
Aβ-Induced Alterations in Membrane Lipids Occur before Synaptic Loss Appears

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. Primary Hippocampal Neurone Cultures
2.3. Primary Glial Cell Cultures
2.4. Aβ Treatment
2.5. Synaptic Study
Immunofluorescence
2.6. Lipidomic Study
2.6.1. Samples Collection for Lipid Analysis
2.6.2. Lipid Extraction
2.6.3. High-Performance Thin-Layer Chromatography
2.6.4. Tandem Mass Spectrometry
2.7. Data Analysis
3. Results
3.1. Synaptic Loss after Aβ(1–40) and Aβ(1–42) Treatment
3.2. Alterations in Cellular Lipids of Hippocampal Neurones and Glial Cells after Aβ(1–40) and Aβ(1–42) Treatment
3.2.1. Disruption in Cellular Lipids of Hippocampal Neurones and Glial Cells after Aβ(1–40) and Aβ(1–42) Treatment Using High-Performance Thin-Layer Chromatography (HPTLC)
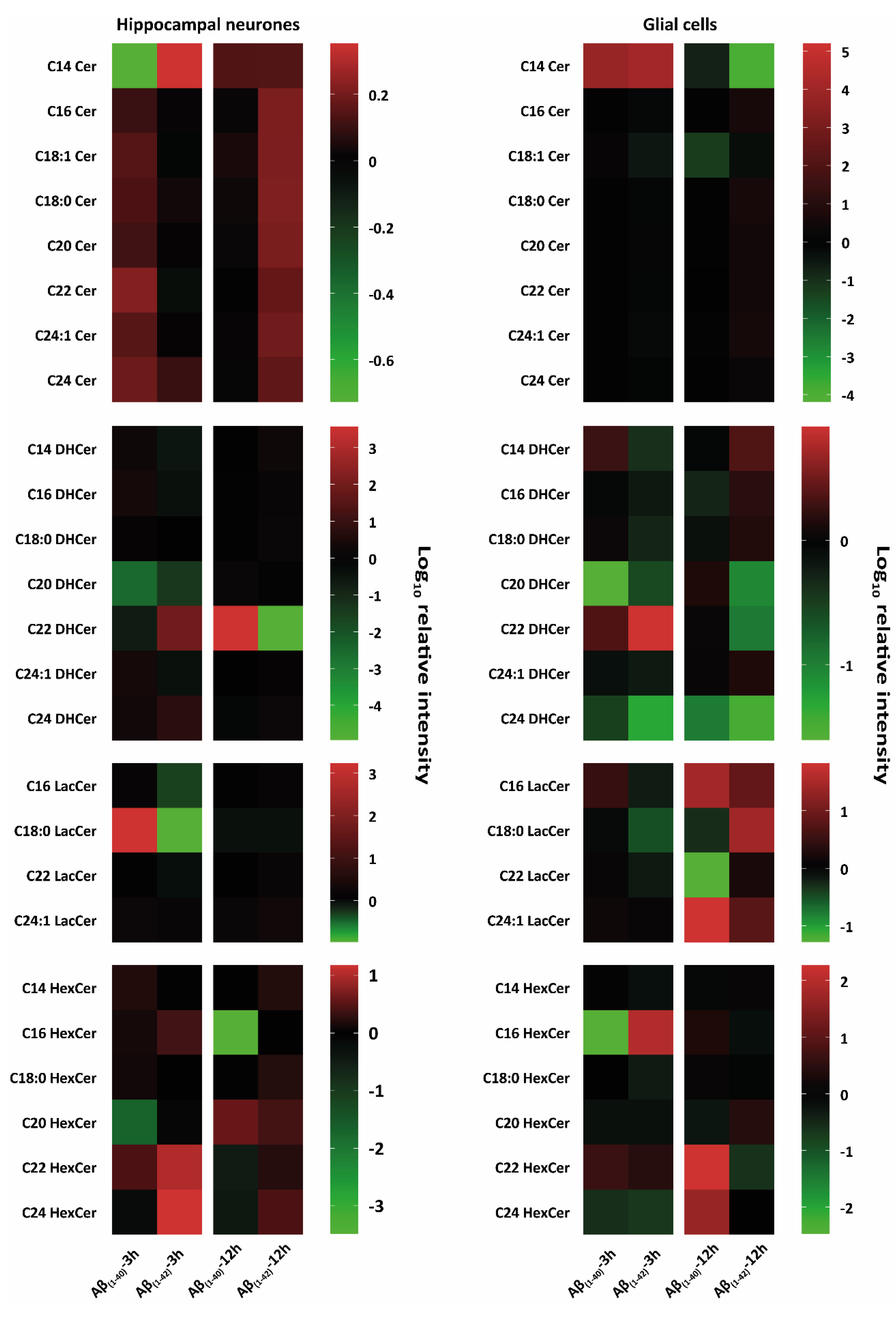
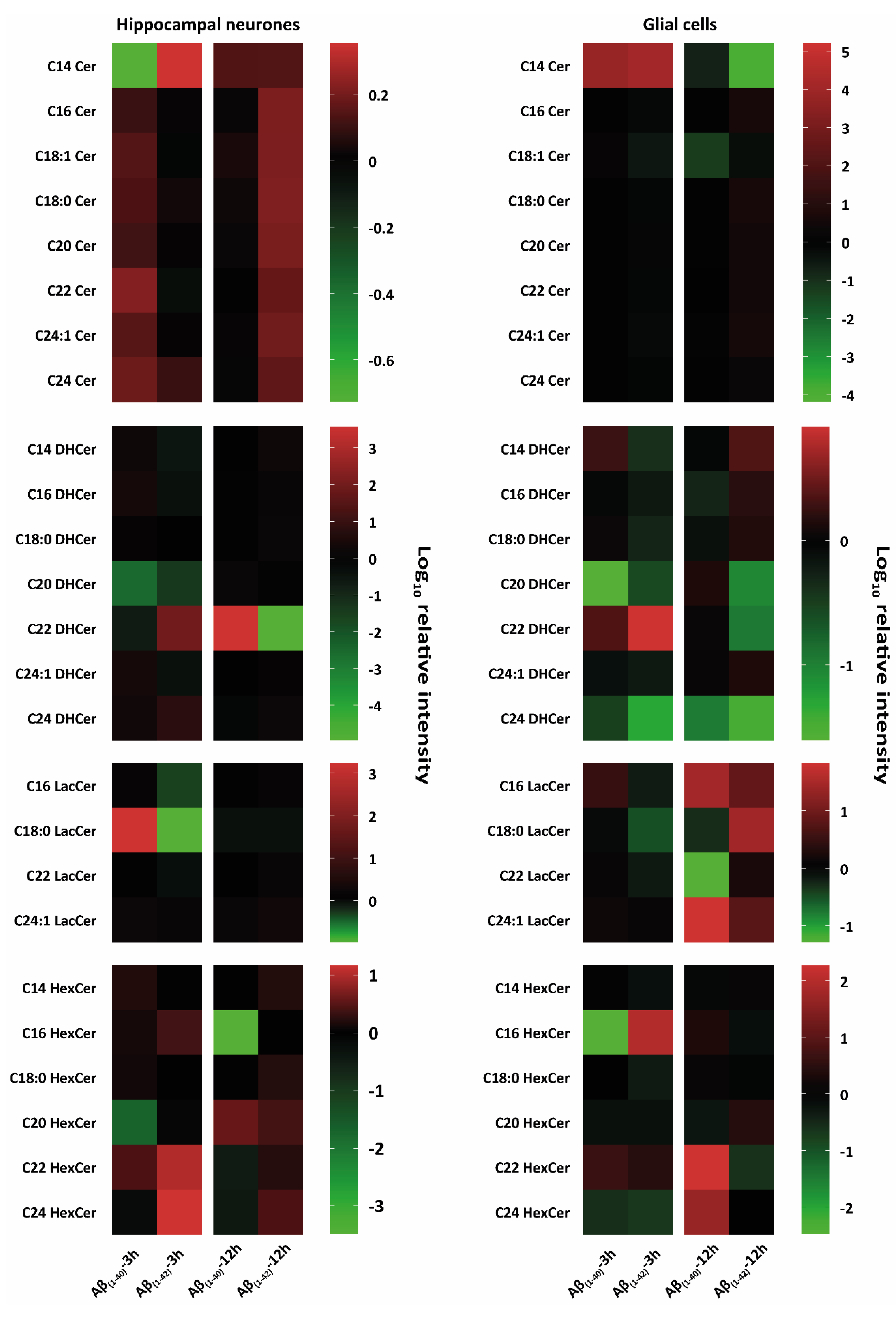
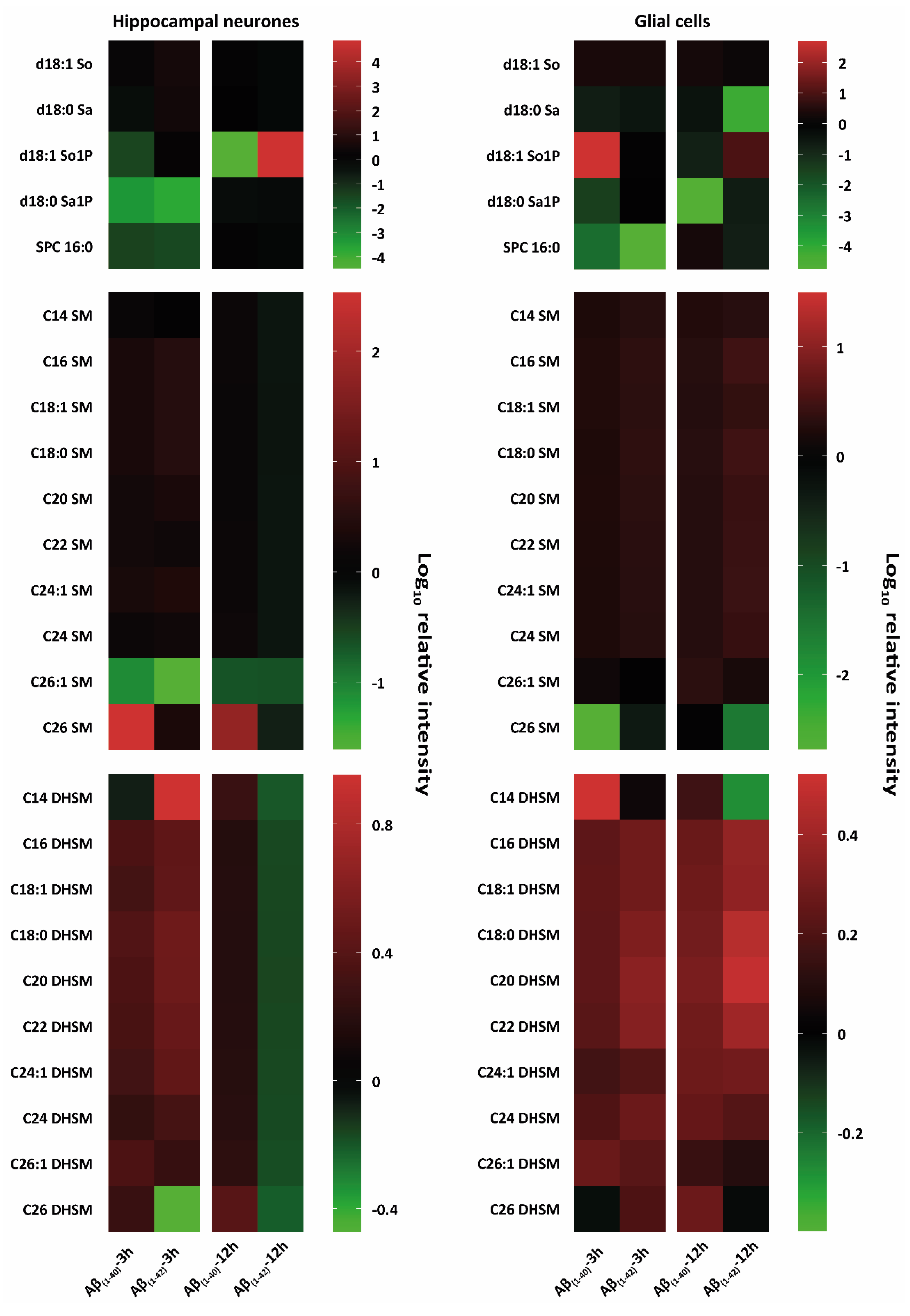
3.2.2. Specific Identification of Various Lipid Isoforms after Aβ(1–40) and Aβ(1–42) Treatment from Hippocampal Neurones and Glial Cells Using Tandem Mass Spectrometry Analysis
Ceramides (Cer)
Dihydroceramides (DHCer)
Lactosylceramides (LacCer)
Monohexosylceramides (HexCer)
Sphingosine (So) and Sphingosine-1-Phosphate (So1P)
Sphinganine (Sa) and Sphinganine-1-Phosphate (Sa1P)
Sphingosylphosphorylcholine (SPC)
Sphingomyelines (SM)
Dihydrosphingomyelines (DHSM)
Phosphatidylcholines (PC) and Lyso-Phosphatidylcholines (LPC)
Lyso-Phosphatidylethanolamine (LPE)
Lyso-Phosphatidylglycerol (LPG)
Lyso-Phosphatidylserine (LPS)
Lyso-Platelet-Activating Factor (Lyso-PAF)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| POPC | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DMPG | 1,2-Dimyristoyl-sn-glycero-3-phosphoglycerol |
| nSMase | Neutral isoform sphingomyelinase |
| aSMase | Acid sphingomyelinase |
