
Tumor Necrosis Factor-α Induces a Preeclamptic-like Phenotype in Placental Villi via Sphingosine Kinase 1 Activation

 ,
, 
Abstract
1. Introduction
2. Results
2.1. SphK1 Expression in Placentas from Normal and PE Pregnancies
2.2. Experimental Villous Explant Model of Syncytial Self-Regeneration
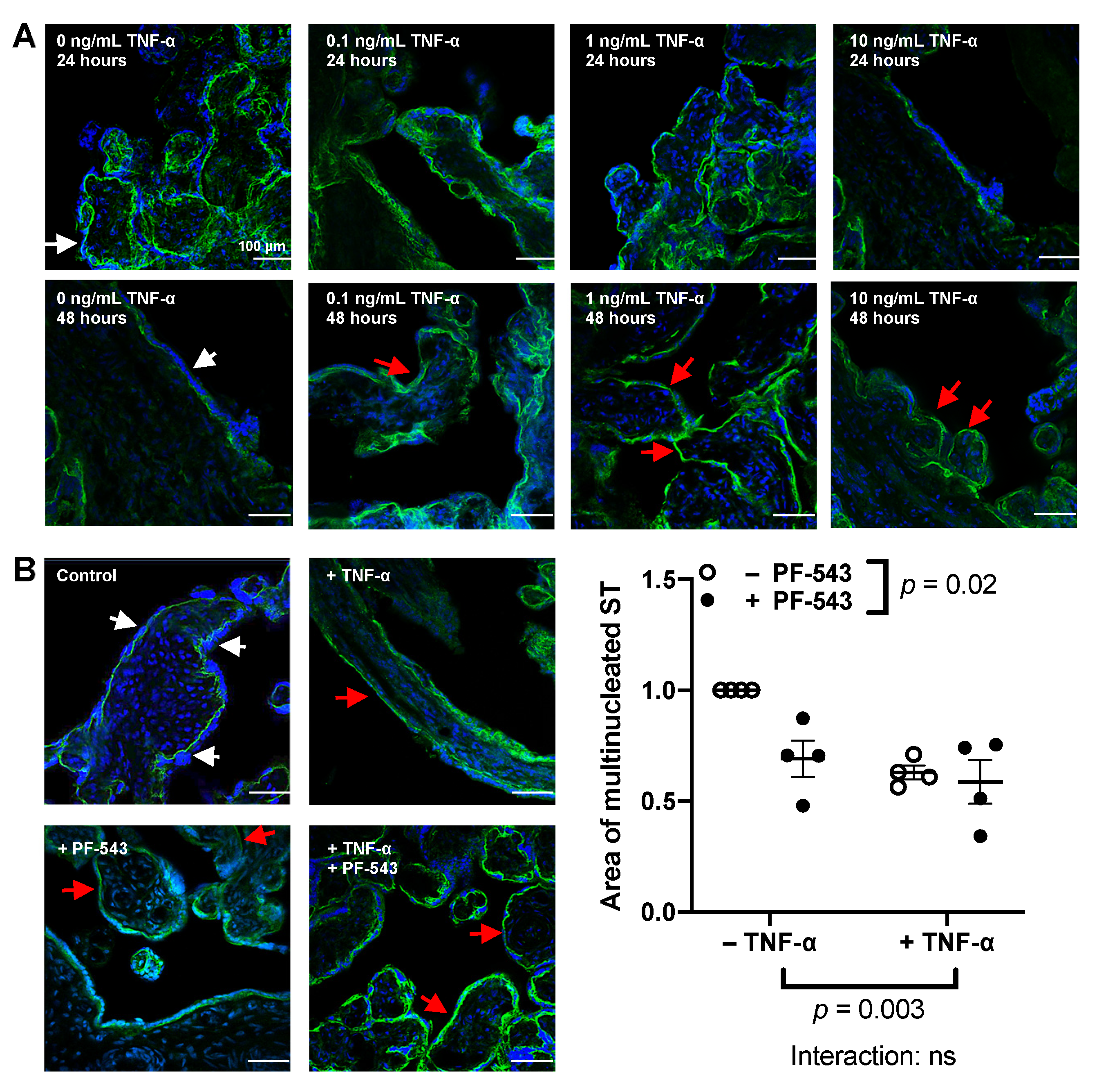
2.3. Effect of TNF-α on the Syncytium and its Interaction with SphK1
2.4. Effect of TNF-α on Placental Cytokine and Factor Release and Its Dependence on SphK1 Activity
2.5. PF-543 Dose Response in Villous Explants
3. Discussion
4. Materials and Methods
4.1. Placenta Samples
4.2. SphK1 Immunohistochemistry of Placental Sections
4.3. RNA Isolation
4.4. Quantitative RT-PCR
4.5. Placental Explant Culture
4.6. Treatments and Experimental Protocols
4.7. Trophoblast Fusion and E-Cadherin Staining
4.8. BCA Protein Assay
4.9. Lactate Dehydrogenase (LDH) Quantification
4.10. Quantification of the Beta-Subunit of Human Chorionic Gonadotropin (β-hCG)
4.11. Quantification of Human Placental Alkaline Phosphatase Activity (PLAP)
4.12. Quantification of Cytokines
4.13. Statistics and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harmon, Q.E.; Huang, L.; Umbach, D.M.; Klungsøyr, K.; Engel, S.M.; Magnus, P.; Skjærven, R.; Zhang, J.; Wilcox, A.J. Risk of Fetal Death With Preeclampsia. Obstet. Gynecol. 2015, 125, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Kuklina, E.V.; Ayala, C.; Callaghan, W.M. Hypertensive disorders and severe obstetric morbidity in the United States. Obstet. Gynecol. 2009, 113, 1299–1306. [Google Scholar] [CrossRef]
- Turco, M.Y.; Moffett, A. Development of the human placenta. Development 2019, 146, dev163428. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.S.; Hu, J.; Ren, C.E.; Chen, H.; Li, J.; Varvoutis, M.S.; Leaphart, L.W.; Byck, D.B.; Zhu, X.; Jiang, S.W. Morphological changes of placental syncytium and their implications for the pathogenesis of preeclampsia. Cell Mol. Life Sci. 2016, 73, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Lok, C.A.R.; Van der Post, J.A.M.; Sturk, A.; Sargent, I.L.; Nieuwland, R. The functions of microparticles in preeclampsia. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2011, 1, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Tannetta, D.S.; Dragovic, R.A.; Gardiner, C.; Redman, C.W.; Sargent, I.L. Characterisation of syncytiotrophoblast vesicles in normal pregnancy and pre-eclampsia: Expression of Flt-1 and endoglin. PLoS ONE 2013, 8, e56754. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Knofler, M. Human tumour necrosis factor: Physiological and pathological roles in placenta and endometrium. Placenta 2009, 30, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Shaarawy, M.; Nagui, A.R. Enhanced expression of cytokines may play a fundamental role in the mechanisms of immunologically mediated recurrent spontaneous abortion. Acta Obstet. Gynecol. Scand. 