
SMRT and Illumina RNA-Seq Identifies Potential Candidate Genes Related to the Double Flower Phenotype and Unveils SsAP2 as a Key Regulator of the Double-Flower Trait in Sagittaria sagittifolia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Floral Morphological Characteristics of Single and Double Flowers of S. sagittifolia
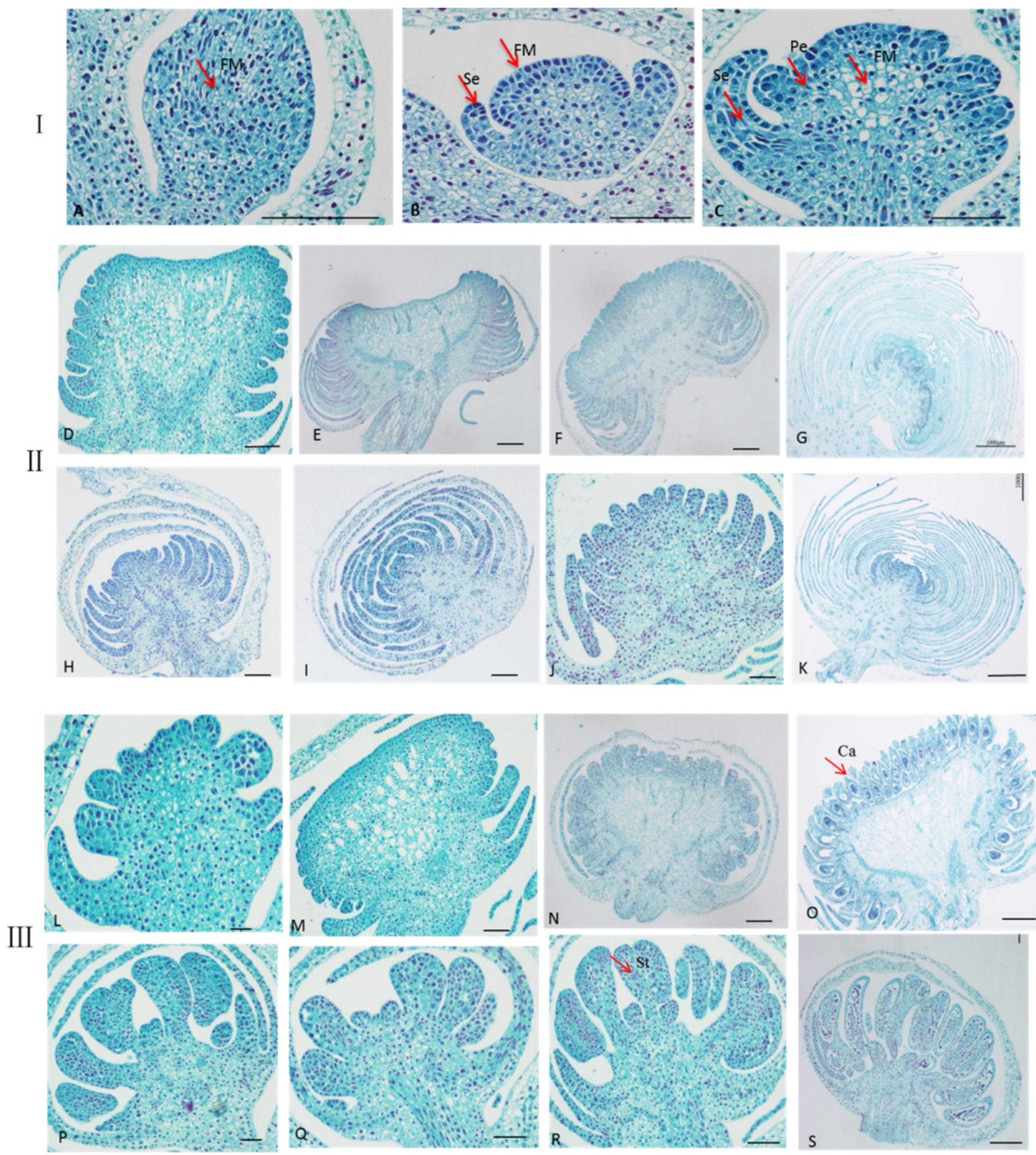