References
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid beta peptides. J. Physiol. 2006, 575, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sosa, L.J.; Cáceres, A.; Dupraz, S.; Oksdath, M.; Quiroga, S.; Lorenzo, A. The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. J. Neurochem. 2017, 143, 11–29. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Deciphering the Molecular Basis of Memory Failure in Alzheimer’s Disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Schultz, C.; Dehghani, F.; Yamaguchi, H.; Braak, H.; Braak, E. Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol. 2000, 100, 608–617. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Hayashi, M.K.; Tang, C.; Verpelli, C.; Narayanan, R.; Stearns, M.H.; Xu, R.-M.; Li, H.; Sala, C.; Hayashi, Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 2009, 137, 159–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Ferrera, P.; Arias, C. Amyloid-β protein modulates insulin signaling in presynaptic terminals. Neurochem. Res. 2012, 37, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Sokolow, S.; Luu, S.H.; Nandy, K.; Miller, C.A.; Vinters, H.V.; Poon, W.W.; Gylys, K.H. Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer’s disease cortex. Neurobiol. Dis. 2012, 45, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Sekino, Y.; Tanaka, S.; Mizui, T.; Kishi, S.; Shirao, T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 6586–6595. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Almeida, C.G.; Kearney, P.F.; Yu, F.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 3592–3599. [Google Scholar] [CrossRef]
