
Unilateral Acute Renal Ischemia-Reperfusion Injury Induces Cardiac Dysfunction through Intracellular Calcium Mishandling

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
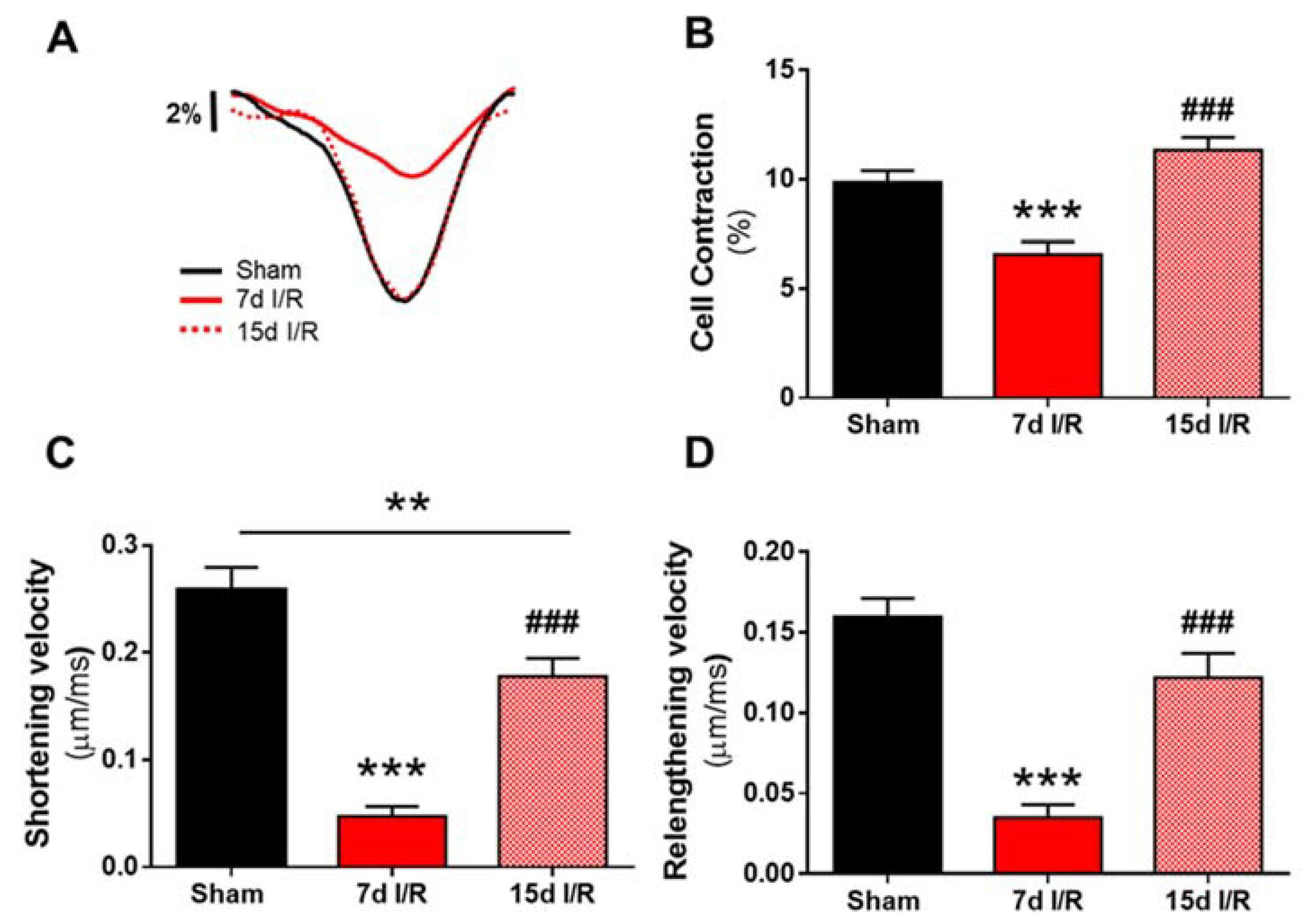
2.1. Renal Ischemia and Reperfusion Induces Contractile Dysfunction
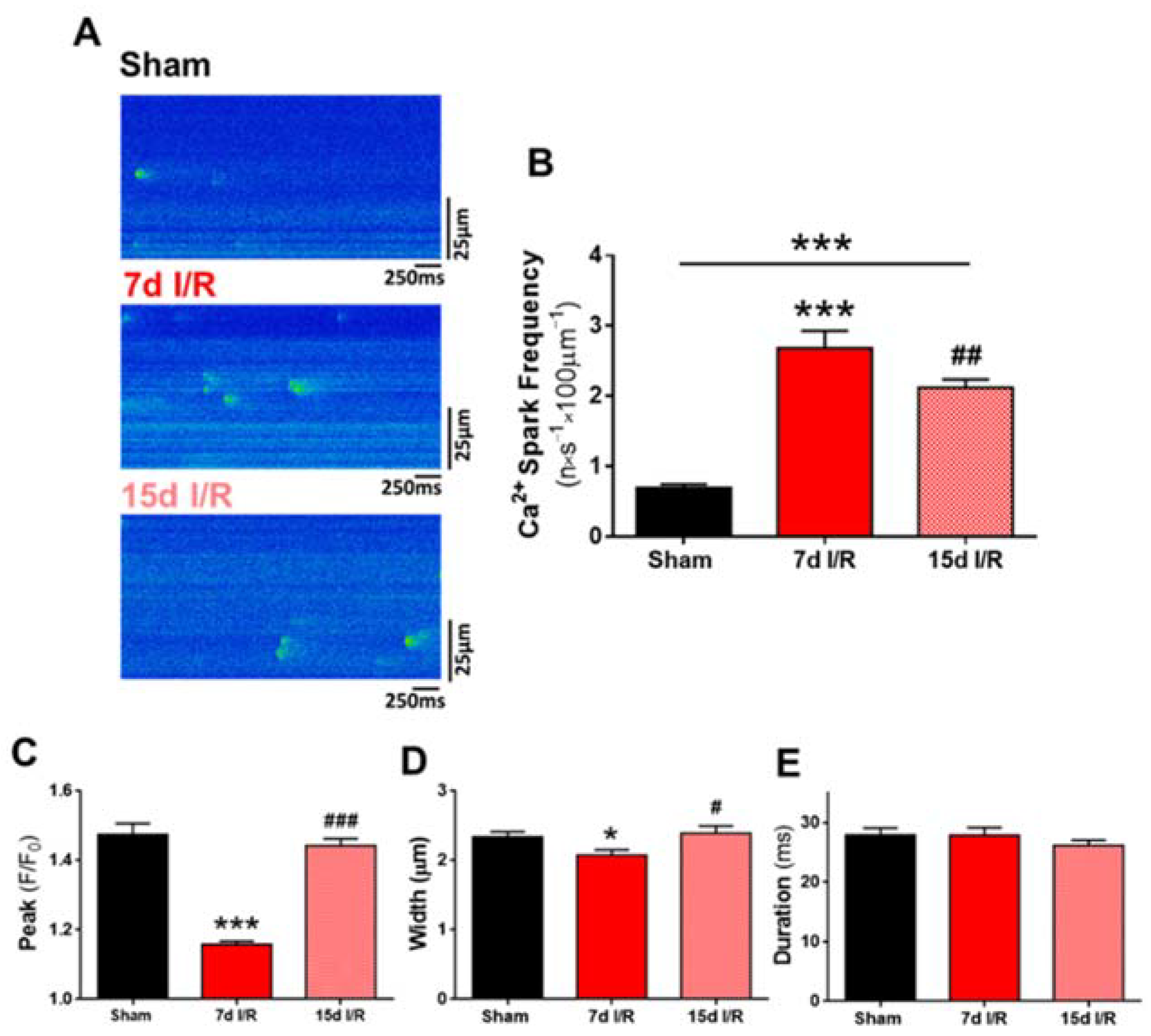
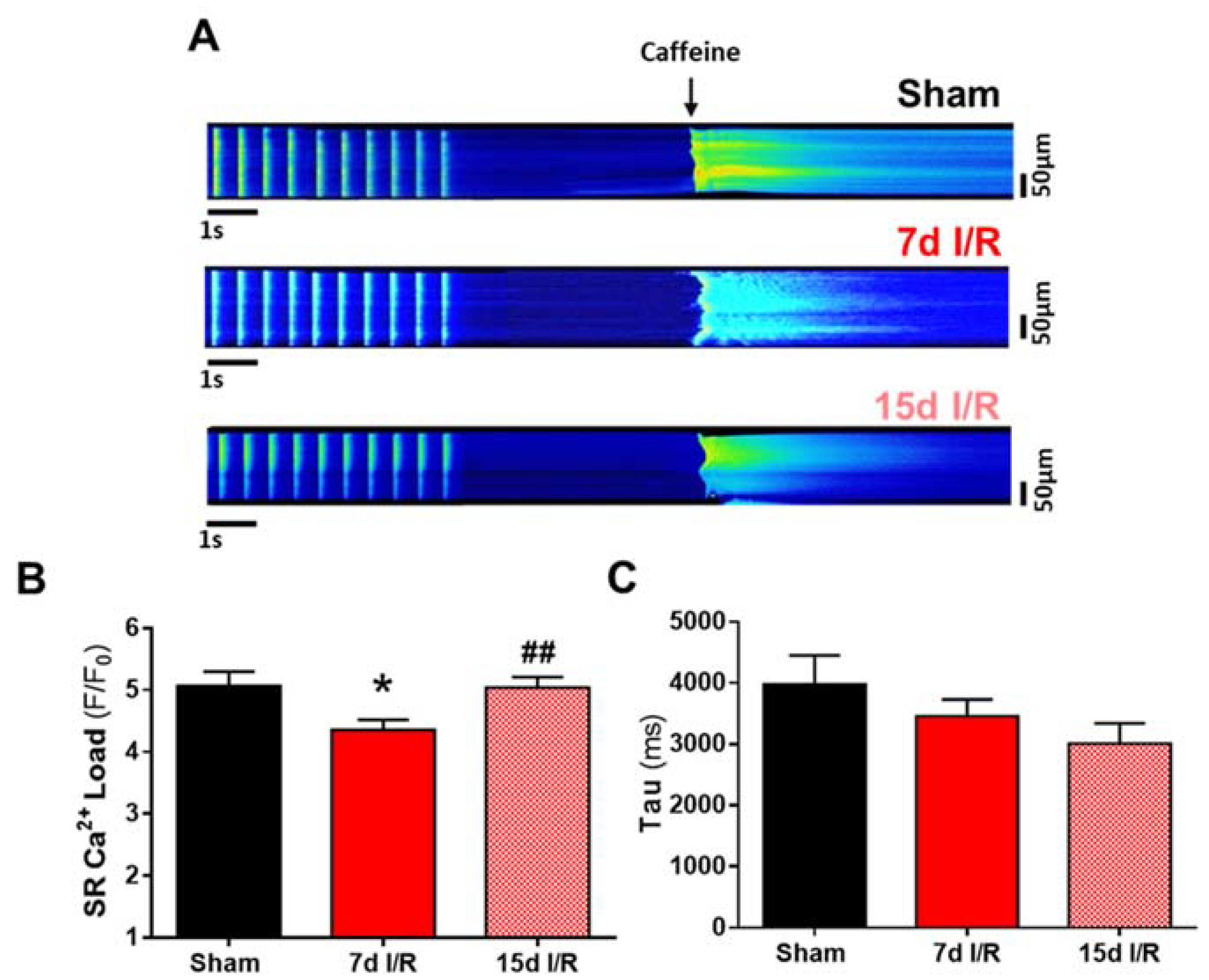
2.2. Systolic Ca2+ Release Deteriorates 7 Days following Renal Ischemia and Reperfusion
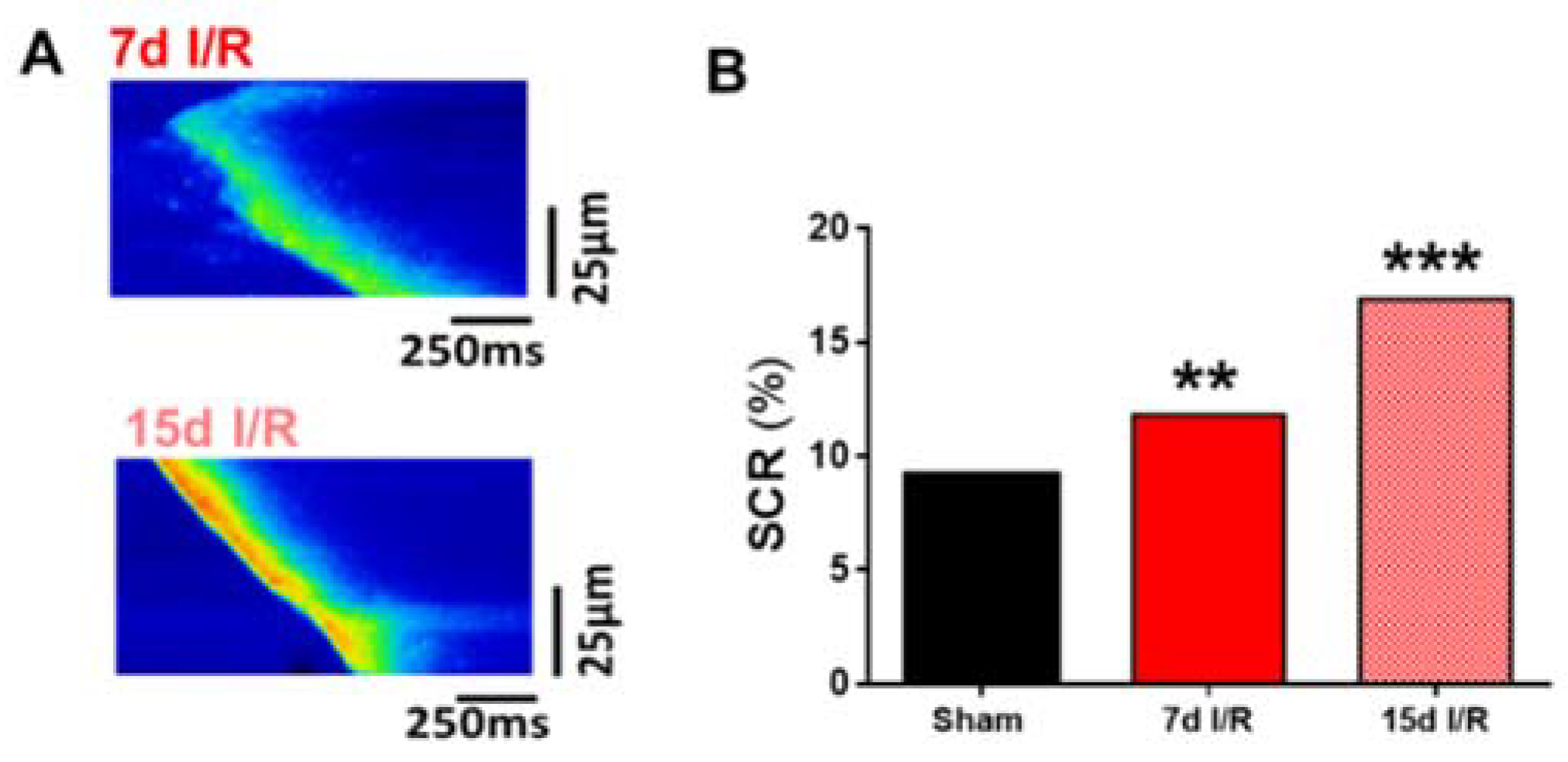
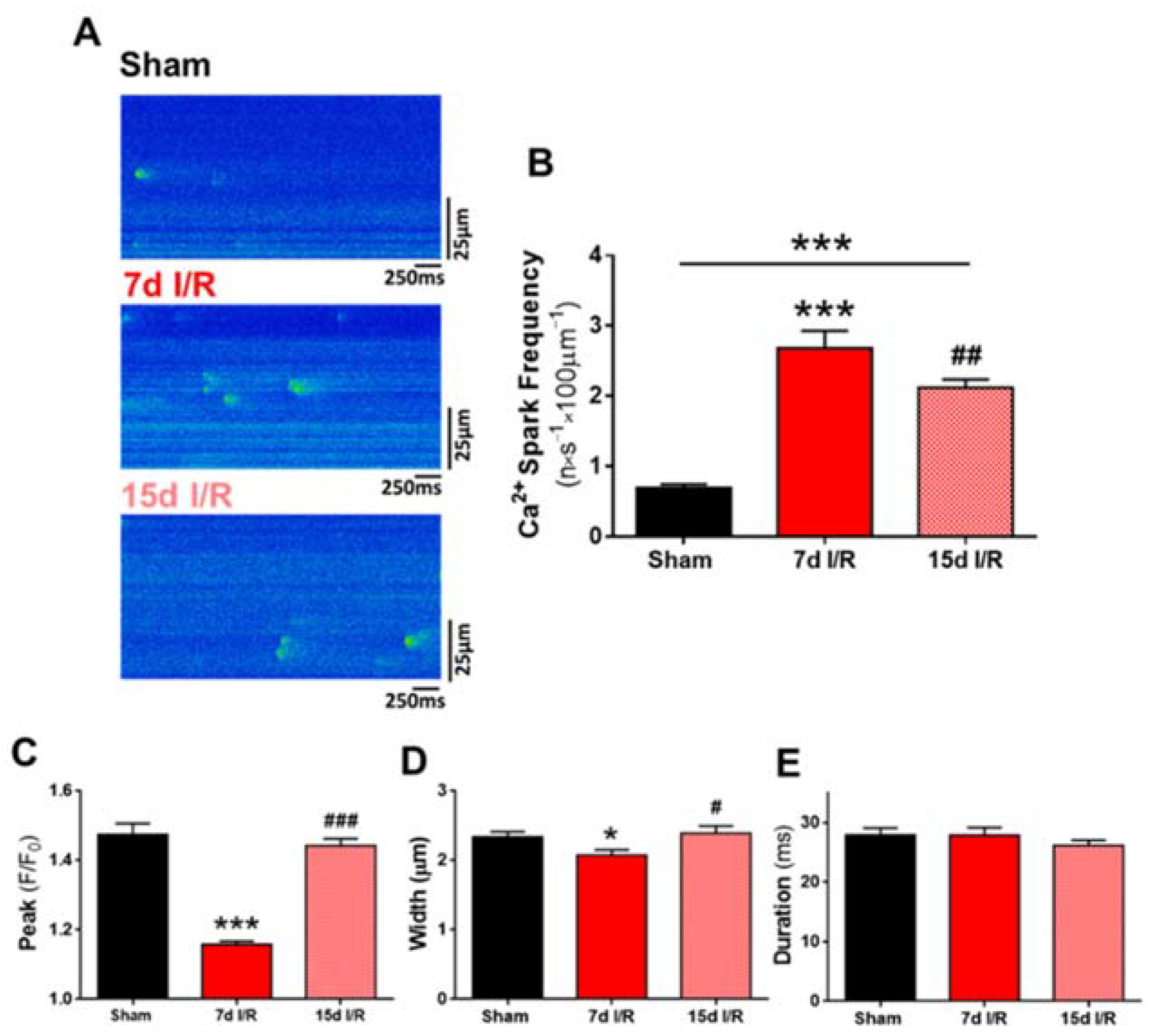
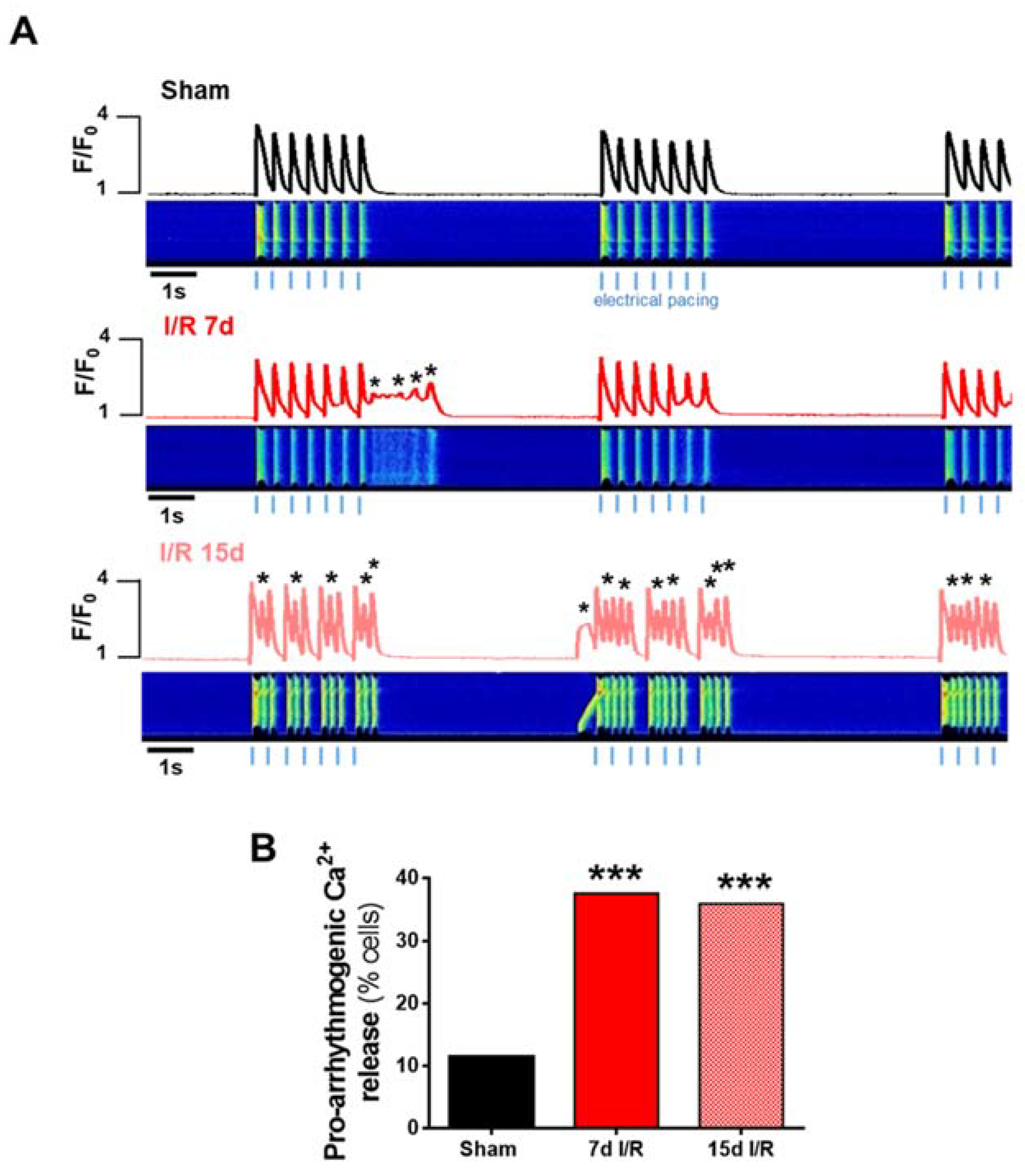
2.3. Acute Renal Failure Induces Diastolic Ca2+ Release from the Sarcoplasmic Reticulum and Triggers Pro-Arrhythmic Ca2+ Events 7 Days of Renal Ischemia and Reperfusion That Remain Elevated after 15 Days
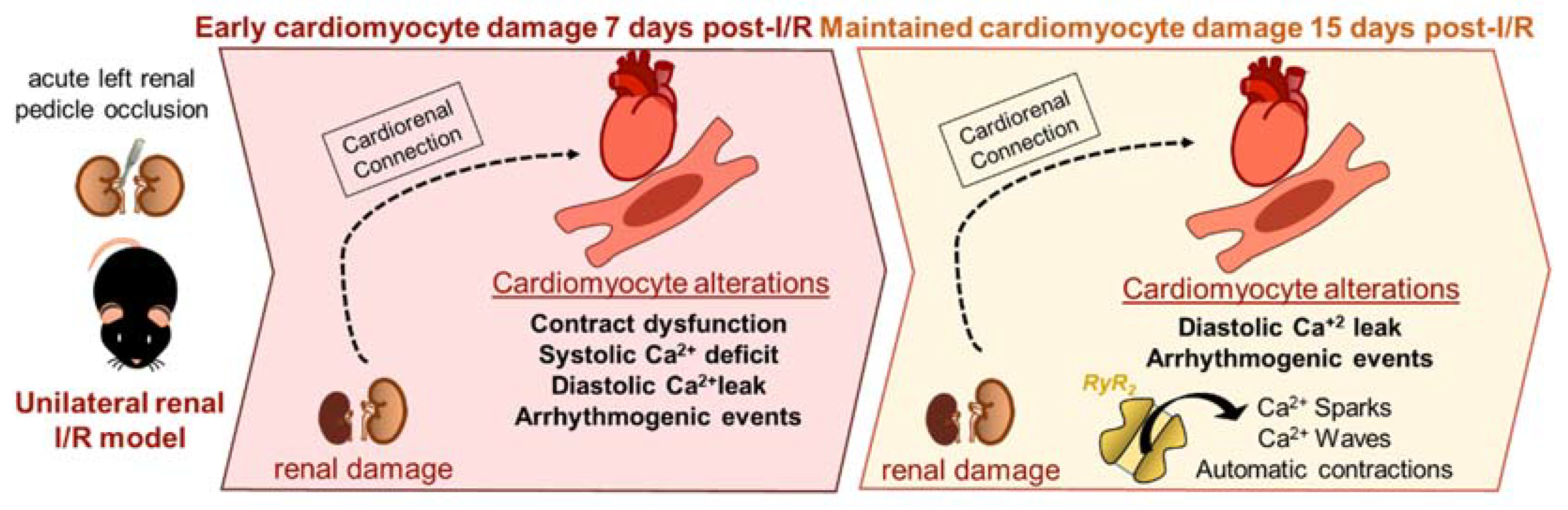
3. Discussion
4. Materials and Methods
4.1. Animal Procedures
4.2. Experimental Renal Ischemia/Reperfusion
4.3. Biochemical Assays
4.4. Isolation of Adult Ventricular Cardiomyocytes
4.5. Intracellular Ca2+ Imaging
4.6. Immunobloting
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARF | acute renal failure |
| BUN | blood urea nitrogen |
| BW | body weight |
| Ca2+ | calcium |
| CRS | cardiorenal syndrome |
| EC | excitation-contraction |
| FGF-23 | fibroblast growth factor-23 |
| HF | heart failure |
| HW | heart weight |
| I/R | ischemia and reperfusion |
| IRI | ischemia reperfusion kidney injury |
| LKW | left kidney weight |
| RKW | right kidney weight |
| RyR2 | ryanodine receptor |
| SERCA2a | sarcoplasmic/endoplasmic reticulum Ca2+-adenosine triphosphatase 2a |
| SCR | spontaneous calcium release |
| SR | sarcoplasmic reticulum |
| TL | tibia length |
References
