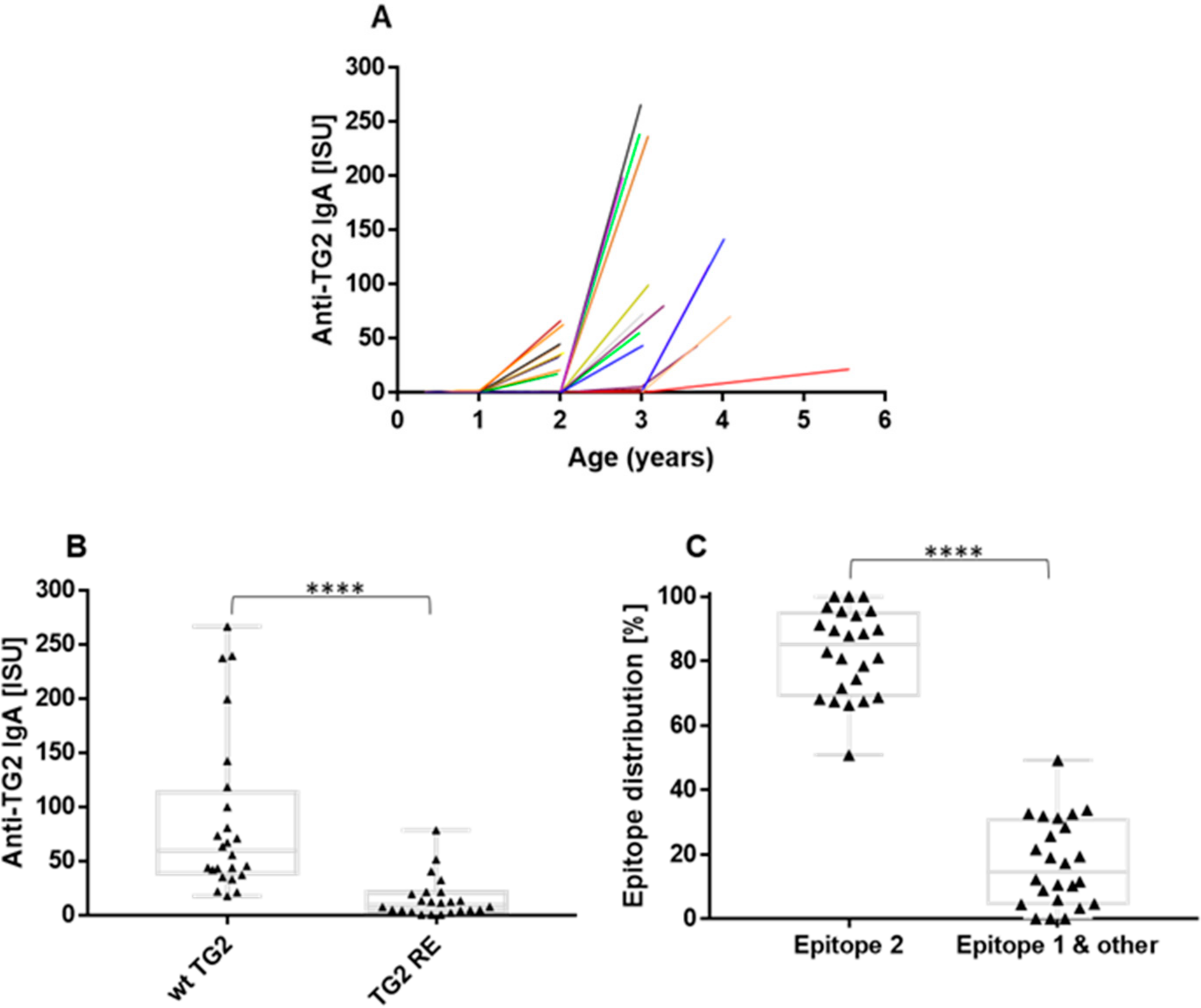
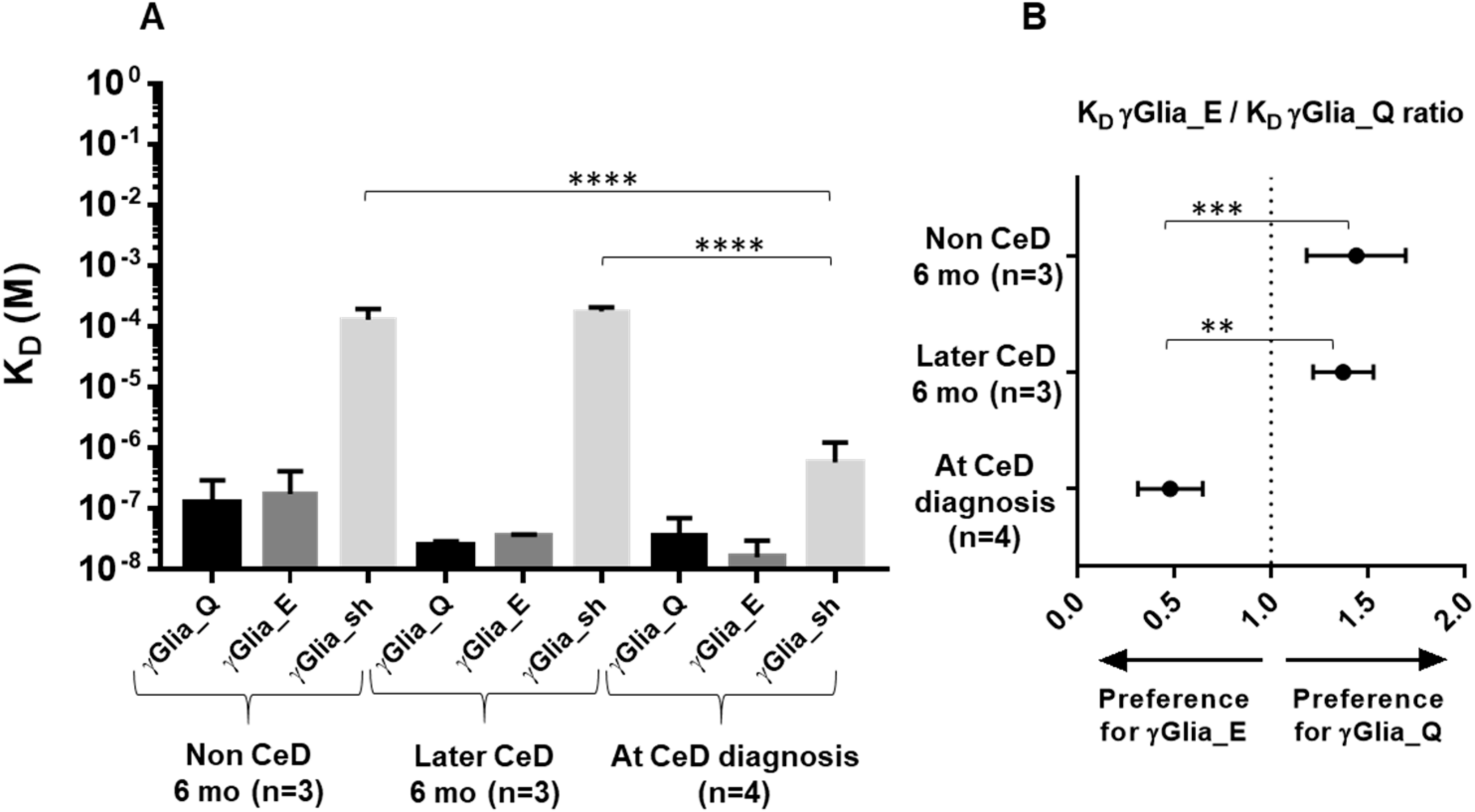
2.3. Increase in Binding Strength of Anti-Gliadin Antibodies for the Deamidated PEQPFP Core Epitope at the Manifestation of CeD
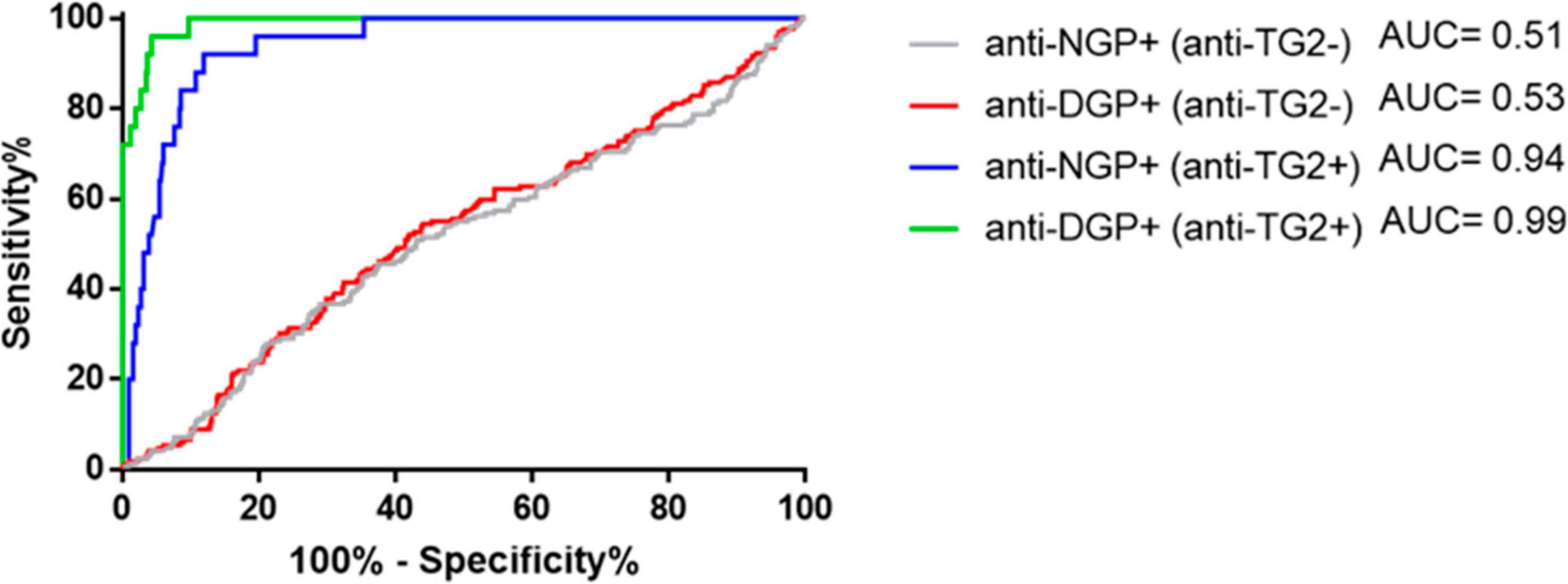
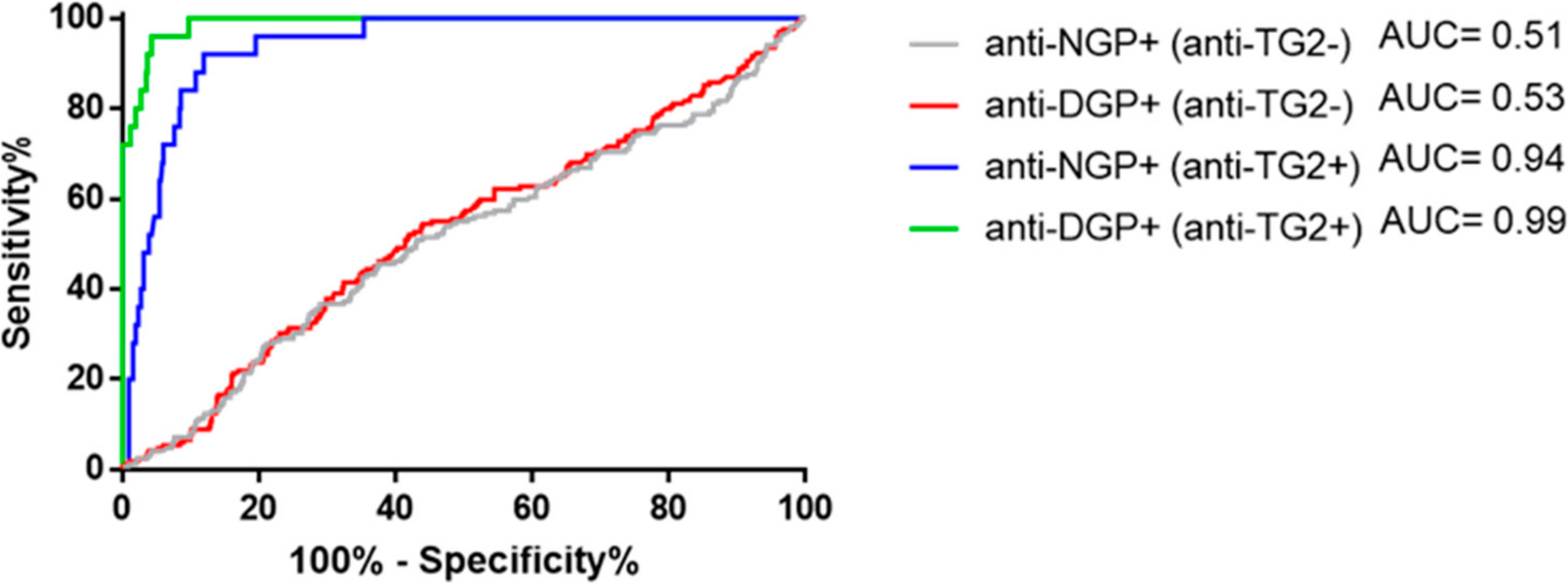
2.4. Presence of Gliadin-Specific Antibodies in the Absence of Anti-TG2 Positivity Does Not Predict Having or Later Developing CeD
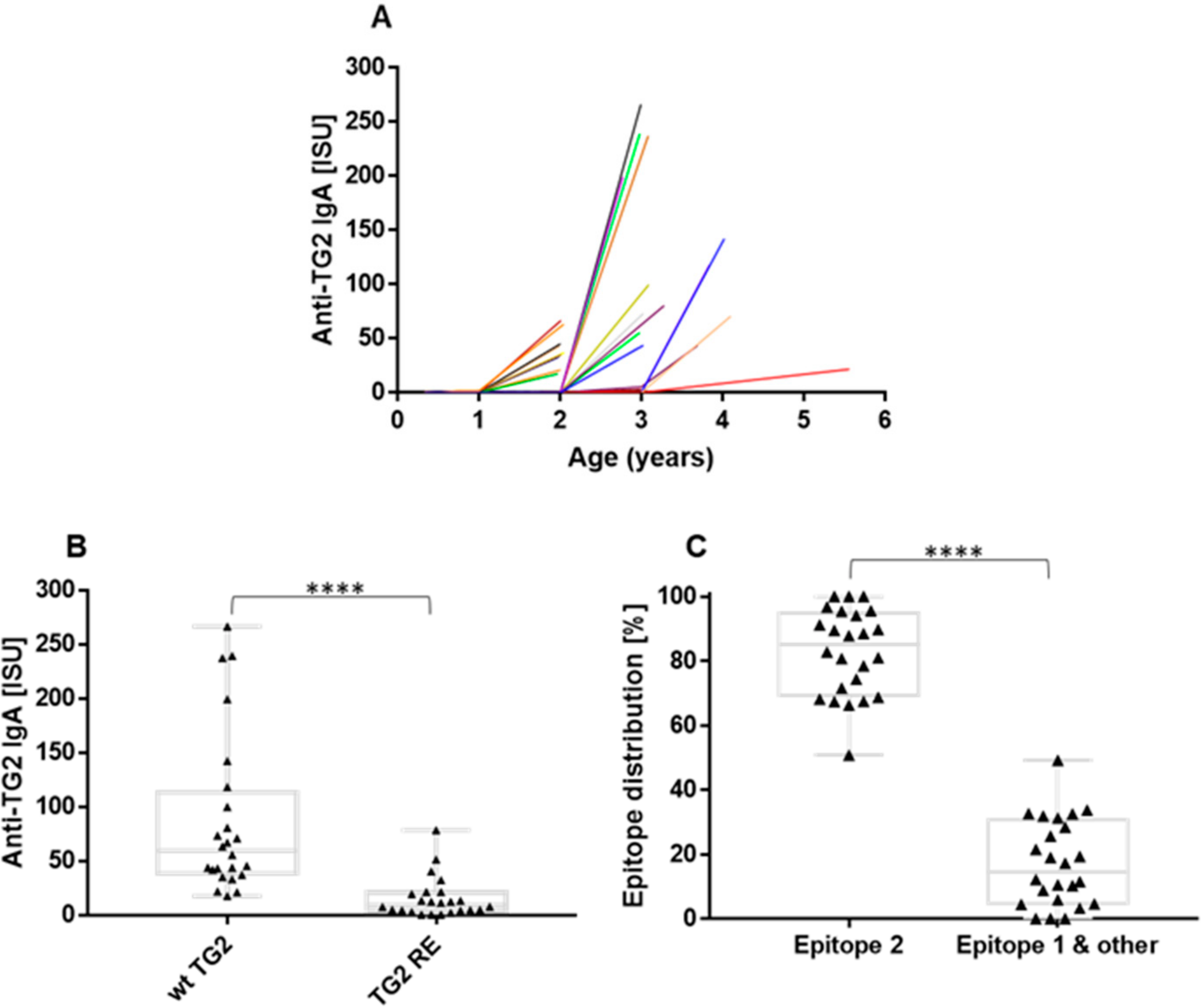
2.5. The Anti-TG2 Immune Reaction Predominantly Targets TG2’s Celiac Epitope 2
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Peptides and Reagents
4.3. Microarray Detection of Serum Antibodies
4.4. Affinity Purification of Gliadin-Specific Antibodies from Patient Serum Samples
4.5. Determination of the Binding Kinetics of Affinity-Purified Antibodies by Bli System
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sollid, L.M.; Jabri, B. Triggers and drivers of autoimmunity: Lessons from coeliac disease. Nat. Rev. Immunol. 2013, 13, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Qiao, S.-W.; Arentz-Hansen, H.; Molberg, Ø.; Gray, G.M.; Sollid, L.M.; Khosla, C. Identification and Analysis of Multivalent Proteolytically Resistant Peptides from Gluten: Implications for Celiac Sprue. J. Proteome Res. 2005, 4, 1732–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollid, L.M.; Qiao, S.-W.; Anderson, R.P.; Gianfrani, C.; Koning, F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 2012, 64, 455–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trynka, G.; Hunt, K.A.; Bockett, N.A.; Romanos, J.; Mistry, V.; Szperl, A.; Bakker, S.F.; Bardella, M.T.; Bhaw-Rosun, L.; Castillejo, G.; et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011, 43, 1193–1201. [Google Scholar] [CrossRef] [Green Version]