1997, 76, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Christiaens, I.; Zaragoza, D.B.; Guilbert, L.; Robertson, S.A.; Mitchell, B.F.; Olson, D.M. Inflammatory processes in preterm and term parturition. J. Reprod. Immunol. 2008, 79, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Alijotas-Reig, J.; Esteve-Valverde, E.; Ferrer-Oliveras, R.; Llurba, E.; Gris, J.M. Tumor Necrosis Factor-Alpha and Pregnancy: Focus on Biologics. An Updated and Comprehensive Review. Clin. Rev. Allergy Immunol. 2017, 53, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, N.S.; Thomson, S.E.; Heffernan, S.J.; Lim, S.; Thompson, J.; Ogle, R.; McKenzie, P.; Kirwan, P.J.; Makris, A.; Hennessy, A. Tumor necrosis factor alpha induces a model of preeclampsia in pregnant baboons (Papio hamadryas). Cytokine 2011, 56, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Leisser, C.; Saleh, L.; Haider, S.; Husslein, H.; Sonderegger, S.; Knöfler, M. Tumour necrosis factor-alpha impairs chorionic gonadotrophin beta-subunit expression and cell fusion of human villous cytotrophoblast. Mol. Hum. Reprod. 2006, 12, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Siwetz, M.; Blaschitz, A.; El-Heliebi, A.; Hiden, U.; Desoye, G.; Huppertz, B.; Gauster, M. TNF-α alters the inflammatory secretion profile of human first trimester placenta. Lab. Investig. 2016, 96, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Thathiah, A.; Brayman, M.; Dharmaraj, N.; Julian, J.J.; Lagow, E.L.; Carson, D.D. Tumor necrosis factor α stimulates MUC1 synthesis and ectodomain release in a human uterine epithelial cell line. Endocrinology 2004, 145, 4192–4203. [Google Scholar] [CrossRef]
- Yamashita, M.; Passegué, E. TNF-α coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell 2019, 25, 357–372.e357. [Google Scholar] [CrossRef]
- Annibaldi, A.; Meier, P. Checkpoints in TNF-induced cell death: Implications in inflammation and cancer. Trends Mol. Med. 2018, 24, 49–65. [Google Scholar] [CrossRef]
- Lankford, K.L.; Arroyo, E.J.; Kocsis, J.D. Chronic TNFα Exposure Induces Robust Proliferation of Olfactory Ensheathing Cells, but not Schwann Cells. Neurochem. Res. 2017, 42, 2595–2609. [Google Scholar] [CrossRef]
- Peng, W.C.; Logan, C.Y.; Fish, M.; Anbarchian, T.; Aguisanda, F.; Álvarez-Varela, A.; Wu, P.; Jin, Y.; Zhu, J.; Li, B. Inflammatory cytokine TNFα promotes the long-term expansion of primary hepatocytes in 3D culture. Cell 2018, 175, 1607–1619.e1615. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, J.; Zhao, J.; Hou, D.; Zhang, H.; Li, L.; Zou, D.; Hu, J.; Zhang, Y.; Jing, Z. MiR-18a-downregulated RORA inhibits the proliferation and tumorigenesis of glioma using the TNF-α-mediated NF-κB signaling pathway. EBioMedicine 2020, 52, 102651. [Google Scholar] [CrossRef]
