
Urinary Proteomic Signature in Acute Decompensated Heart Failure: Advances into Molecular Pathophysiology

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics of the ADHF Patient Population
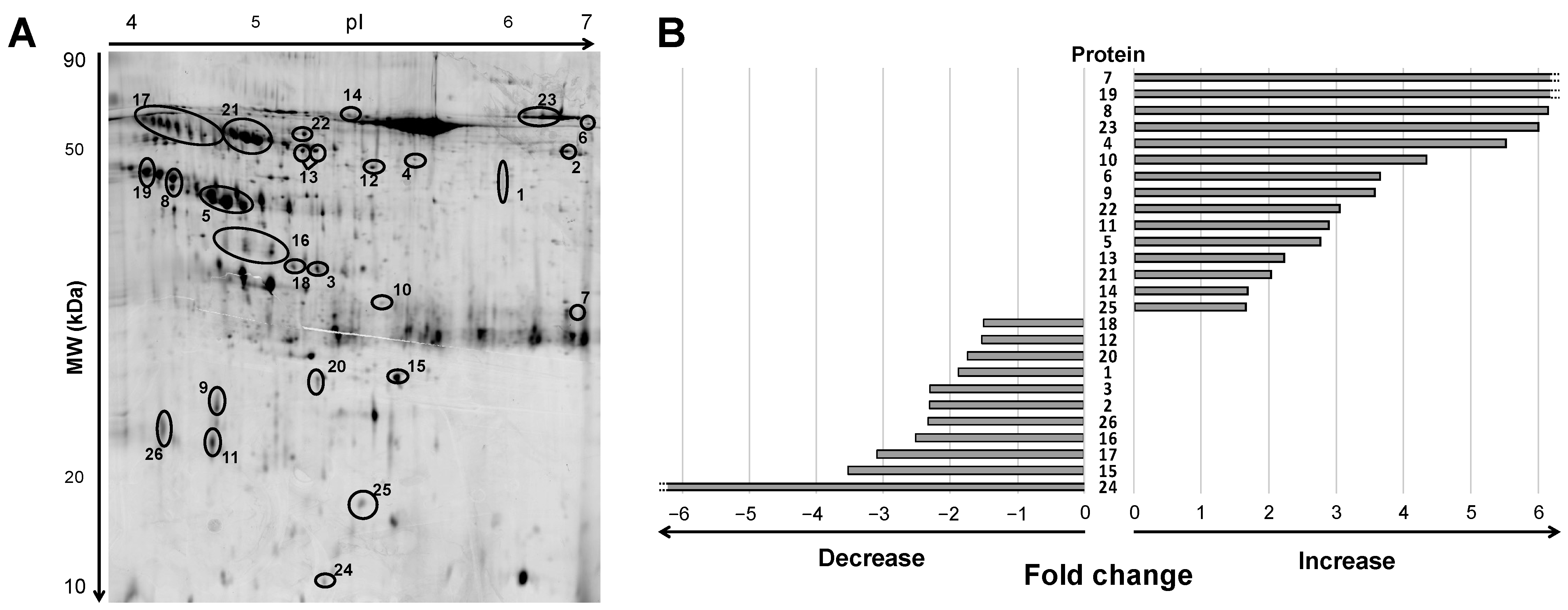
2.2. Urine Protein Signature in ADHF Patients by 2DE Mass Spectrometry
2.3. Changes in the ADHF Urine Protein Signature and Relation to Kidney and Heart Function
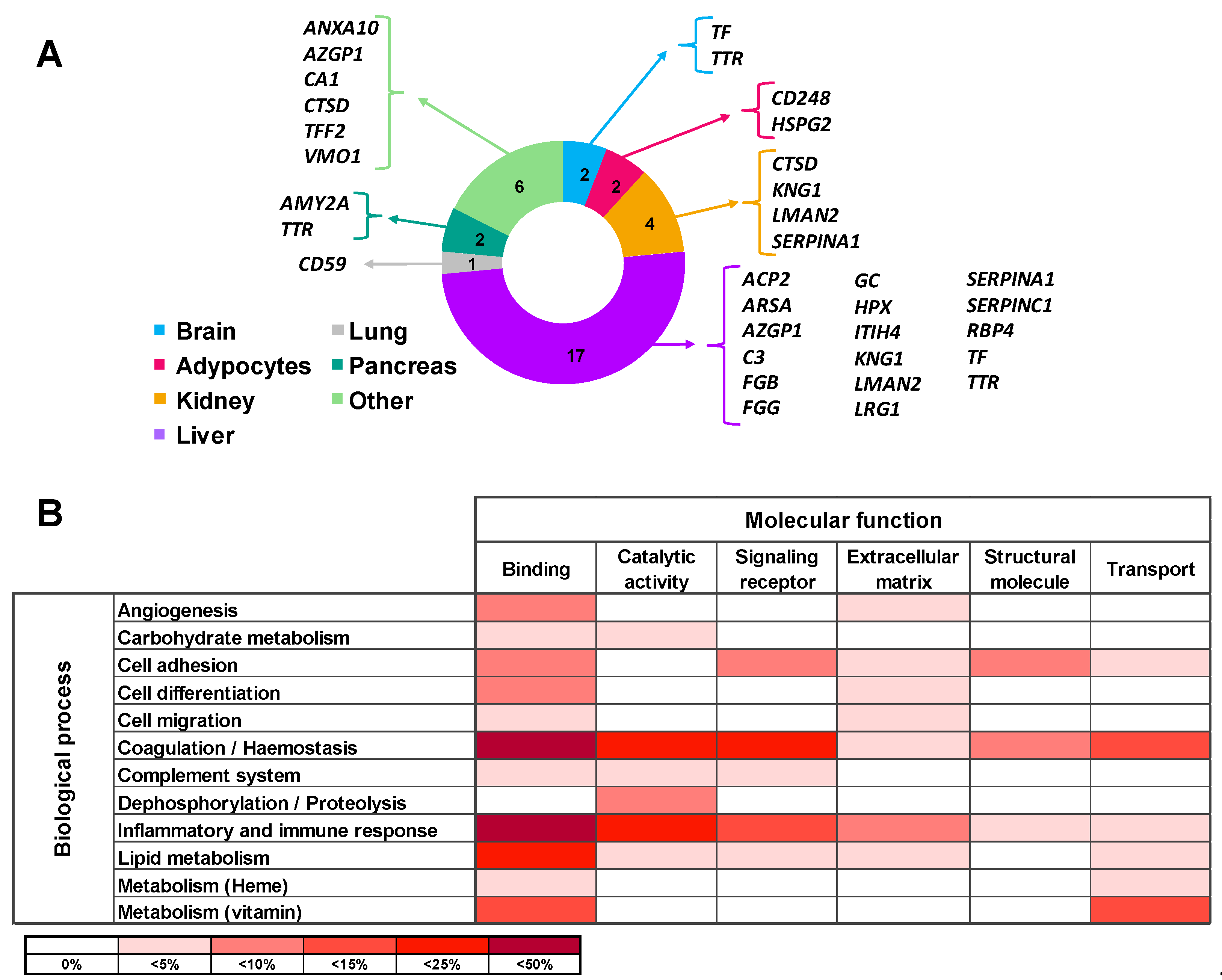
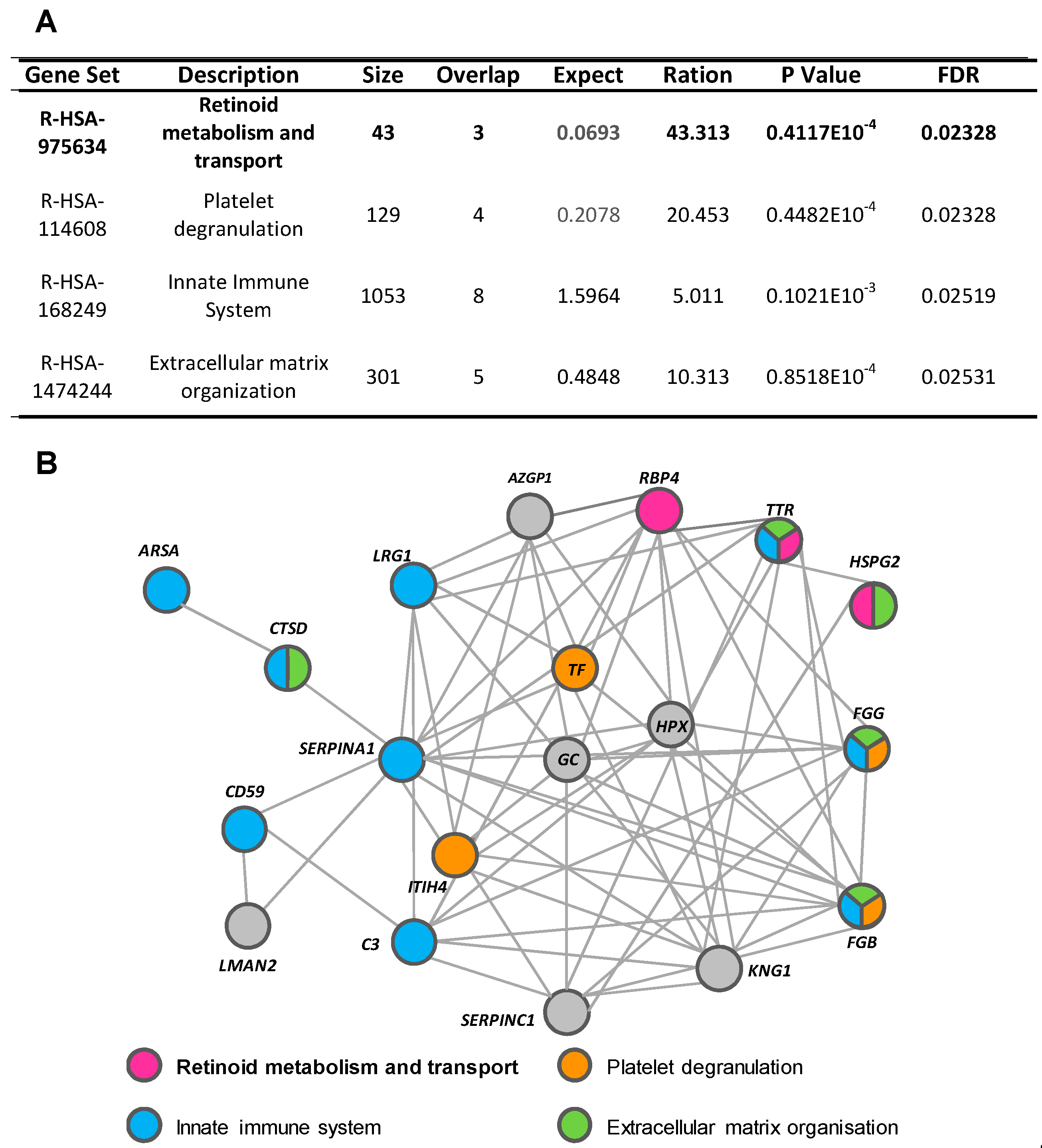
2.4. Functional Characteristics and Pathway Analysis of the Differential Urine Protein Signature in ADHF
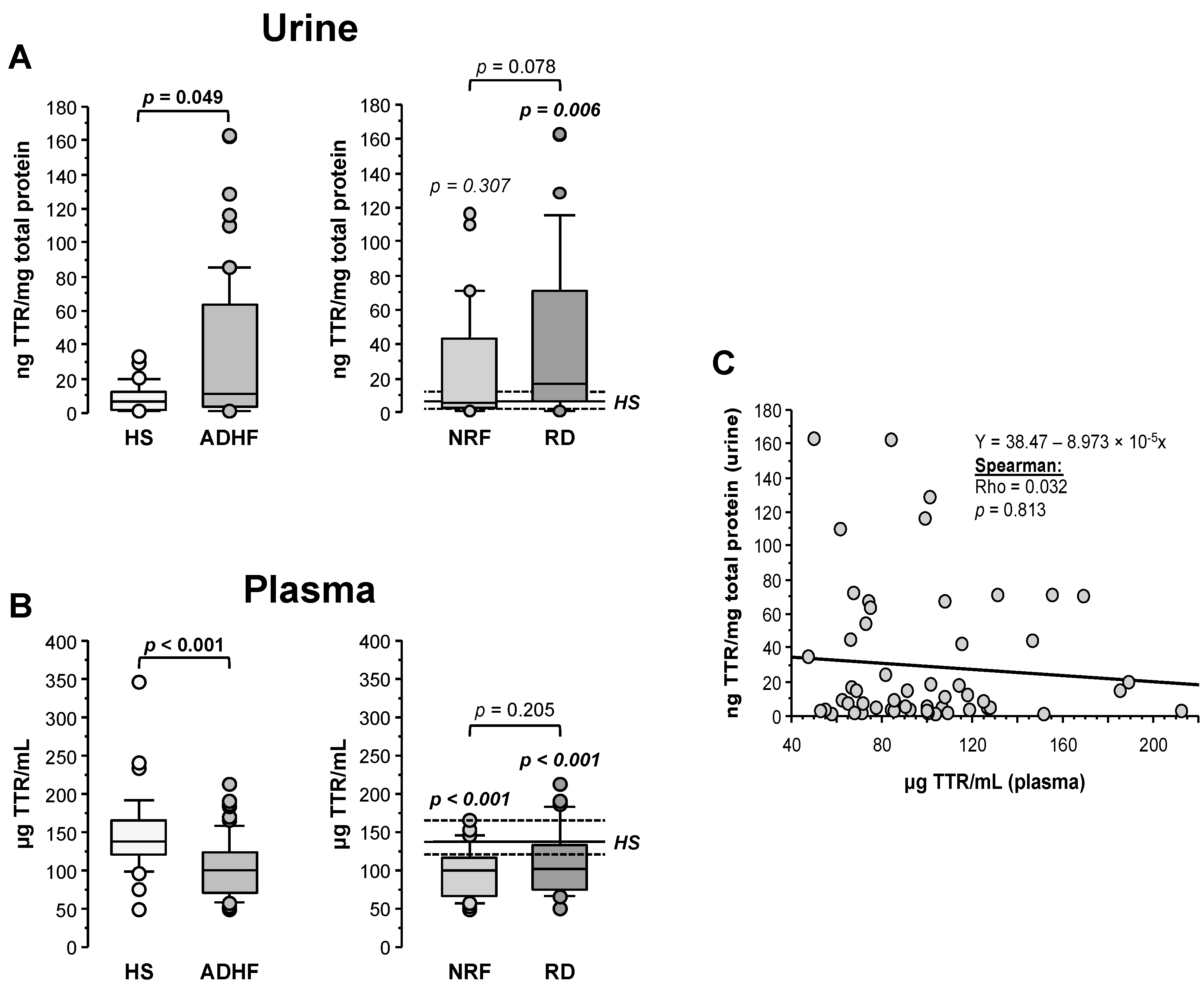
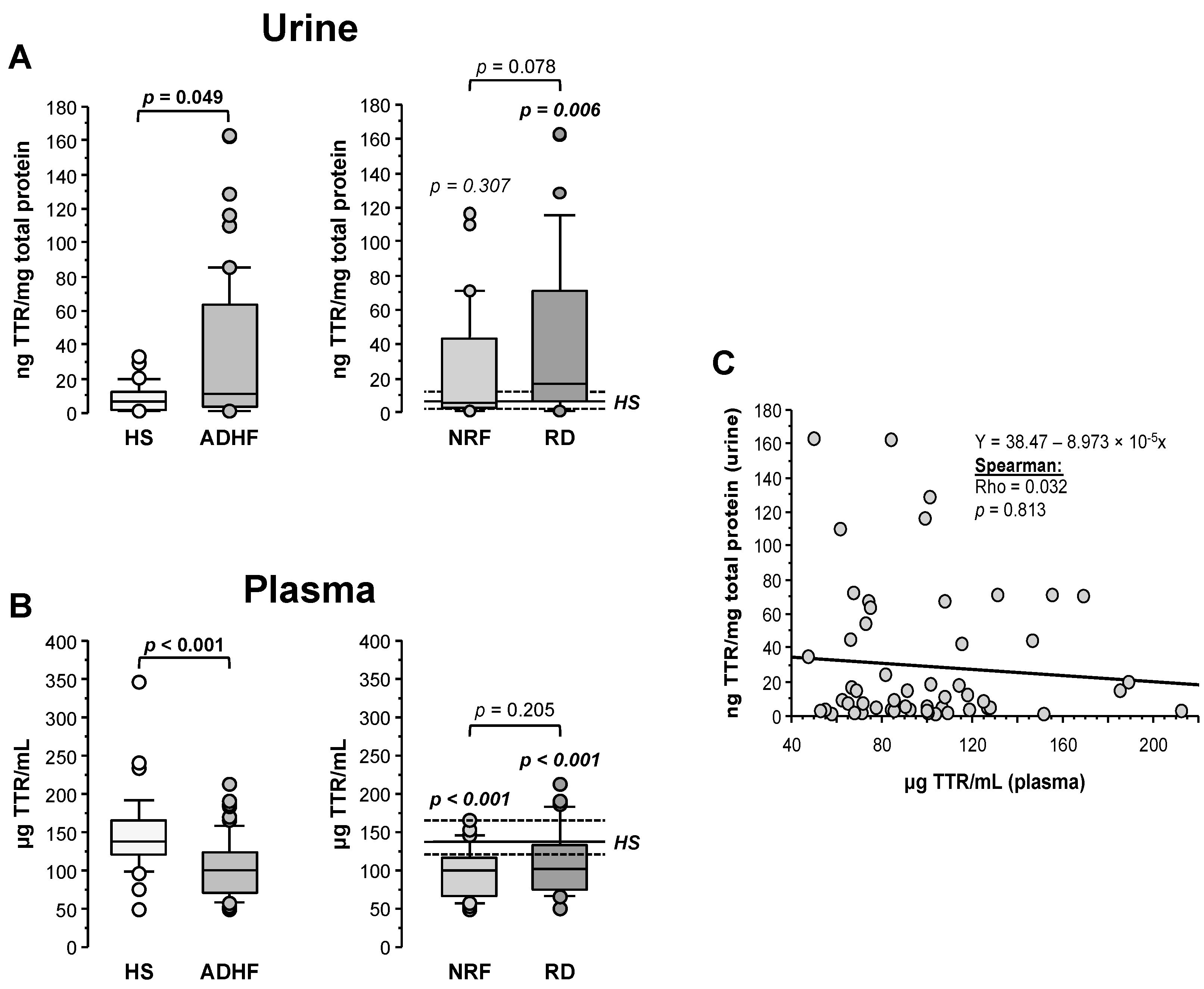
2.5. Urine and Plasma Transthyretin Levels in ADHF Patients
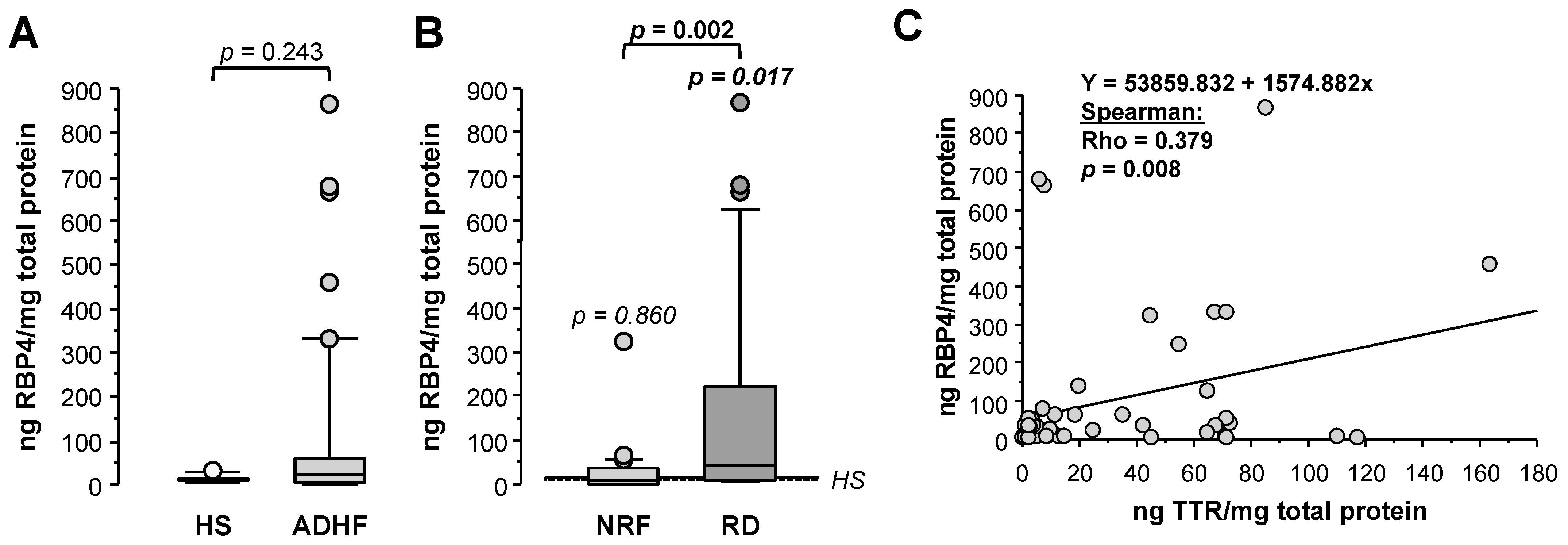
2.6. Changes in Urine RBP4 Directly Correlate with TTR and Are Related to Renal Function
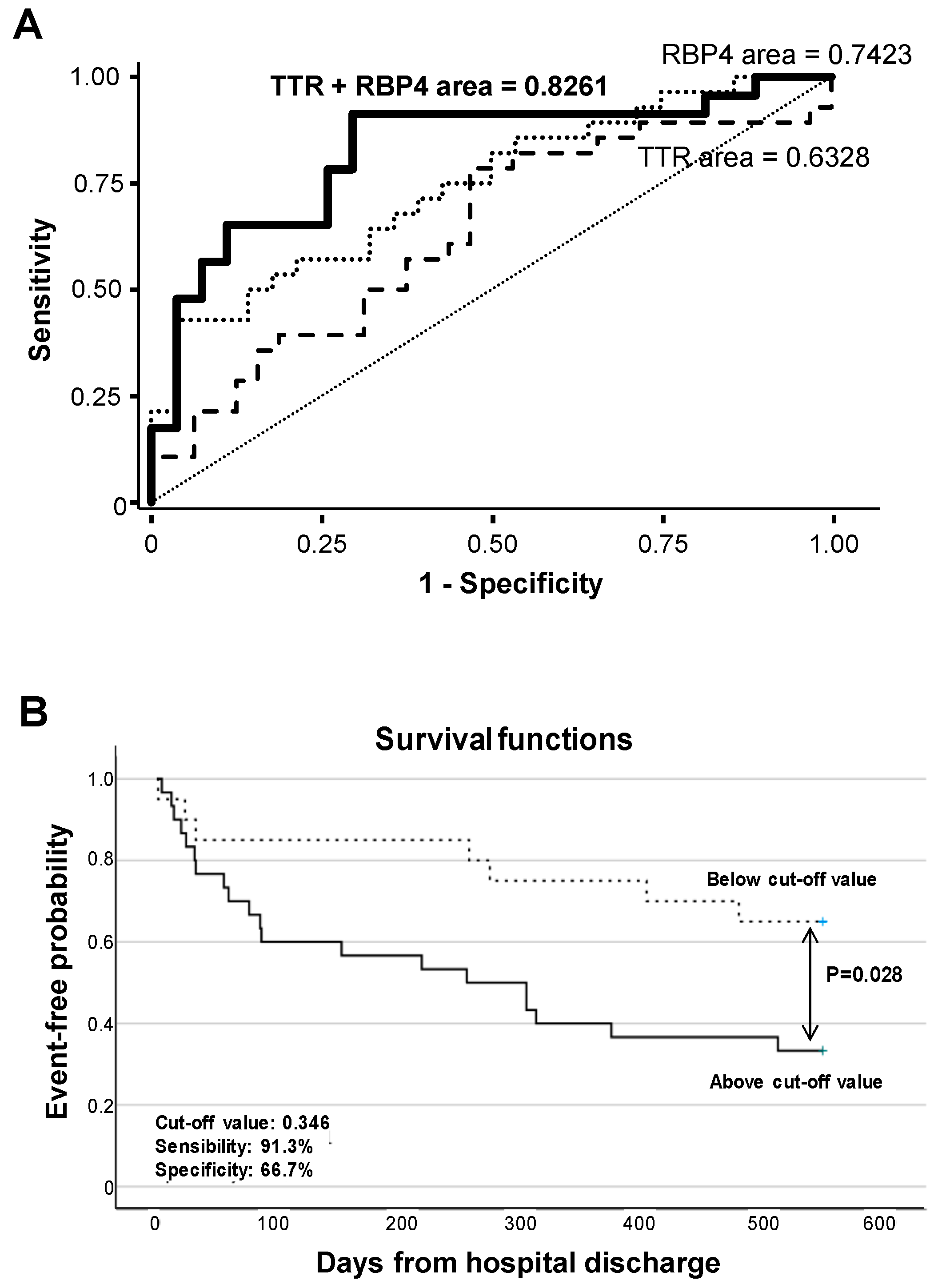
2.7. RBP4 and TTR Levels in Urine at Hospital Admission Relate with Disease Evolution within 18-Month Follow-Up
3. Discussion
4. Materials and Methods
4.1. Study Population and Study Design
4.2. Biological Samples
4.3. Two-Dimmensional Gel Electrophoresis and MALDI-TOF/TOF MS
4.4. In Silico Analysis
4.5. Enzyme-Linked Immunosorbent Assays
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emdin, M.; Vittorini, S.; Passino, C.; Clerico, A. Old and new biomarkers of heart failure. Eur. J. Heart Fail. 2009, 11, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, M.S.; Brutsaert, D.; Dickstein, K.; Drexler, H.; Follath, F.; Harjola, V.-P.; Hochadel, M.; Komajda, M.; Lassus, J.; Luis Lopez-Sendon, J.; et al. EuroHeart Failure Survey II (EHFS II): A survey on hospitalized acute heart failure patients: Description of population. Eur. Heart J. 2006, 27, 2725–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lala, A.; McNulty, S.E.; Mentz, R.J.; Dunlay, S.M.; Vader, J.M.; AbouEzzeddine, O.F.; DeVore, A.D.; Khazanie, P.; Redfield, M.M.; Goldsmith, S.R.; et al. Relief and recurrence of congestion during and after hospitalization for acute heart failure insights from diuretic optimization strategy evaluation in acute decompensated heart failure (DOSE-AHF) and cardiorenal rescue study in acute decompensated heart. Circ. Heart Fail. 2015, 8, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Fonarow, G.C.; Adams, K.F.; Abraham, W.T.; Yancy, C.W.; Boscardin, W.J. Risk stratification for in-hospital mortality in acutely decompensated heart failure. J. Am. Med. Assoc. 2005, 293, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, C.; Bellasi, A.; Di Lullo, L. Implication of Acute Kidney Injury in Heart Failure. Heart Fail. Clin. 2019, 15, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Al-Naher, A.; Wright, D.; Devonald, M.A.J.; Pirmohamed, M. Renal function monitoring in heart failure – what is the optimal frequency? A narrative review. Br. J. Clin. Pharmacol. 2018, 84, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Virzì, G.M.; Clementi, A.; Battaglia, G.G.; Ronco, C. Multi-Omics Approach: New Potential Key Mechanisms Implicated in Cardiorenal Syndromes. Cardiorenal Med. 2019, 9, 201–211. [Google Scholar] [CrossRef]