- Klöck, C.; DiRaimondo, T.R.; Khosla, C. Role of transglutaminase 2 in celiac disease pathogenesis. Semin. Immunopathol. 2012, 34, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vader, L.W.; De Ru, A.; Van Der Wal, Y.; Kooy, Y.M.; Benckhuijsen, W.; Mearin, M.L.; Drijfhout, J.W.; van Veelen, P.; Koning, F. Specificity of Tissue Transglutaminase Explains Cereal Toxicity in Celiac Disease. J. Exp. Med. 2002, 195, 643–649. [Google Scholar] [CrossRef]
- Dørum, S.; Arntzen, M.; Qiao, S.-W.; Holm, A.; Koehler, C.J.; Thiede, B.; Sollid, L.M.; Fleckenstein, B. The Preferred Substrates for Transglutaminase 2 in a Complex Wheat Gluten Digest Are Peptide Fragments Harboring Celiac Disease T-Cell Epitopes. PLoS ONE 2010, 5, e14056. [Google Scholar] [CrossRef] [Green Version]
- Dørum, S.; Steinsbø, Ø.; Bergseng, E.; Arntzen, M.; de Souza, G.; Sollid, L.M. Gluten-specific antibodies of celiac disease gut plasma cells recognize long proteolytic fragments that typically harbor T-cell epitopes. Sci. Rep. 2016, 6, 25565. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.-W.; Bergseng, E.; Molberg, Ø.; Jung, G.; Fleckenstein, B.; Sollid, L.M. Refining the Rules of Gliadin T Cell Epitope Binding to the Disease-Associated DQ2 Molecule in Celiac Disease: Importance of Proline Spacing and Glutamine Deamidation. J. Immunol. 2005, 175, 254–261. [Google Scholar] [CrossRef]