2.2. Observation the Development of Single and Double Flowers Morphology of S. sagittifolia under Light Microscope
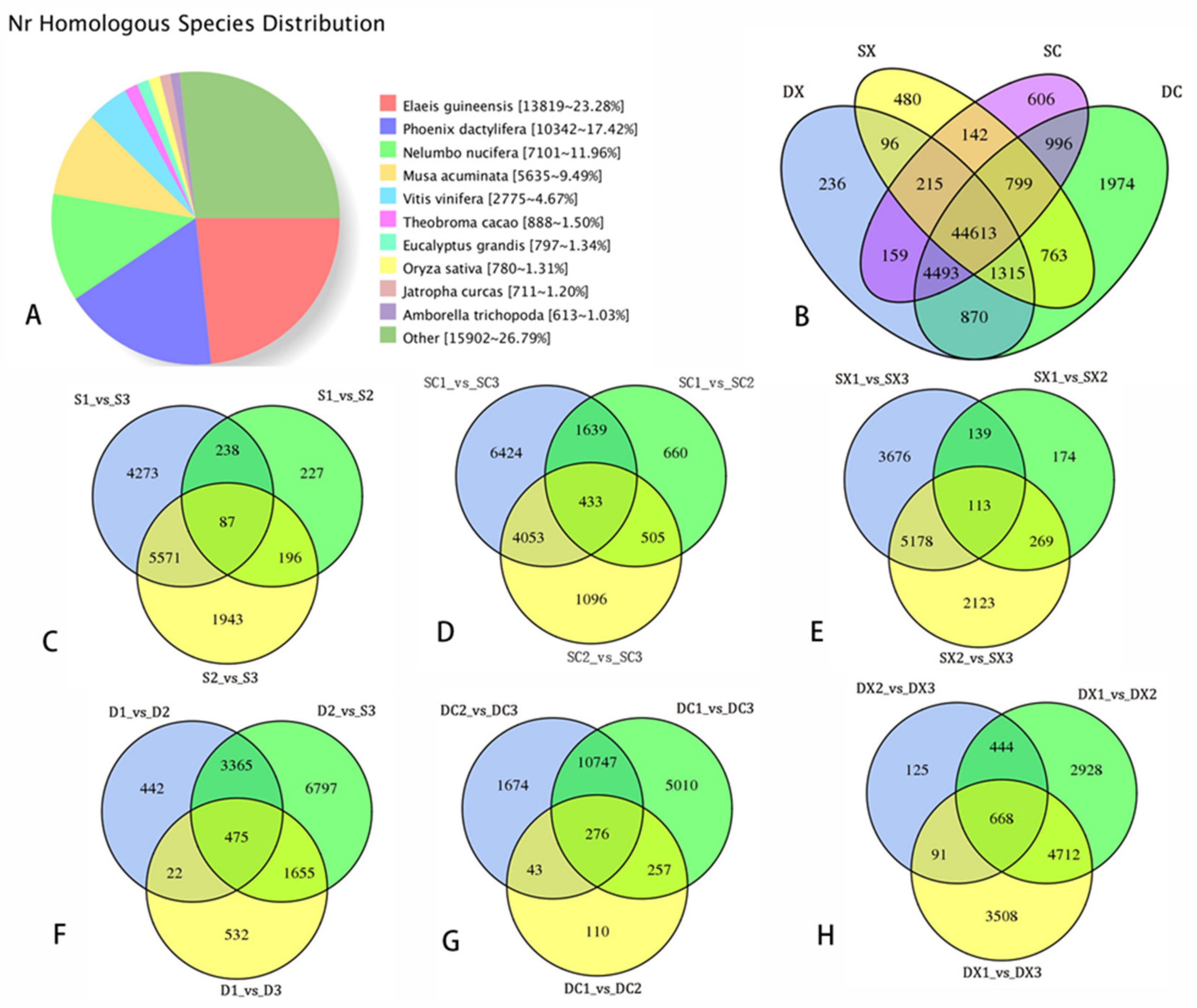