- Klementieva, O.; Willén, K.; Martinsson, I.; Israelsson, B.; Engdahl, A.; Cladera, J.; Uvdal, P.; Gouras, G.K. Pre-plaque conformational changes in Alzheimer’s disease-linked Aβ and APP. Nat. Commun. 2017, 8, 14726. [Google Scholar] [CrossRef]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurother. J. Am. Soc. Exp. NeuroTher. 2015, 12, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.-M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Novo, M.; Freire, S.; Al-Soufi, W. Critical aggregation concentration for the formation of early Amyloid-β (1-42) oligomers. Sci. Rep. 2018, 8, 1783. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.O.W.; Grimm, H.S.; Pätzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschäpe, J.-A.; Strooper, B.D.; Müller, U.; Shen, J.; et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Huang, Y.; Li, B.; Gong, C.-X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Kim, C.; Jang, H.Y.; Mook-Jung, I. Inhibition of Cholesterol Biosynthesis Reduces γ-Secretase Activity and Amyloid-β Generation. J. Alzheimer’s Dis. JAD 2016, 51, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Ellis, B.C.; Saunders, A.J.; Kovacs, D.M. Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J. Biol. Chem. 2003, 278, 19777–19783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Korshavn, K.J.; Satriano, C.; Lin, Y.; Zhang, R.; Dulchavsky, M.; Bhunia, A.; Ivanova, M.I.; Lee, Y.-H.; La Rosa, C.; Lim, M.H.; et al. Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-β. J. Biol. Chem. 2017, 292, 4638–4650. [Google Scholar] [CrossRef] [Green Version]
- Niu, Z.; Zhao, W.; Zhang, Z.; Xiao, F.; Tang, X.; Yang, J. The molecular structure of Alzheimer β-amyloid fibrils formed in the presence of phospholipid vesicles. Angew. Chem. Int. Ed. Engl. 2014, 53, 9294–9297. [Google Scholar] [CrossRef]