- Ráki, M.; Dahal-Koirala, S.; Yu, H.; Korponay-Szabó, I.R.; Gyimesi, J.; Castillejo, G.; Jahnsen, J.; Qiao, S.-W.; Sollid, L.M. Similar Responses of Intestinal T Cells From Untreated Children and Adults With Celiac Disease to Deamidated Gluten Epitopes. Gastroenterology 2017, 153, 787–798e4. [Google Scholar] [CrossRef] [Green Version]
- Osman, A.A.; Günnel, T.; Dietl, A.; Uhlig, H.H.; Amin, M.; Fleckenstein, B.; Richter, T.; Mothes, T. B cell epitopes of gliadin. Clin. Exp. Immunol. 2000, 121, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Schwertz, E.; Kahlenberg, F.; Sack, U.; Richter, T.; Stern, M.; Conrad, K.; Zimmer, K.-P.; Mothes, T. Serologic Assay Based on Gliadin-Related Nonapeptides as a Highly Sensitive and Specific Diagnostic Aid in Celiac Disease. Clin. Chem. 2004, 50, 2370–2375. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B.; Focke-Tejkl, M.; Swoboda, I.; Constantin, C.; Mittermann, I.; Pahr, S.; Vogelsang, H.; Huber, W.-D.; Valenta, R. A combined biochemical, biophysical and immunological approach towards the identification of celiac disease-specific wheat antigens. Amino Acids 2013, 45, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Iversen, R.; Di Niro, R.; Stamnaes, J.; Lundin, K.E.A.; Wilson, P.C.; Sollid, L.M. Transglutaminase 2–Specific Autoantibodies in Celiac Disease Target Clustered, N-Terminal Epitopes Not Displayed on the Surface of Cells. J. Immunol. 