- Pedroza-Díaz, J.; Röthlisberger, S. Advances in urinary protein biomarkers for urogenital and non-urogenital pathologies. Biochem. Med. 2015, 25, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Benabdelkamel, H.; Masood, A.; Ekhzaimy, A.A.; Alfadda, A.A. Proteomics profiling of the urine of patients with hyperthyroidism after anti-thyroid treatment. Molecules 2021, 26, 1991. [Google Scholar] [CrossRef]
- Levent, P.; Kocaturk, M.; Akgun, E.; Saril, A.; Cevik, O.; Baykal, A.T.; Tanaka, R.; Ceron, J.J.; Yilmaz, Z. Platelet proteome changes in dogs with congestive heart failure. BMC Vet. Res. 2020, 16, 1–13. [Google Scholar] [CrossRef]
- Petra, E.; He, T.; Lygirou, V.; Latosinska, A.; Mischak, H.; Vlahou, A.; Jankowski, J. Urine peptidome analysis in cardiorenal syndrome reflects molecular processes. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ura, B.; Biffi, S.; Monasta, L.; Arrigoni, G.; Battisti, I.; Di Lorenzo, G.; Romano, F.; Aloisio, M.; Celsi, F.; Addobbati, R.; et al. Two dimensional-difference in gel electrophoresis (2D-DIGE) proteomic approach for the identification of biomarkers in endometrial cancer serum. Cancers 2021, 13, 3639. [Google Scholar] [CrossRef] [PubMed]
- Cubedo, J.; Padró, T.; García-Moll, X.; Pintó, X.; Cinca, J.; Badimon, L. Serum proteome in acute myocardial infarction. Clin. Investig. Arterioscler. 2011, 23, 147–154. [Google Scholar] [CrossRef]
- Cubedo, J.; Padró, T.; Badimon, L. Coordinated proteomic signature changes in immune response and complement proteins in acute myocardial infarction: The implication of serum amyloid P-component. Int. J. Cardiol. 2013, 168, 5196–5204. [Google Scholar] [CrossRef] [PubMed]
- Chiva-Blanch, G.; Peña, E.; Cubedo, J.; García-Arguinzonis, M.; Pané, A.; Gil, P.A.; Perez, A.; Ortega, E.; Padró, T.; Badimon, L. Molecular mapping of platelet hyperreactivity in diabetes: The stress proteins complex HSPA8/Hsp90/CSK2α and platelet aggregation in diabetic and normal platelets. Transl. Res. 2021, 235, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Akashi, M.; Minami, Y.; Haruki, S.; Jujo, K.; Hagiwara, N. Prognostic implications of prealbumin level on admission in patients with acute heart failure referred to a cardiac intensive care unit. J. Cardiol. 2019, 73, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Franco, J.; Formiga, F.; Trullas, J.C.; Salamanca Bautista, P.; Conde, A.; Manzano, L.; Quirós, R.; Franco, Á.G.; Ezquerro, A.M.; Montero-Pérez-Barquero, M. Impact of prealbumin on mortality and hospital readmission in patients with acute heart failure. Eur. J. Intern. Med. 2017, 43, 36–41. [Google Scholar] [CrossRef]