- Li, X.; Ren, G.; Cai, C.; Yang, X.; Nie, L.; Jing, X.; Li, C. TNF-α regulates the osteogenic differentiation of bone morphogenetic factor 9 adenovirus-transduced rat follicle stem cells via Wnt signaling. Mol. Med. Rep. 2020, 22, 3141–3150. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Kanterman, J.; Ish-Shalom, E.; Elnekave, M.; Horwitz, E.; Baniyash, M. Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity 2013, 38, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Du, F.; Wang, X. TNF-alpha Induces Two Distinct Caspase-8 Activation Pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Romanowska-Próchnicka, K.; Felis-Giemza, A.; Olesińska, M.; Wojdasiewicz, P.; Paradowska-Gorycka, A.; Szukiewicz, D. The Role of TNF-α and Anti-TNF-α Agents during Preconception, Pregnancy, and Breastfeeding. Int. J. Mol. Sci. 2021, 22, 2922. [Google Scholar] [CrossRef]
- Park, E.S.; Choi, S.; Shin, B.; Yu, J.; Yu, J.; Hwang, J.M.; Yun, H.; Chung, Y.H.; Choi, J.S.; Choi, Y.; et al. Tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting protein (TRIP) negatively regulates the TRAF2 ubiquitin-dependent pathway by suppressing the TRAF2-sphingosine 1-phosphate (S1P) interaction. J. Biol. Chem. 2015, 290, 9660–9673. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Gamble, J.R.; Rye, K.A.; Wang, L.; Hii, C.S.; Cockerill, P.; Khew-Goodall, Y.; Bert, A.G.; Barter, P.J.; Vadas, M.A. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 14196–14201. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, L.; Gamble, J.R.; Vadas, M.A. Activation of sphingosine kinase by tumor necrosis factor-alpha inhibits apoptosis in human endothelial cells. J. Biol. Chem. 1999, 274, 34499–34505. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, L.; Moretti, P.A.; Albanese, N.; Chai, F.; Pitson, S.M.; D’Andrea, R.J.; Gamble, J.R.; Vadas, M.A. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J. Biol. Chem. 2002, 277, 7996–8003. [Google Scholar] [CrossRef]
- Fakhr, Y.; Brindley, D.N.; Hemmings, D.G. Physiological and pathological functions of sphingolipids in pregnancy. Cell. Signal. 2021, 85, 110041. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef]
- Smith, S.; Francis, R.; Guilbert, L.; Baker, P.N. Growth factor rescue of cytokine mediated trophoblast apoptosis. Placenta 2002, 23, 322–330. [Google Scholar] [CrossRef]
- Straszewski-Chavez, S.L.; Visintin, I.P.; Karassina, N.; Los, G.; Liston, P.; Halaban, R.; Fadiel, A.; Mor, G. XAF1 mediates tumor necrosis factor-alpha-induced apoptosis and X-linked inhibitor of apoptosis cleavage by acting through the mitochondrial pathway. J. Biol. Chem. 2007, 282, 13059–13072. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Banno, Y.; Nagaki, M.; Brenner, D.A.; Naiki, T.; Nozawa, Y.; Nakashima, S.; Moriwaki, H. TNF-alpha-induced sphingosine 1-phosphate inhibits apoptosis through a phosphatidylinositol 3-kinase/Akt pathway in human hepatocytes. J. Immunol. 2001, 167, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Bielawski, J.; Porcelli, A.M.; Reames, D.L.; Johnson, K.R.; Morrow, J.; Chalfant, C.E.; Obeid, L.M.; Hannun, Y.A. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-α. FASEB J. 2003, 17, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Yatomi, Y. Sphingosine 1-phosphate and atherosclerosis. J. Atheroscler. Thromb. 2017, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef]