2013, 190, 5981–5991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Vecsei, Z.; Kiraly, R.; Bagossi, P.; Toth, B.; Dahlbom, I.; Caja, S.; Csosz, E.; Lindfors, K.; Sblattero, D.; Nemes, E.; et al. A single conformational transglutaminase 2 epitope contributed by three domains is critical for celiac antibody binding and effects. Proc. Natl. Acad. Sci. USA 2012, 109, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simell, S.; Hoppu, S.; Hekkala, A.; Simell, T.; Ståhlberg, M.-R.; Viander, M.; Yrjänäinen, H.; Grönlund, J.; Markula, P.; Simell, V.; et al. Fate of Five Celiac Disease-Associated Antibodies During Normal Diet in Genetically At-Risk Children Observed from Birth in a Natural History Study. Am. J. Gastroenterol. 2007, 102, 2026–2035. [Google Scholar] [CrossRef]
- Amarri, S.; Alvisi, P.; De Giorgio, R.; Gelli, M.C.; Cicola, R.; Tovoli, F.; Sassatelli, R.; Caio, G.; Volta, U. Antibodies to Deamidated Gliadin Peptides: An Accurate Predictor of Coeliac Disease in Infancy. J. Clin. Immunol. 2013, 33, 1027–1030. [Google Scholar] [CrossRef]
- Liu, E.; Lee, H.-S.; Aronsson, C.A.; Hagopian, W.A.; Koletzko, S.; Rewers, M.J.; Eisenbarth, G.S.; Bingley, P.J.; Bonifacio, E.; Simell, V.; et al. Risk of Pediatric Celiac Disease According to HLA Haplotype and Country. N. Engl. J. Med. 2014, 371, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, E.; Castellaneta, S.; Francavilla, R.; Pulvirenti, A.; Tonutti, E.; Amarri, S.; Barbato, M.; Barbera, C.; Barera, G.; Bellantoni, A.; et al. Introduction of Gluten, HLA Status, and the Risk of Celiac Disease in Children. N. Engl. J. Med. 2014, 371, 1295–1303. [Google Scholar] [CrossRef] [Green Version]