- Wang, W.; Wang, C.S.; Ren, D.; Li, T.; Yao, H.C.; Ma, S.J. Low serum prealbumin levels on admission can independently predict in-hospital adverse cardiac events in patients with acute coronary syndrome. Medicine 2018, 97, 1–5. [Google Scholar] [CrossRef]
- Li, O.; Geng, X.; Ma, Q.; Wang, W.; Liu, R.; Yin, Z.; Wang, S.; Cai, G.; Chen, X.; Hong, Q. Gene Microarray Integrated with High-Throughput Proteomics for the Discovery of Transthyretin in Rhabdomyolysis-Induced Acute Kidney Injury. Cell. Physiol. Biochem. 2017, 43, 1673–1688. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, L.H.; Ingenbleek, Y. Transthyretin: Its response to malnutrition and stress injury. Clinical usefulness and economic implications. Clin. Chem. Lab. Med. 2002, 40, 1344–1348. [Google Scholar] [CrossRef]
- Dellière, S.; Cynober, L. Is transthyretin a good marker of nutritional status? Clin. Nutr. 2017, 36, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Harpole, M.; Davis, J.; Espina, V. Current state of the art for enhancing urine biomarker discovery Current state of the art for enhancing urine biomarker discovery. Expert Rev. Proteom. 2016, 9450, 609–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wang, W.; Chen, Z.; Xu, F.; Zheng, Y. Proteomics applications in biomarker discovery and pathogenesis for abdominal aortic aneurysm. Expert Rev. Proteom. 2021, 18, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Laribi, S.; Mebazaa, A. Cardiohepatic syndrome: Liver injury in decompensated heart failure. Curr. Heart Fail. Rep. 2014, 11, 236–240. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Kellum, J.A.; Haase, M.; Müller, C.; Damman, K.; Murray, P.T.; Cruz, D.; House, A.A.; Schmidt-Ott, K.M.; Vescovo, G.; et al. Pathophysiology of the cardiorenal syndromes: Executive summary from the eleventh consensus conference of the acute dialysis quality initiative (ADQI). Contrib. Nephrol. 2013, 182, 82–98. [Google Scholar] [CrossRef]
- Biegus, J.; Zymliński, R.; Sokolski, M.; Siwołowski, P.; Gajewski, P.; Nawrocka-Millward, S.; Poniewierka, E.; Jankowska, E.A.; Banasiak, W.; Ponikowski, P. Impaired hepato-renal function defined by the MELD XI score as prognosticator in acute heart failure. Eur. J. Heart Fail. 2016, 18, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Kawahira, M.; Tamaki, S.; Yamada, T.; Watanabe, T.; Morita, T.; Furukawa, Y.; Kawasaki, M.; Kikuchi, A.; Kawai, T.; Seo, M.; et al. Prognostic value of impaired hepato-renal function and liver fibrosis in patients admitted for acute heart failure. ESC Heart Fail. 2021, 8, 1274–1283. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Dufour, J.F.; Petta, S.; Romero-Gomez, M.; Allison, M.; Oliveira, C.P.; Francque, S.; Van Gaal, L.; Schattenberg, J.M.; et al. Age as a Confounding Factor for the Accurate Non-Invasive Diagnosis of Advanced NAFLD Fibrosis. Am. J. Gastroenterol. 2017, 112, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Panchani, N.; Schulz, P.; Van Zyl, J.; Felius, J.; Baxter, R.; Yoon, E.T.; Baldawi, H.; Bindra, A.; Asrani, S.K. Liver stiffness and prediction of cardiac outcomes in patients with acute decompensated heart failure. Clin. Transplant. 2021, e14545. [Google Scholar] [CrossRef]
- Santander, C.; Brandan, E. Betaglycan induces TGF-β signaling in a ligand-independent manner, through activation of the p38 pathway. Cell. Signal. 2006, 18, 1482–1491. [Google Scholar] [CrossRef]
- Liu, C.; Lim, S.T.; Teo, M.H.Y.; Tan, M.S.Y.; Kulkarni, M.D.; Qiu, B.; Li, A.; Lal, S.; Dos Remedios, C.G.; Tan, N.S.; et al. Collaborative Regulation of LRG1 by TGF-β1 and PPAR-β/δ Modulates Chronic Pressure Overload-Induced Cardiac Fibrosis. Circ. Heart Fail. 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Sörensen-Zender, I.; Bhayana, S.; Susnik, N.; Rolli, V.; Batkai, S.; Baisantry, A.; Bahram, S.; Sen, P.; Teng, B.; Lindner, R.; et al. Zinc-a2-Glycoprotein Exerts Antifibrotic Effects in Kidney and Heart. J. Am. Soc. Nephrol. 2015, 26, 2659–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeropoulos, A.P.; Tang, W.H.W.; Hsu, A.; Felker, G.M.; Hernandez, A.F.; Troughton, R.W.; Voors, A.A.; Anker, S.D.; Metra, M.; McMurray, J.J.V.; et al. High-sensitivity C-reactive protein in acute heart failure: Insights from the ASCEND-HF trial. J. Card. Fail. 2014, 20, 319–326. [Google Scholar] [CrossRef]
- Pascual-Figal, D.A.; Bayes-Genis, A.; Asensio-Lopez, M.C.; Hernández-Vicente, A.; Garrido-Bravo, I.; Pastor-Perez, F.; Díez, J.; Ibáñez, B.; Lax, A. The Interleukin-1 Axis and Risk of Death in Patients With Acutely Decompensated Heart Failure. J. Am. Coll. Cardiol. 2019, 73, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, M.; Obermeier, F.; Schreiter, K.; Spottl, T.; Falk, W.; Schölmerich, J.; Herfarth, H.; Saftig, P.; Rogler, G. Cathepsin D is up-regulated in inflammatory bowel disease macrophages. Clin. Exp. Immunol. 2004, 136, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Voors, A.A.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; van der Harst, P.; Hillege, H.L.; Lang, C.C.; ter Maaten, J.M.; Ng, L.; et al. A systems BIOlogy Study to TAilored Treatment in Chronic Heart Failure: Rationale, design, and baseline characteristics of BIOSTAT-CHF. Eur. J. Heart Fail. 2016, 18, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Hoes, M.F.; Tromp, J.; Ouwerkerk, W.; Bomer, N.; Oberdorf-Maass, S.U.; Samani, N.J.; Ng, L.L.; Lang, C.C.; van der Harst, P.; Hillege, H.; et al. The role of cathepsin D in the pathophysiology of heart failure and its potentially beneficial properties: A translational approach. Eur. J. Heart Fail. 2020, 22, 2102–2111. [Google Scholar] [CrossRef]