- Gao, Y.; Zeng, Z.; Li, T.; Xu, S.; Wang, X.; Chen, Z.; Lin, C. Polydatin inhibits mitochondrial dysfunction in the renal tubular epithelial cells of a rat model of sepsis-induced acute kidney injury. Anesth. Analg. 2015, 121, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Benck, U.; Schnuelle, P.; Krüger, B.; Nowak, K.; Riester, T.; Mundt, H.; Lutz, N.; Jung, M.; Birck, R.; Krämer, B.K.; et al. Excellent graft and patient survival after renal transplantation from donors after brain death with acute kidney injury: A case-control study. Int. Urol. Nephrol. 2015, 47, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-B.; Zhang, Y.P.; Zhang, J.; Zhang, Y.B. Evaluation of Vitamin C supplementation on kidney function and vascular reactivity following renal ischemic injury in mice. Kidney Blood Press. Res. 2016, 41, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, S.; Marks, A.; Fluck, N.; Levin, A.; Prescott, G.; Black, C. Intermediate and Long-term Outcomes of Survivors of Acute Kidney Injury Episodes: A Large Population-Based Cohort Study. Am. J. Kidney Dis. 2017, 69, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Doi, K. Kidney-Heart Interactions in Acute Kidney Injury. Nephron 2016, 134, 141–144. [Google Scholar] [CrossRef]
- Groeneveld, A.B.J.; Tran, D.D.; van der Meulen, J.; Nauta, J.J.P.; Thijs, L. Acute Renal Failure in the Medical Intensive Care Unit: Predisposing, Complicating Factors and Outcome. Nephron 1991, 59, 602–610. [Google Scholar] [CrossRef]
- Ronco, C.; Bellasi, A.; Di Lullo, L. Cardiorenal Syndrome: An Overview. Adv. Chronic Kidney Dis. 2018, 25, 382–390. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Trentin-Sonoda, M.; Da Silva, R.C.; Kmit, F.V.; Abrahão, M.V.; Cahli, G.M.; Brasil, G.V.; Muzi-Filho, H.; Silva, P.A.; Tovar-Moll, F.F.; Vieyra, A.; et al. Knockout of toll-like receptors 2 and 4 prevents renal ischemia-reperfusion-induced cardiac hypertrophy in mice. PLoS ONE 2015, 10, e0139350. [Google Scholar] [CrossRef] [Green Version]