- Willén, K.; Sroka, A.; Takahashi, R.H.; Gouras, G.K. Heterogeneous Association of Alzheimer’s Disease-Linked Amyloid-β and Amyloid-β Protein Precursor with Synapses. J. Alzheimer’s Dis. JAD 2017, 60, 511–524. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, J.R.; Chan, E.S.; Poore, C.P.; Pareek, T.K.; Cheong, W.F.; Shui, G.; Tang, N.; Low, C.-M.; Wenk, M.R.; Kesavapany, S. Cdk5/p25-Induced Cytosolic PLA2-Mediated Lysophosphatidylcholine Production Regulates Neuroinflammation and Triggers Neurodegeneration. J. Neurosci. 2012, 32, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Verstraelen, P.; Barriga, G.G.-D.; Verschuuren, M.; Asselbergh, B.; Nuydens, R.; Larsen, P.H.; Timmermans, J.-P.; De Vos, W.H. Systematic quantification of synapses in primary neuronal culture. Iscience 2020, 23, 101542. [Google Scholar] [CrossRef]
- de Ménorval, M.-A.; Mir, L.M.; Fernández, M.L.; Reigada, R. Effects of Dimethyl Sulfoxide in Cholesterol-Containing Lipid Membranes: A Comparative Study of Experiments In Silico and with Cells. PLoS ONE 2012, 7, e41733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bate, C.; Williams, A. Amyloid-β(1-40) inhibits amyloid-β(1-42) induced activation of cytoplasmic phospholipase A2 and synapse degeneration. J. Alzheimer’s Dis. JAD 2010, 21, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Sun, Y.; Guo, Y.; Chen, Y.; Yu, B.; Zhang, H.; Wu, J.; Yu, X.; Kong, W.; Wu, H. Comparison of neurotoxicity of different aggregated forms of Aβ40, Aβ42 and Aβ43 in cell cultures. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2017, 23, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 12, 1. [Google Scholar] [CrossRef]
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C. Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 1–10. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxidative Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.-L.; Xiu, J.; Shan, K.-R.; Xiao, Y.; Gu, R.; Liu, R.-Y.; Guan, Z.-Z. Oxidative stress induced by beta-amyloid peptide(1-42) is involved in the altered composition of cellular membrane lipids and the decreased expression of nicotinic receptors in human SH-SY5Y neuroblastoma cells. Neurochem. Int. 2005, 46, 613–621. [Google Scholar] [CrossRef]
- Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.-L.; Oster, T.; Pillot, T. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef]
- Zhang, Y.; Ranta, F.; Tang, C.; Shumilina, E.; Mahmud, H.; Föller, M.; Ullrich, S.; Häring, H.-U.; Lang, F. Sphingomyelinase dependent apoptosis following treatment of pancreatic beta-cells with amyloid peptides Abeta(1-42) or IAPP. Apoptosis Int. J. Program. Cell Death 2009, 14, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Michno, W.; Kaya, I.; Nyström, S.; Guerard, L.; Nilsson, K.P.R.; Hammarström, P.; Blennow, K.; Zetterberg, H.; Hanrieder, J. Multimodal Chemical Imaging of Amyloid Plaque Polymorphism Reveals Aβ Aggregation Dependent Anionic Lipid Accumulations and Metabolism. Anal. Chem. 2018, 90, 8130–8138. [Google Scholar] [CrossRef] [PubMed]
- Czubowicz, K.; Cieślik, M.; Pyszko, J.; Strosznajder, J.B.; Strosznajder, R.P. Sphingosine-1-Phosphate and Its Effect on Glucose Deprivation/Glucose Reload Stress: From Gene Expression to Neuronal Survival. Mol. Neurobiol. 2015, 51, 1300–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Jiang, X.; Zhu, Z.; Qin, H.; Dinkins, M.B.; Kong, J.-N.; Leanhart, S.; Wang, R.; Elsherbini, A.; Bieberich, E.; et al. Lipid transporter Spns2 promotes microglia pro-inflammatory activation in response to amyloid-beta peptide. Glia 2019, 67, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Lee, S.J.; Lee, J.; Myung, C.-S.; Park, W.-K.; Lim, H.-J.; Lee, G.H.; Kong, J.Y.; Cho, H. Sphingosylphosphorylcholine attenuated β-amyloid production by reducing BACE1 expression and catalysis in PC12 cells. Neurochem. Res. 2011, 36, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Lee, C.H. Role of Sphingosylphosphorylcholine in Tumor and Tumor Microenvironment. Cancers 2019, 11, 1696. [Google Scholar] [CrossRef] [Green Version]