- Vriezinga, S.L.; Auricchio, R.; Bravi, E.; Castillejo, G.; Chmielewska, A.; Crespo-Escobar, P.; Kolaček, S.; Koletzko, S.; Korponay-Szabo, I.R.; Mummert, E.; et al. Randomized Feeding Intervention in Infants at High Risk for Celiac Disease. N. Engl. J. Med. 2014, 371, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Van De Perre, P. Transfer of antibody via mother’s milk. Vaccine 2003, 21, 3374–3376. [Google Scholar] [CrossRef]
- Diós, Á.; Elek, R.; Szabó, I.; Horváth, S.; Gyimesi, J.; Király, R.; Werkstetter, K.; Koletzko, S.; Fésüs, L.; Korponay-Szabó, I.R. Gamma-gliadin specific celiac disease antibodies recognize p31-43 and p57-68 alpha gliadin peptides in deamidation related manner as a result of cross-reaction. Amino Acids 2021, 53, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Snir, O.; Chen, X.; Gidoni, M.; Du Pré, M.F.; Zhao, Y.; Steinsbø, Ø.; Lundin, K.E.; Yaari, G.; Sollid, L.M. Stereotyped antibody responses target posttranslationally modified gluten in celiac disease. JCI Insight 2017, 2, 93961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korponay-Szabo, I.R.; Halttunen, T.; Szalai, Z.; Laurila, K.; Király, R.; Kovács, J.B.; Fésüs, L.; Mäki, M. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut 2004, 53, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, I.; Secondo, A.; Lepretti, M.; Paolella, G.; Auricchio, S.; Barone, M.V.; Esposito, C. Gliadin Peptides Induce Tissue Transglutaminase Activation and ER-Stress through Ca2+ Mobilization in Caco-2 Cells. PLoS ONE 2012, 7, e45209. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Auricchio, R.; De Musis, C.; Discepolo, V.; Miele, E.; Jabri, B.; Troncone, R.; Auricchio, S.; et al. P31–43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: A role in celiac disease. Sci. Rep. 2018, 8, 10821. [Google Scholar] [CrossRef] [Green Version]
- Aronsson, C.A.; Lee, H.-S.; Segerstad, E.M.H.A.; Uusitalo, U.; Yang, J.; Koletzko, S.; Liu, E.; Kurppa, K.; Bingley, P.J.; Toppari, J.; et al. Association of Gluten Intake During the First 5 Years of Life With Incidence of Celiac Disease Autoimmunity and Celiac Disease Among Children at Increased Risk. JAMA J. Am. Med. Assoc. 2019, 322, 514–523. [Google Scholar] [CrossRef]
- Fleckenstein, B.; Qiao, S.-W.; Larsen, M.R.; Jung, G.; Roepstorff, P.; Sollid, L.M. Molecular Characterization of Covalent Complexes between Tissue Transglutaminase and Gliadin Peptides. J. Biol. Chem. 2004, 279, 17607–17616. [Google Scholar] [CrossRef] [Green Version]
- Lexhaller, B.; Ludwig, C.; Scherf, K.A. Comprehensive Detection of Isopeptides between Human Tissue Transglutaminase and Gluten Peptides. Nutrients 2019, 11, 2263. [Google Scholar] [CrossRef] [Green Version]
- Iversen, R.; Amundsen, S.F.; Kleppa, L.; du Pré, M.F.; Stamnæs, J.; Sollid, L.M. Evidence That Pathogenic Transglutaminase 2 in Celiac Disease Derives From Enterocytes. Gastroenterology 2020, 159, 788–790. [Google Scholar] [CrossRef]