- Junho, C.V.C.; Panico, K.; Nakama, K.K.; Sonoda, M.T.; Christoffolete, M.A.; Beserra, S.S.; Roman-Campos, D.; Carneiro-Ramos, M.S. Time Course of Gene Expression Profile in Renal Ischemia and Reperfusion Injury in Mice. Transplant. Proc. 2020, 52, 2970–2976. [Google Scholar] [CrossRef] [PubMed]
- Neres-Santos, R.S.; Junho, C.V.C.; Panico, K.; Caio-Silva, W.; Pieretti, J.C.; Tamashiro, J.A.; Seabra, A.B.; Ribeiro, C.A.J.; Carneiro-Ramos, M.S. Mitochondrial Dysfunction in Cardiorenal Syndrome 3: Renocardiac Effect of Vitamin C. Cells 2021, 10, 3029. [Google Scholar] [CrossRef] [PubMed]
- Caio-Silva, W.; da Silva Dias, D.; Junho, C.V.C.; Panico, K.; Neres-Santos, R.S.; Pelegrino, M.T.; Pieretti, J.C.; Seabra, A.B.; De Angelis, K.; Carneiro-Ramos, M.S. Characterization of the Oxidative Stress in Renal Ischemia/Reperfusion-Induced Cardiorenal Syndrome Type 3. BioMed Res. Int. 2020, 2020, 1605358. [Google Scholar] [CrossRef] [PubMed]
- Cirino-Silva, R.; Kmit, F.V.; Trentin-Sonoda, M.; Nakama, K.K.; Panico, K.; Alvim, J.M.; Dreyer, T.R.; Martinho-Silva, H.; Carneiro-Ramos, M.S. Renal ischemia/reperfusion-induced cardiac hypertrophy in mice: Cardiac morphological and morphometric characterization. JRSM Cardiovasc. Dis. 2017, 6, 204800401668944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon, M.M.L.; Trentin-Sonoda, M.; Panico, K.; Schleier, Y.; Duque, T.; Moreno-Loaiza, O.; de Yurre, A.R.; Ferreira, F.; Caio-Silva, W.; Coury, P.R.; et al. Cardiac arrhythmias after renal I/R depend on IL-1β. J. Mol. Cell. Cardiol. 2019, 131, 101–111. [Google Scholar] [CrossRef]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef]