- McGinley, M.P.; Cohen, J.A. Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet 2021, 398, 1184–1194. [Google Scholar] [CrossRef]
- Charkiewicz, K.; Goscik, J.; Blachnio-Zabielska, A.; Raba, G.; Sakowicz, A.; Kalinka, J.; Chabowski, A.; Laudanski, P. Sphingolipids as a new factor in the pathomechanism of preeclampsia—Mass spectrometry analysis. PLoS ONE 2017, 12, e0177601. [Google Scholar] [CrossRef]
- Dobierzewska, A.; Soman, S.; Illanes, S.E.; Morris, A.J. Plasma cross-gestational sphingolipidomic analyses reveal potential first trimester biomarkers of preeclampsia. PLoS ONE 2017, 12, e0175118. [Google Scholar] [CrossRef]
- Johnstone, E.D.; Chan, G.; Sibley, C.P.; Davidge, S.T.; Lowen, B.; Guilbert, L.J. Sphingosine-1-phosphate inhibition of placental trophoblast differentiation through a G(i)-coupled receptor response. J. Lipid Res. 2005, 46, 1833–1839. [Google Scholar] [CrossRef]
- Singh, A.T.; Dharmarajan, A.; Aye, I.L.; Keelan, J.A. Sphingosine-sphingosine-1-phosphate pathway regulates trophoblast differentiation and syncytialization. Reprod. Biomed. Online 2012, 24, 224–234. [Google Scholar] [CrossRef][Green Version]
- Benton, S.J.; Leavey, K.; Grynspan, D.; Cox, B.J.; Bainbridge, S.A. The clinical heterogeneity of preeclampsia is related to both placental gene expression and placental histopathology. Am. J. Obstet. Gynecol. 2018, 219, 604.e601–604.e625. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef]
- Pchejetski, D.; Nunes, J.; Coughlan, K.; Lall, H.; Pitson, S.M.; Waxman, J.; Sumbayev, V.V. The involvement of sphingosine kinase 1 in LPS-induced Toll-like receptor 4-mediated accumulation of HIF-1α protein, activation of ASK1 and production of the pro-inflammatory cytokine IL-6. Immunol. Cell Biol. 2011, 89, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Snider, A.J.; Alexa Orr Gandy, K.; Obeid, L.M. Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 2010, 92, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Eisenhoffer, G.T.; Loftus, P.D.; Yoshigi, M.; Otsuna, H.; Chien, C.B.; Morcos, P.A.; Rosenblatt, J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Charnock-Jones, D.S.; Skepper, J.N.; Burton, G.J. Secretion of tumor necrosis factor-alpha from human placental tissues induced by hypoxia-reoxygenation causes endothelial cell activation in vitro: A potential mediator of the inflammatory response in preeclampsia. Am. J. Pathol. 2004, 164, 1049–1061. [Google Scholar] [CrossRef]
- Dobierzewska, A.; Palominos, M.; Sanchez, M.; Dyhr, M.; Helgert, K.; Venegas-Araneda, P.; Tong, S.; Illanes, S.E. Impairment of Angiogenic Sphingosine Kinase-1/Sphingosine-1-Phosphate Receptors Pathway in Preeclampsia. PLoS ONE 2016, 11, e0157221. [Google Scholar] [CrossRef]