- Camarca, A.; Auricchio, R.; Picascia, S.; Fierro, O.; Maglio, M.; Miele, E.; Malamisura, B.; Greco, L.; Troncone, R.; Gianfrani, C. Gliadin-reactive T cells in Italian children from preventCD cohort at high risk of celiac disease. Pediatr. Allergy Immunol. 2017, 28, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B.; Focke-Tejkl, M.; Weber, M.; Pahr, S.; Baar, A.; Atreya, R.; Neurath, M.F.; Vogelsang, H.; Huber, W.-D.; Valenta, R. Usefulness of recombinant γ-gliadin 1 for identifying patients with celiac disease and monitoring adherence to a gluten-free diet. J. Allergy Clin. Immunol. 2015, 136, 1607–1618e3. [Google Scholar] [CrossRef] [Green Version]
- Lupinek, C.; Wollmann, E.; Baar, A.; Banerjee, S.; Breiteneder, H.; Broecker, B.M.; Bublin, M.; Curin, M.; Flicker, S.; Garmatiuk, T.; et al. Advances in allergen-microarray technology for diagnosis and monitoring of allergy: The MeDALL allergen-chip. Methods 2014, 66, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Hiller, R.; Laffer, S.; Harwanegg, C.; Huber, M.; Schmidt, W.M.; Twardosz, A.; Barletta, B.; Becker, W.M.; Blaser, K.; Breiteneder, H.; et al. Microarrayed allergen molecules: Diagnostic gatekeepers for allergy treatment. FASEB J. 2002, 16, 414–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchan, K.; Ergülen, E.; Király, R.; Simon-Vecsei, Z.; Fuxreiter, M.; Fésüs, L. Identification of a specific one amino acid change in recombinant human transglutaminase 2 that regulates its activity and calcium sensitivity. Biochem. J. 2013, 455, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husby, S.; Koletzko, S.; Korponay-Szabó, I.; Kurppa, K.; Mearin, M.L.; Ribes-Koninckx, C.; Shamir, R.; Troncone, R.; Auricchio, R.; Castillejo, G.; et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diós, Á.; Srinivasan, B.; Gyimesi, J.; Werkstetter, K.; Valenta, R.; Koletzko, S.; Korponay-Szabó, I.R. Changes