- Dridi, H.; Kushnir, A.; Zalk, R.; Yuan, Q.; Melville, Z.; Marks, A.R. Intracellular calcium leak in heart failure and atrial fibrillation: A unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 2020, 17, 732–747. [Google Scholar] [CrossRef]
- Kansakar, U.; Varzideh, F.; Jankauskas, S.S.; Gambardella, J.; Trimarco, B.; Santulli, G. Advances in the understanding of excitation-contraction coupling: The pulsing quest for drugs against heart failure and arrhythmias. Eur Heart J. Cardiovasc. Pharmacother. 2021, 7, e91–e93. [Google Scholar] [CrossRef]
- Ruiz-Hurtado, G.; Li, L.; Fernández-Velasco, M.; Rueda, A.; Lefebvre, F.; Wang, Y.; Mateo, P.; Cassan, C.; Gellen, B.; Benitah, J.P.; et al. Reconciling depressed Ca2+ sparks occurrence with enhanced RyR2 activity in failing mice cardiomyocytes. J. Gen. Physiol. 2015, 146, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Val-Blasco, A.; Piedras, M.J.G.M.; Ruiz-Hurtado, G.; Suarez, N.; Prieto, P.; Gonzalez-Ramos, S.; Gómez-Hurtado, N.; Delgado, C.; Pereira, L.; Benito, G.; et al. Role of NOD1 in Heart Failure Progression via Regulation of Ca2+ Handling. J. Am. Coll. Cardiol. 2017, 69, 423–433. [Google Scholar] [CrossRef]
- Di Fonso, A.; Pietrangelo, L.; D’Onofrio, L.; Michelucci, A.; Boncompagni, S.; Protasi, F. Ageing Causes Ultrastructural Modification to Calcium Release Units and Mitochondria in Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 8364. [Google Scholar] [CrossRef]
- Eckardt, K.U.; Kasiske, B.L. Improving Global Outcomes. Nat. Rev. Nephrol. 2009, 5, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Pavez-Giani, M.G.; Sánchez-Aguilera, P.I.; Bomer, N.; Miyamoto, S.; Booij, H.G.; Giraldo, P.; Oberdorf-Maass, S.U.; Nijholt, K.T.; Yurista, S.R.; Milting, H.; et al. ATPase Inhibitory Factor-1 Disrupts Mitochondrial Ca2+ Handling and Promotes Pathological Cardiac Hypertrophy through CaMKIIδ. Int. J. Mol. Sci. 2021, 22, 4427. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol. 2018, 1067, 373–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Morales, J.C.; Xia, Y.; Renzo, T.J.; Zhang, X.H.; Morad, M. Mutation in RyR2-FKBP Binding site alters Ca2+ signaling modestly but increases “arrhythmogenesis” in human stem cells derived cardiomyocytes. Cell Calcium. 2022, 101, 102500. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, A.; Santulli, G.; Reiken, S.R.; Coromilas, E.; Godfrey, S.J.; Brunjes, D.L.; Colombo, P.C.; Yuzefpolskaya, M.; Sokol, S.I.; Kitsis, R.N.; et al. Ryanodine Receptor Calcium Leak in Circulating B-Lymphocytes as a Biomarker in Heart Failure. Circulation 2018, 138, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M. Forging Forward with 10 Burning Questions on FGF23 in Kidney Disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Neyra, J.A.; Moe, O.W.; Hu, M.C. Fibroblast growth factor 23 and acute kidney injury. Pediatr. Nephrol. 2015, 30, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Leaf, D.E.; Jacob, K.A.; Srivastava, A.; Chen, M.E.; Christov, M.; Jüppner, H.; Sabbisetti, V.S.; Martin, A.; Wolf, M.; Waikar, S.S. Fibroblast Growth Factor 23 Levels Associate with AKI and Death in Critical Illness. J. Am. Soc. Nephrol. 2017, 28, 1877–1885. [Google Scholar] [CrossRef]
- Memmos, E.; Papagianni, A. New Insights into the Role of FGF-23 and Klotho in Cardiovascular Disease in Chronic Kidney Disease Patients. Curr. Vasc. Pharmacol. 2020, 19, 55–62. [Google Scholar] [CrossRef]