- Chen, Q.; Yin, Q.; Song, J.; Liu, C.; Chen, H.; Li, S. Identification of monocyte-associated genes as predictive biomarkers of heart failure after acute myocardial infarction. BMC Med. Genomics 2021, 14, 1–13. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Jin, Y.P.; Qin, G.M.; Wang, J.H. Association of platelet-monocyte aggregates with platelet activation, systemic inflammation, and myocardial injury in patients with non-ST elevation acute coronary syndromes. Clin. Cardiol. 2007, 30, 26–31. [Google Scholar] [CrossRef]
- Peacock, W.F.; De Marco, T.; Fonarow, G.C.; Diercks, D.; Wynne, J.; Apple, F.S.; Wu, A.H.B. Cardiac Troponin and Outcome in Acute Heart Failure. N. Engl. J. Med. 2008, 358, 2117–2126. [Google Scholar] [CrossRef] [Green Version]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin amyloid cardiomyopathy: JACC State of the Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, H.; Zhang, L.; Cao, Y.; Bao, J.Z.; Liu, Z.X.; Wang, L.S.; Yang, Q.; Lu, X. Retinol-binding protein 4 induces cardiomyocyte hypertrophy by activating TLR4/MyD88 pathway. Endocrinology 2016, 157, 2282–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majerczyk, M.; Choręza, P.; Mizia-Stec, K.; Bożentowicz-Wikarek, M.; Brzozowska, A.; Arabzada, H.; Owczarek, A.J.; Szybalska, A.; Grodzicki, T.; Więcek, A.; et al. Plasma Level of Retinol-Binding Protein 4, N-Terminal proBNP and Renal Function in Older Patients Hospitalized for Heart Failure. Cardiorenal Med. 2018, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Bobbert, P.; Weithäuser, A.; Andres, J.; Bobbert, T.; Kühl, U.; Schultheiss, H.P.; Rauch, U.; Skurk, C. Increased plasma retinol binding protein 4 levels in patients with inflammatory cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 1163–1168. [Google Scholar] [CrossRef]
- Leite, A.R.; Neves, J.S.; Borges-Canha, M.; Vale, C.; Von Hafe, M.; Carvalho, D.; Leite-Moreira, A. Evaluation of thyroid function in patients hospitalized for acute heart failure. Int. J. Endocrinol. 2021, 2021, 6616681. [Google Scholar] [CrossRef]
- Schmidt-Ott, U.M.; Ascheim, D.D. Thyroid Hormone and Heart Failure. Curr. Heart Fail. Rep. 2006, 3, 114–119. [Google Scholar] [CrossRef]
- Cubedo, J.; Padró, T.; Alonso, R.; Cinca, J.; Mata, P.; Badimon, L. Differential proteomic distribution of TTR (pre-albumin) forms in serum and HDL of patients with high cardiovascular risk. Atherosclerosis 2012, 222, 263–269. [Google Scholar] [CrossRef]