in Non-Deamidated versus Deamidated Epitope Targeting and Disease Prediction during the Antibody Response to Gliadin and Transglutaminase of Infants at Risk for Celiac Disease. Int. J. Mol. Sci. 2022, 23, 2498. https://doi.org/10.3390/ijms23052498
Diós Á, Srinivasan B, Gyimesi J, Werkstetter K, Valenta R, Koletzko S, Korponay-Szabó IR. Changes in Non-Deamidated versus Deamidated Epitope Targeting and Disease Prediction during the Antibody Response to Gliadin and Transglutaminase of Infants at Risk for Celiac Disease. International Journal of Molecular Sciences. 2022; 23(5):2498. https://doi.org/10.3390/ijms23052498
Chicago/Turabian StyleDiós, Ádám, Bharani Srinivasan, Judit Gyimesi, Katharina Werkstetter, Rudolf Valenta, Sibylle Koletzko, and Ilma R. Korponay-Szabó. 2022. "Changes in Non-Deamidated versus Deamidated Epitope Targeting and Disease Prediction during the Antibody Response to Gliadin and Transglutaminase of Infants at Risk for Celiac Disease" International Journal of Molecular Sciences 23, no. 5: 2498. https://doi.org/10.3390/ijms23052498
APA StyleDiós, Á., Srinivasan, B., Gyimesi, J., Werkstetter, K., Valenta, R., Koletzko, S., & Korponay-Szabó, I. R. (2022). Changes in Non-Deamidated versus Deamidated Epitope Targeting and Disease Prediction during the Antibody Response to Gliadin and Transglutaminase of Infants at Risk for Celiac Disease. International Journal of Molecular Sciences, 23(5), 2498. https://doi.org/10.3390/ijms23052498