- Navarro-García, J.A.; Delgado, C.; Fernández-Velasco, M.; Val-Blasco, A.; Rodríguez-Sánchez, E.; Aceves-Ripoll, J.; Gómez-Hurtado, N.; Bada-Bosch, T.; Mérida-Herrero, E.; Hernández, E.; et al. Fibroblast growth factor-23 promotes rhythm alterations and contractile dysfunction in adult ventricular cardiomyocytes. Nephrol. Dial. Transplant. 2019, 34, 1864–1875. [Google Scholar] [CrossRef]
- Navarro-García, J.A.; Fernández-Velasco, M.; Delgado, C.; Delgado, J.F.; Kuro-o, M.; Ruilope, L.M.; Ruiz-Hurtado, G. PTH, vitamin D, and the FGF-23–klotho axis and heart: Going beyond the confines of nephrology. Eur. J. Clin. Investig. 2018, 48. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.-H.; Chen, Y.-C.; Lin, Y.-K.; Shiu, R.-J.; Chao, T.-F.; Chen, S.-A.; Chen, Y.-J. FGF-23 dysregulates calcium homeostasis and electrophysiological properties in HL-1 atrial cells. Eur. J. Clin. Investig. 2014, 44, 795–801. [Google Scholar] [CrossRef]
- Geach, T. FGF-23 associated with incident AF—A link with CKD? Nat. Rev. Cardiol. 2014, 11, 436. [Google Scholar] [CrossRef]
- Lindner, M.; Mehel, H.; David, A.; Leroy, C.; Burtin, M.; Friedlander, G.; Terzi, F.; Mika, D.; Fischmeister, R.; Prié, D. Fibroblast growth factor 23 decreases PDE4 expression in heart increasing the risk of cardiac arrhythmia; Klotho opposes these effects. Basic Res. Cardiol. 2020, 115, 51. [Google Scholar] [CrossRef]
- Navarro-García, J.A.; Rueda, A.; Romero-García, T.; Aceves-Ripoll, J.; Rodríguez-Sánchez, E.; González-Lafuente, L.; Zaragoza, C.; Fernández-Velasco, M.; Kuro-o, M.; Ruilope, L.M.; et al. Enhanced Klotho availability protects against cardiac dysfunction induced by uraemic cardiomyopathy by regulating Ca2+ handling. Br. J. Pharmacol. 2020, 177, 4701–4719. [Google Scholar] [CrossRef]