- Liao, J.; Zheng, Y.; Hu, M.; Xu, P.; Lin, L.; Liu, X.; Wu, Y.; Huang, B.; Ye, X.; Li, S. Impaired Sphingosine-1-Phosphate Synthesis Induces Preeclampsia by Deactivating Trophoblastic YAP (Yes-Associated Protein) Through S1PR2 (Sphingosine-1-Phosphate Receptor-2)-Induced Actin Polymerizations. Hypertension 2021, 79, 399–412. [Google Scholar] [CrossRef]
- Wick, S.M.; Duniec, J. Effects of various fixatives on the reactivity of plant cell tubulin and calmodulin in immunofluorescence microscopy. Protoplasma 1986, 133, 1–18. [Google Scholar] [CrossRef]
- Billich, A.; Ettmayer, P. Fluorescence-based assay of sphingosine kinases. Anal. Biochem. 2004, 326, 114–119. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Olson, D.M.; van Bennekom, M.; Brindley, D.N.; Hemmings, D.G. Increased expression of enzymes for sphingosine 1-phosphate turnover and signaling in human decidua during late pregnancy. Biol. Reprod. 2010, 82, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Yui, J.; Hemmings, D.; Garcia-Lloret, M.; Guilbert, L.J. Expression of the human p55 and p75 tumor necrosis factor receptors in primary villous trophoblasts and their role in cytotoxic signal transduction. Biol. Reprod. 1996, 55, 400–409. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Knofler, M.; Mosl, B.; Bauer, S.; Griesinger, G.; Husslein, P. TNF-alpha/TNFRI in primary and immortalized first trimester cytotrophoblasts. Placenta 2000, 21, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.T.; Dharmarajan, A.; Aye, I.L.M.H.; Keelan, J.A. Ceramide biosynthesis and metabolism in trophoblast syncytialization. Mol. Cell. Endocrinol. 2012, 362, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Kim, Y.-W.; Inagaki, Y.; Hwang, Y.-A.; Mitsutake, S.; Ryu, Y.-W.; Lee, W.K.; Ha, H.-J.; Park, C.-S.; Igarashi, Y. Synthesis and evaluation of sphingoid analogs as inhibitors of sphingosine kinases. Bioorganic Med. Chem. 2005, 13, 3475–3485. [Google Scholar] [CrossRef]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Urbańska, K.; Orzechowski, A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 2085. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-y.; Roh, C.-R. Autophagy in the placenta. Obs. Gynecol. Sci. 2017, 60, 241–259. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, W.; Liu, S.; Wu, W.; Qin, M.; Huang, J. Activation of the SphK1/ERK/p-ERK pathway promotes autophagy in colon cancer cells. Oncol. Lett. 2018, 15, 9719–9724. [Google Scholar] [CrossRef]
- Melland-Smith, M.; Ermini, L.; Chauvin, S.; Craig-Barnes, H.; Tagliaferro, A.; Todros, T.; Post, M.; Caniggia, I. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy 2015, 11, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Stefan, B.; Susanne, E.U.; Horst-Dieter, R.; Myriam, R.; Mathias, B.; Heinrich, H.D.M.; Thomas, E.S.; Megan, M.; Gerhard, S.; Gerhard, W.; et al. Comparison of the Effects of Early Pregnancy with Human Interferon, Alpha 2 (IFNA2), on Gene Expression in Bovine Endometrium. Biol. Reprod. 2011, 86. [Google Scholar] [CrossRef]