- Chaikijurajai, T. Wilson Reappraisal of Inflammatory Biomarkers in Heart Failure. Curr Heart Fail Rep. 2020, 17, 9–19. [Google Scholar] [CrossRef]
- Johnson, A.M.; Merlini, G.; Sheldon, J.; Ichihara, K. Clinical indications for plasma protein assays: Transthyretin (prealbumin) in inflammation and malnutrition - International federation of clinical chemistry and laboratory medicine (IFCC): IFCC scientific division committee on plasma proteins (C-PP). Clin. Chem. Lab. Med. 2007, 45, 419–426. [Google Scholar] [CrossRef]
- Clark, M.A.; Hentzen, B.T.H.; Plank, L.D.; Hill, G.L. Sequential changes in insulin-like growth factor 1, plasma proteins, and total body protein in severe sepsis and multiple injury. J. Parenter. Enter. Nutr. 1996, 20, 363–370. [Google Scholar] [CrossRef]
- Greve, A.M.; Christoffersen, M.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Association of Low Plasma Transthyretin Concentration with Risk of Heart Failure in the General Population. JAMA Cardiol. 2021, 6, 258–266. [Google Scholar] [CrossRef]
- Judge, D.P.; Heitner, S.B.; Falk, R.H.; Maurer, M.S.; Shah, S.J.; Witteles, R.M.; Grogan, M.; Selby, V.N.; Jacoby, D.; Hanna, M.; et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Porcari, A.; Merlo, M.; Rapezzi, C.; Sinagra, G. Transthyretin amyloid cardiomyopathy: An uncharted territory awaiting discovery. Eur. J. Intern. Med. 2020, 82, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Raghu, P.; Sivakumar, B. Interactions amongst plasma retinol-binding protein, transthyretin and their ligands: Implications in vitamin A homeostasis and transthyretin amyloidosis. Biochim. Biophys. Acta Proteins Proteom. 2004, 1703, 1–9. [Google Scholar] [CrossRef]
- Perduca, M.; Nicolis, S.; Mannucci, B.; Galliano, M.; Monaco, H.L. Human plasma retinol-binding protein (RBP4) is also a fatty acid-binding protein. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Tarwater, K. Estimated glomerular filtration rate explained. Mo. Med. 2011, 108, 29–32. [Google Scholar] [CrossRef]
- Cubedo, J.; Padró, T.; Alonso, R.; Mata, P.; Badimon, L. ApoL1 levels in high density lipoprotein and cardiovascular event presentation in patients with familial hypercholesterolemia. J. Lipid Res. 2016, 57, 1059–1073. [Google Scholar] [CrossRef] [Green Version]
- García-Arguinzonis, M.; Padró, T.; Lugano, R.; Llorente-Cortes, V.; Badimon, L. Low-density lipoproteins induce heat shock protein 27 dephosphorylation, oligomerization, and subcellular relocalization in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1212–1219. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with PANTHER Classification System. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Jupe, S.; Fabregat, A.; Hermjakob, H. Expression data analysis with Reactome. Curr. Protoc. Bioinform. 2015, 49, 8.20.1–8.20.9. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Protein name | Gene name | Swiss Prot number | Up/down regulation | Experimental pI | Molecular weight (KDa) | MS or MS/MS | MASCOT Score | Coverage |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Lysosomal acid phosphatase | ACP2 | P11117 | ↓ | 5.80 | 45.5 | MS | 63 | 7 |