- Cheek, T.R.; Moreton, R.B.; Berridge, M.J.; Stauderman, K.A.; Murawsky, M.M.; Bootman, M.D. Quantal Ca2+ release from caffeine-sensitive stores in adrenal chromaffin cells. J. Biol. Chem. 1993, 268, 27076–27083. [Google Scholar] [CrossRef]
- Rodríguez-Sánchez, E.; Navarro-García, J.A.; González-Lafuente, L.; Aceves-Ripoll, J.; Vázquez-Sánchez, S.; Poveda, J.; Mercado-García, E.; Corbacho-Alonso, N.; Calvo-Bonacho, E.; Fernández-Velasco, M.; et al. Oxidized Low-Density Lipoprotein Associates with Ventricular Stress in Young Adults and Triggers Intracellular Ca2+ Alterations in Adult Ventricular Cardiomyocytes. Antioxidants 2020, 9, 1213. [Google Scholar] [CrossRef]
- González-Lafuente, L.; Navarro-García, J.A.; Rodríguez-Sánchez, E.; Aceves-Ripoll, J.; Poveda, J.; Vázquez-Sánchez, S.; Mercado-García, E.; Fernández-Velasco, M.; Kuro-o, M.; Liaño, F.; et al. Interplay between mineral bone disorder and cardiac damage in acute kidney injury: From Ca2+ mishandling and preventive role of Klotho in mice to its potential mortality prediction in human. Transl. Res. 2022, in press. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sham | 7 d I/R | 15 d I/R |
|---|---|---|---|
| BW (g) | 24.0 ± 0.5 | 22.2 ± 0.6 | 23.2 ± 0.7 |
| TL (mm) | 16.6 ± 0.13 | 16.2 ± 0.12 | 16.2 ± 0.21 |
| HW (g) | 0.160 ± 0.005 | 0.177 ± 0.009 | 0.179 ± 0.007 |
| LKW (g) | 0.151 ± 0.004 | 0.148 ± 0.004 | 0.115 ± 0.006 ***, ### |
| RKW (g) | 0.146 ± 0.009 | 0.167 ± 0.005 | 0.178 ± 0.007 * |
| HW/BW (mg/g) | 6.7 ± 0.21 | 7.97 ± 0.37 * | 7.77 ± 0.32 ** |
| HW/TL (mg/mm) | 96.9 ± 3.09 | 109.1 ± 6.15 * | 110.2 ± 3.34 * |
| LKW/BW (mg/g) | 6.3 ± 0.10 | 6.7 ± 0.28 | 5.0 ± 0.25 ***, ### |
| LKW/TL (mg/mm) | 90.9 ± 2.08 | 91.1 ± 2.83 | 70.7 ± 3.28 ***, ### |
| RKW/BW (mg/g) | 6.01 ± 0.37 | 7.56 ± 0.36 * | 7.70 ± 0.35 ** |
| RKW/TL (mg/mm) | 88.3 ± 5.63 | 102.7 ± 3.68 | 109.2 ± 3.85 * |