- Elias, K.M.; Harvey, R.A.; Hasselblatt, K.T.; Seckl, M.J.; Berkowitz, R.S. Type I interferons modulate methotrexate resistance in gestational trophoblastic neoplasia. Am. J. Reprod. Immunol. 2017, 77, e12666. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.E.; Cartwright, J.E.; Begum, R.; Laing, K.; Thilaganathan, B.; Whitley, G.S. Trophoblast-Induced Changes in C-X-C Motif Chemokine 10 Expression Contribute to Vascular Smooth Muscle Cell Dedifferentiation During Spiral Artery Remodeling. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e93–e101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Huang, F.; Chai, X.; Yuan, W.; Ding, H.; Wu, X. The role of IP-10 and its receptor CXCR3 in early pregnancy. Mol. Immunol. 2021, 140, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Qiao, F.; Yin, X. Hypoxia Induces FGF2 Production by Vascular Endothelial Cells and Alters MMP9 and TIMP1 Expression in Extravillous Trophoblasts and Their Invasiveness in a Cocultured Model. J. Reprod. Dev. 2011, 57, 84–91. [Google Scholar] [CrossRef]
- Holleran, G. The Role of Angiopoietins and Their Mediators in Symptomatic Small Bowel Angiodysplasia; Identifying Novel Diagnostic and Therapeutic Targets in Chronic Anaemia and Obscure Gastrointestinal Bleeding. Ph.D. Thesis, Trinity College Dublin School of Medicine, Dublin, Ireland, 2021. [Google Scholar]
- Aggarwal, R.; Jain, A.K.; Mittal, P.; Kohli, M.; Jawanjal, P.; Rath, G. Association of pro-and anti-inflammatory cytokines in preeclampsia. J. Clin. Lab. Anal. 2019, 33, e22834. [Google Scholar] [CrossRef] [PubMed]
- Kalkunte, S.; Nevers, T.; Norris, W.E.; Sharma, S. Vascular IL-10: A protective role in preeclampsia. J. Reprod. Immunol. 2011, 88, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.A.; Rubin, D.T. New targets in inflammatory bowel disease therapy: 2021. Curr. Opin. Gastroenterol. 2021, 37, 357–363. [Google Scholar] [CrossRef]
- Mulla, M.J.; Brosens, J.J.; Chamley, L.W.; Giles, I.; Pericleous, C.; Rahman, A.; Joyce, S.K.; Panda, B.; Paidas, M.J.; Abrahams, V.M. ORIGINAL ARTICLE: Antiphospholipid Antibodies Induce a Pro-Inflammatory Response in First Trimester Trophoblast Via the TLR4/MyD88 Pathway. Am. J. Reprod. Immunol. 2009, 62, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Usatyuk, P.V.; Ebenezer, D.L.; Natarajan, V. ID: 111: The s1p transporter, spns2, mediates hgf-induced lamellipodia formation and migration of human lung endothelial cells. J. Investig. Med. 2016, 64, 964. [Google Scholar] [CrossRef]
- León, Y.; Magariños, M.; Varela-Nieto, I. Ceramide Kinase Inhibition Blocks IGF-1-Mediated Survival of Otic Neurosensory Progenitors by Impairing AKT Phosphorylation. Front. Cell Dev. Biol. 2021, 9, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Lisabeth, E.M.; Neubig, R.R. Transforming Growth Factor β1 Increases Expression of Contractile Genes in Human Pulmonary Arterial Smooth Muscle Cells by Potentiating Sphingosine-1-Phosphate Signaling. Mol. Pharmacol. 2021, 100, 83. [Google Scholar] [CrossRef] [PubMed]