| 2 | Pancreatic α-amylase | AMY2A | P04746 | ↓ | 6.45 | 51.6 | MS/MS | 61 | - |
| 3 | Annexin A10 | ANXA10 | Q9UJ72 | ↓ | 5.20 | 35.2 | MS | 60 | 19 |
| 4 | Arylsulfatase A | ARSA | P15289 | ↑ | 5.50 | 49.6 | MS | 67 | 10 |
| 5 | Zinc-α-2-glycoprotein | AZGP1 | P25311 | ↑ | 4.8-5.1 | 41.6-43.5 | MS | 66 | 11 |
| 6 | Complement C3 | C3 | P01024 | ↑ | 6.75 | 54.8 | MS | 62 | 3 |
| 7 | Carbonic anhydrase 1 | CA1 | P00915 | ↑ | 6.70 | 30.1 | MS/MS | 71 | - |
| 8 | Endosialin | CD248 | Q9HCU0 | ↑ | 4.70 | 44.6-45.6 | MS/MS | 54 | |
| 9 | CD59 glycoprotein | CD59 | P13987 | ↑ | 4.90 | 22.7 | MS/MS | 82 | - |
| 10 | Cathepsin D | CTSD | P07339 | ↑ | 5.40 | 31.2 | MS/MS | 58 | - |
| 11 | Fibrinogen β-chain | FGB | P02675 | ↓ | 4.90 | 18.7-19.8 | MS/MS | 55 | |
| 12 | Fibrinogen γ-chain | FGG | P02679 | ↑ | 5.30-5.35 | 48.0-48.2 | MS/MS | 55 | - |
| 13 | Vitamin D binding protein | GC | P02774 | ↑ | 5.20 | 50.5 | MS | 94 | 17 |
| 14 | Hemopexin | HPX | P02790 | ↑ | 5.30-5.35 | 55.4 | MS/MS | 70 | |
| 15 | Basement membrane-specific heparan sulfate proteoglycan core protein | HSPG2 | P98160 | ↓ | 5.40 | 24.9 | MS/MS | 102 | - |
| 16 | Inter-alpha-trypsin inhibitor heavy chain H4 | ITIH4 | Q14624 | ↓ | 4.9-5.1 | 36.6-37.2 | MS | 74 | 10 |
| 17 | Kininogen-1 | KNG1 | P01042 | ↓ | 4.7-4.9 | 50.5-53.3 | MS | 63 | 8 |
| 18 | Vesicular integral-membrane protein VIP36 | LMAN2 | Q12907 | ↓ | 5.20 | 35.2 | MS/MS | 60 | - |
| 19 | Leucine-rich alpha-2-glycoprotein | LRG1 | P02750 | ↑ | 4.60 | 47.4 | MS/MS | 79 | - |
| 20 | Retinol binding protein | RBP4 | P02753 | ↓ | 5.20 | 24.7-25.2 | MS/MS | 66 | - |
| 21 | Alpha-1-antitrypsin | SERPINA1 | P01009 | ↑ | 5.00-5.10 | 51.4-52.2 | MS | 130 | 19 |
| 22 | Antithrombin III | SERPINC1 | P01008 | ↑ | 5.20 | 52.6 | MS/MS | 78 | - |
| 23 | Serotransferrin | TF | P02787 | ↑ | 6.00-6.40 | 56.5 | MS | 234 | 26 |
| 24 | Trefoil factor 2 | TFF2 | Q03403 | ↓ | 5.20 | 11.1 | MS/MS | 83 | - |
| 25 | Transthyretin | TTR | P02766 | ↑ | 5.30 | 15.9 | MS/MS | 61 | - |
| 26 | Vitelline membrane outer layer protein 1 homolog | VMO1 | Q7Z5L0 | ↓ | 4.65 | 21.2 | MS/MS | 87 | - |
| Protein a | Gene | HS (N=6) | ADHF (N=17) | p Value b | Q Value c |
|---|---|---|---|---|---|
| 1 | ACP2 | 0.89 [0.51–1.10] | 0.37 [0.24–0.42] | 0.049 | 0.046 |
| 2 | AMY2A | 1.07 [0.92–1.10] | 0.47 [0.31–1.18] | 0.089 | 0.060 |
| 3 | ANXA10 | 0.55 [0.45–0.85] | 0.33 [0.02–0.54] | 0.077 | 0.056 |
| 4 | ARSA | 0.02 [0.001–0.04] | 0.10 [0.06–0.20] | 0.017 | 0.028 |