| Parameters | Sham | 7 d I/R | 15 d I/R |
|---|---|---|---|
| Urea (mg/dL) | 40.9 ± 2.5 | 61.2 ± 8.9 * | 51.6 ± 3.2 |
| BUN | 19.1 ± 1.2 | 28.6 ± 4.2 * | 24.1 ± 1.5 |
| FGF-23 (pg/mL) | 186.6 ± 50.5 | 298.0 ± 65.2 | 759.7 ± 151.1 **, ## |
| Phosphate (mg/dL) | 6.05 ± 1.01 | 4.43 ± 1.01 | 9.33 ± 1.74 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Junho, C.V.C.; González-Lafuente, L.; Navarro-García, J.A.; Rodríguez-Sánchez, E.; Carneiro-Ramos, M.S.; Ruiz-Hurtado, G. Unilateral Acute Renal Ischemia-Reperfusion Injury Induces Cardiac Dysfunction through Intracellular Calcium Mishandling. Int. J. Mol. Sci. 2022, 23, 2266. https://doi.org/10.3390/ijms23042266
Junho CVC, González-Lafuente L, Navarro-García JA, Rodríguez-Sánchez E, Carneiro-Ramos MS, Ruiz-Hurtado G. Unilateral Acute Renal Ischemia-Reperfusion Injury Induces Cardiac Dysfunction through Intracellular Calcium Mishandling. International Journal of Molecular Sciences. 2022; 23(4):2266. https://doi.org/10.3390/ijms23042266
Chicago/Turabian StyleJunho, Carolina Victoria Cruz, Laura González-Lafuente, José Alberto Navarro-García, Elena Rodríguez-Sánchez, Marcela Sorelli Carneiro-Ramos, and Gema Ruiz-Hurtado. 2022. "Unilateral Acute Renal Ischemia-Reperfusion Injury Induces Cardiac Dysfunction through Intracellular Calcium Mishandling" International Journal of Molecular Sciences 23, no. 4: 2266. https://doi.org/10.3390/ijms23042266
APA StyleJunho, C. V. C., González-Lafuente, L., Navarro-García, J. A., Rodríguez-Sánchez, E., Carneiro-Ramos, M. S., & Ruiz-Hurtado, G. (2022). Unilateral Acute Renal Ischemia-Reperfusion Injury Induces Cardiac Dysfunction through Intracellular Calcium Mishandling. International Journal of Molecular Sciences, 23(4), 2266. https://doi.org/10.3390/ijms23042266