2.3. Sequencing Data Statistical Analysis of SMRT and Illumina RNA-Seq
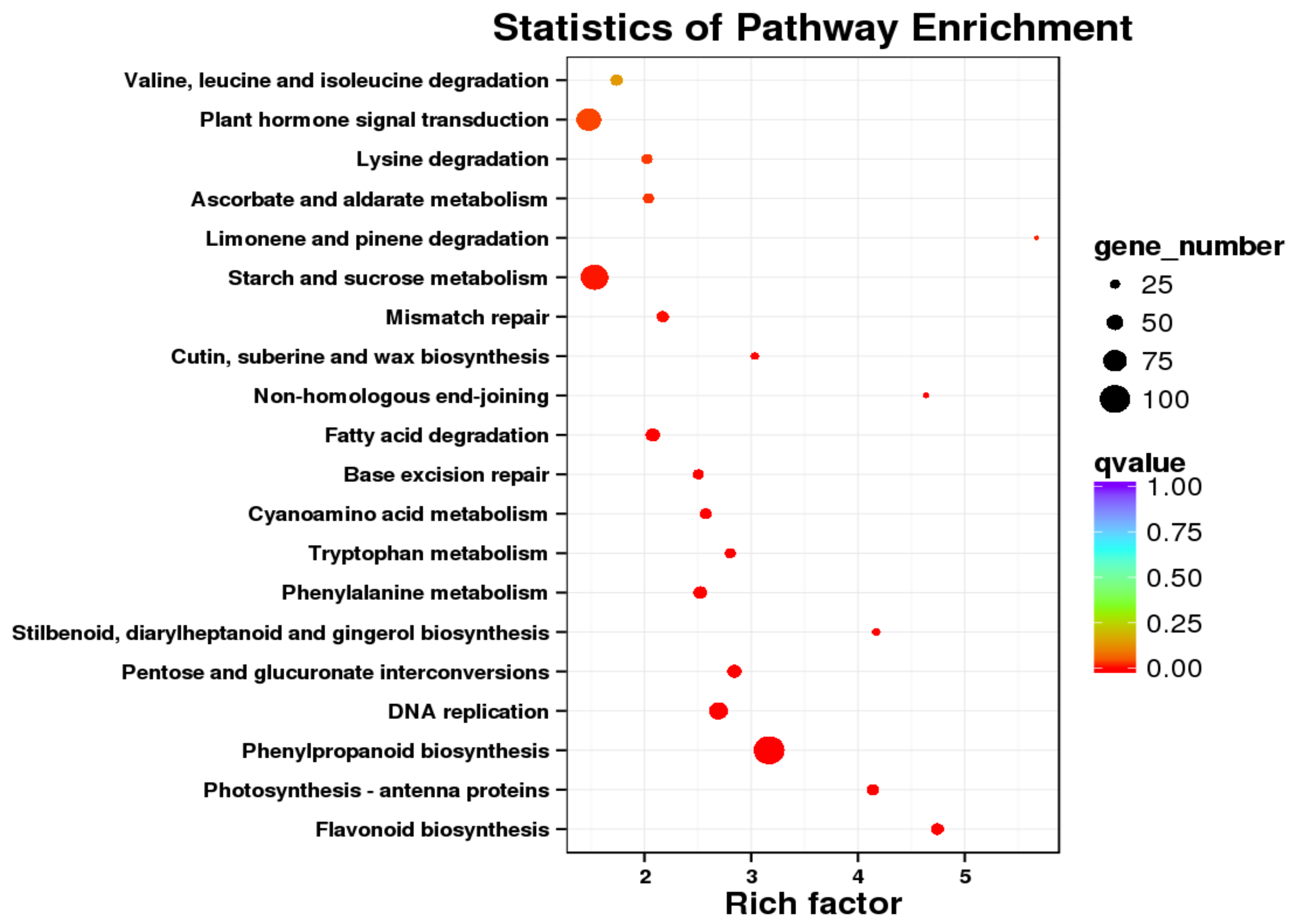
2.4. Differential Expression and Function Annotation of Transcripts