| 5 | AZGP1 | 2.47 [1.61–2.81] | 8.08 [4.92–12.94] | 0.001 | 0.010 |
| 6 | C3 | 0.24 [0.22–0.81] | 0.89 [0.36–1.41] | 0.083 | 0.058 |
| 7 | CA1 | 0.002 [0.001–0.05] | 0.27 [0.18–0.79] | 0.010 | 0.027 |
| 8 | CD248 | 0.18 [0.15–0.19] | 1.12 [0.52–1.65] | 0.003 | 0.013 |
| 9 | CD59 | 0.47 [0.17–1.12] | 1.68 [1.07–2.39] | 0.027 | 0.038 |
| 10 | CTSD | 0.21 [0.16–0.33] | 0.90 [0.21–1.36] | 0.052 | 0.046 |
| 11 | FGB | 3.23 [2.60–3.87] | 2.11 [1.57–2.78] | 0.210 | 0.136 |
| 12 | FGG | 0.19 [0.02–0.28] | 0.58 [0.15–1.75] | 0.062 | 0.051 |
| 13 | GC | 0.77 [0.66–0.98] | 1.71 [0.87–2.55] | 0.042 | 0.044 |
| 14 | HPX | 1.00 [0.63–1.41] | 2.05 [1.00–2.97] | 0.048 | 0.046 |
| 15 | HSPG2 | 3.96 [2.27–6.47] | 1.13 [0.67–2.40] | 0.015 | 0.028 |
| 16 | ITIH4 | 10.66 [7.82–14.06] | 4.25 [2.48–8.04] | 0.006 | 0.021 |
| 17 | KNG1 | 26.14 [18.40–35.01] | 8.73 [3.82–16.11] | 0.003 | 0.013 |
| 18 | LMAN2 | 0.34 [0.28–0.48] | 0.22 [0.07–0.35] | 0.077 | 0.056 |
| 19 | LRG1 | 0.09 [0.07–0.10] | 0.87 [0.35–1.26] | 0.014 | 0.028 |
| 20 | RBP4 | 1.56 [1.41–1.68] | 0.86 [0.71–1.16] | 0.033 | 0.040 |
| 21 | SERPINA1 | 1.58 [1.23–1.83] | 4.53 [3.13–7.41] | 0.013 | 0.028 |
| 22 | SERPINC1 | 0.15 [0.11–0.19] | 0.47 [0.20–0.63] | 0.034 | 0.040 |
| 23 | TF | 1.22 [0.97–1.81] | 7.31 [4.56–7.78] | 0.001 | 0.010 |
| 24 | TFF2 | 0.36 [0.002–0.41] | 0.002 [0.001–0.06] | 0.064 | 0.051 |
| 25 | TTR | 0.40 [0.39–0.67] | 0.88 [0.66–1.45] | 0.031 | 0.040 |
| 26 | VMO1 | 4.03 [2.60–4.57] | 1.74 [1.20–3.17] | 0.023 | 0.035 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Riera, E.; García-Arguinzonis, M.; López, L.; Garcia-Moll, X.; Badimon, L.; Padro, T. Urinary Proteomic Signature in Acute Decompensated Heart Failure: Advances into Molecular Pathophysiology. Int. J. Mol. Sci. 2022, 23, 2344. https://doi.org/10.3390/ijms23042344
Diaz-Riera E, García-Arguinzonis M, López L, Garcia-Moll X, Badimon L, Padro T. Urinary Proteomic Signature in Acute Decompensated Heart Failure: Advances into Molecular Pathophysiology. International Journal of Molecular Sciences. 2022; 23(4):2344. https://doi.org/10.3390/ijms23042344
Chicago/Turabian StyleDiaz-Riera, Elisa, Maísa García-Arguinzonis, Laura López, Xavier Garcia-Moll, Lina Badimon, and Teresa Padro. 2022. "Urinary Proteomic Signature in Acute Decompensated Heart Failure: Advances into Molecular Pathophysiology" International Journal of Molecular Sciences 23, no. 4: 2344. https://doi.org/10.3390/ijms23042344
APA StyleDiaz-Riera, E., García-Arguinzonis, M., López, L., Garcia-Moll, X., Badimon, L., & Padro, T. (2022). Urinary Proteomic Signature in Acute Decompensated Heart Failure: Advances into Molecular Pathophysiology. International Journal of Molecular Sciences, 23(4), 2344. https://doi.org/10.3390/ijms23042344