- Moses, G.S.; Jensen, M.D.; Lue, L.-F.; Walker, D.G.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Secretory PLA2-IIA: A new inflammatory factor for Alzheimer’s disease. J. Neuroinflamm. 2006, 3, 28. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 h Treatment | 12 h Treatment | |
|---|---|---|
| Kruskal–Wallis test | 0.0732 | <0.0001 |
| Dunn’s multiple comparison test | adjusted p-value | adjusted p-value |
| CTRL vs. DMSO | >0.9999 | 0.6106 |
| CTRL vs. Aβ(1–40) | 0.3017 | <0.0001 |
| CTRL vs. Aβ(1–42) | 0.0867 | <0.0001 |
| DMSO vs. Aβ(1–40) | >0.9999 | <0.0001 |
| DMSO vs. Aβ(1–42) | >0.9999 | <0.0001 |
| Aβ(1–40) vs. Aβ(1–42) | >0.9999 | >0.9999 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Bulck, M.; Brandt, N.; Claus, R.A.; Gräler, M.; Bräuer, A.U. Aβ-Induced Alterations in Membrane Lipids Occur before Synaptic Loss Appears. Int. J. Mol. Sci. 2022, 23, 2300. https://doi.org/10.3390/ijms23042300
Van Bulck M, Brandt N, Claus RA, Gräler M, Bräuer AU. Aβ-Induced Alterations in Membrane Lipids Occur before Synaptic Loss Appears. International Journal of Molecular Sciences. 2022; 23(4):2300. https://doi.org/10.3390/ijms23042300
Chicago/Turabian StyleVan Bulck, Michiel, Nicola Brandt, Ralf A. Claus, Markus Gräler, and Anja U. Bräuer. 2022. "Aβ-Induced Alterations in Membrane Lipids Occur before Synaptic Loss Appears" International Journal of Molecular Sciences 23, no. 4: 2300. https://doi.org/10.3390/ijms23042300
APA StyleVan Bulck, M., Brandt, N., Claus, R. A., Gräler, M., & Bräuer, A. U. (2022). Aβ-Induced Alterations in Membrane Lipids Occur before Synaptic Loss Appears. International Journal of Molecular Sciences, 23(4), 2300. https://doi.org/10.3390/ijms23042300