- Drexler, Y.; Molina, J.; Mitrofanova, A.; Fornoni, A.; Merscher, S. Sphingosine-1-Phosphate Metabolism and Signaling in Kidney Diseases. J. Am. Soc. Nephrol. 2021, 32, 9. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, Sphingosine Kinase Isoenzymes with Opposing Functions in Sphingolipid Metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef]
- Amaral, L.M.; Wallace, K.; Owens, M.; LaMarca, B. Pathophysiology and current clinical management of preeclampsia. Curr. Hypertens. Rep. 2017, 19, 1–6. [Google Scholar] [CrossRef]
- Bu, Y.; Wu, H.; Deng, R.; Wang, Y. Therapeutic Potential of SphK1 Inhibitors Based on Abnormal Expression of SphK1 in Inflammatory Immune Related-Diseases. Front. Pharm. 2021, 12, 733387. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Simán, C.M.; Sibley, C.P.; Jones, C.J.; Turner, M.A.; Greenwood, S.L. The functional regeneration of syncytiotrophoblast in cultured explants of term placenta. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R1116–R1122. [Google Scholar] [CrossRef]
- Trowell, O.A. A modified technique for organ culture in vitro. Exp. Cell Res. 1954, 6, 246–248. [Google Scholar] [CrossRef]
- Fakhr, Y.; Saadat, S.; Webster, K.; Hemmings, D.G. Sphingosine 1-Phosphate as a Mediator of Tumour Necrosis Factor-alpha (TNF-alpha) Signaling in Syncytialization. Reprod. Sci. 2020, 27 (Suppl. 1), S-149. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pregnancy | Gestational Age (Weeks) | Infant Weight (g) | Systolic Blood Pressure (mmHg) | Diastolic Blood Pressure (mmHg) | Percent Positive for Proteinuria (%) |
|---|---|---|---|---|---|
| Normal (n = 17) | 38.9 ± 0.3 | 3074 ± 86.8 | 113.8 ± 3.7 | 71.4 ± 3.6 | 0 |
| Preeclampsia (n = 12) | 37.2 ± 0.3 | 2967 ± 274 | 155.3 ± 7.6 | 107.9 ± 15.6 | 100 |
| p-value | 0.003 | ns | p < 0.0001 | 0.008 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakhr, Y.; Koshti, S.; Habibyan, Y.B.; Webster, K.; Hemmings, D.G. Tumor Necrosis Factor-α Induces a Preeclamptic-like Phenotype in Placental Villi via Sphingosine Kinase 1 Activation. Int. J. Mol. Sci. 2022, 23, 3750. https://doi.org/10.3390/ijms23073750
Fakhr Y, Koshti S, Habibyan YB, Webster K, Hemmings DG. Tumor Necrosis Factor-α Induces a Preeclamptic-like Phenotype in Placental Villi via Sphingosine Kinase 1 Activation. International Journal of Molecular Sciences. 2022; 23(7):3750. https://doi.org/10.3390/ijms23073750
Chicago/Turabian StyleFakhr, Yuliya, Saloni Koshti, Yasaman Bahojb Habibyan, Kirsten Webster, and Denise G. Hemmings. 2022. "Tumor Necrosis Factor-α Induces a Preeclamptic-like Phenotype in Placental Villi via Sphingosine Kinase 1 Activation" International Journal of Molecular Sciences 23, no. 7: 3750. https://doi.org/10.3390/ijms23073750
APA StyleFakhr, Y., Koshti, S., Habibyan, Y. B., Webster, K., & Hemmings, D. G. (2022). Tumor Necrosis Factor-α Induces a Preeclamptic-like Phenotype in Placental Villi via Sphingosine Kinase 1 Activation. International Journal of Molecular Sciences, 23(7), 3750. https://doi.org/10.3390/ijms23073750