Obtained by Transcriptome Sequencing
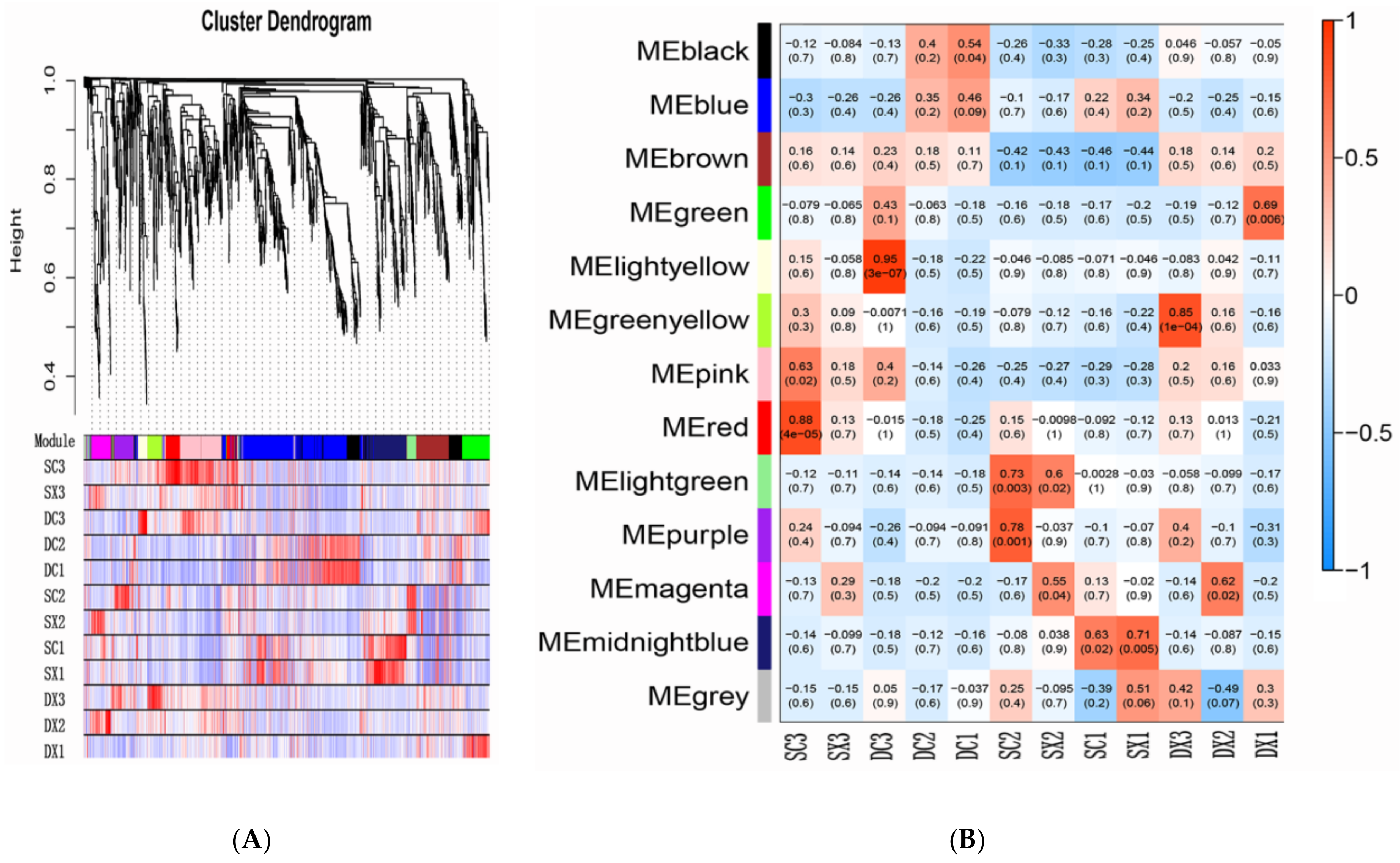
2.5. Transcription Factors and WGCNA
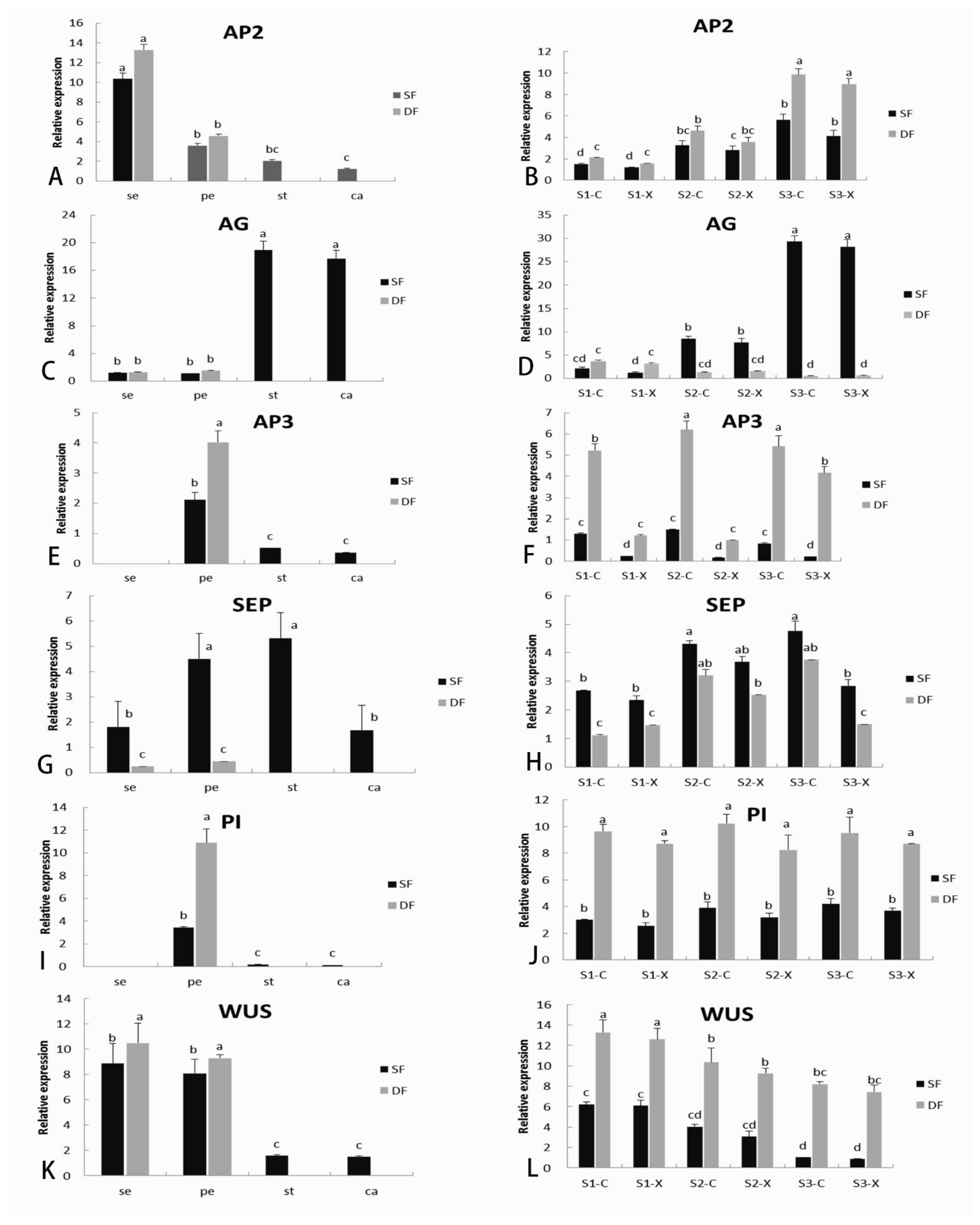
2.6. Identification of DGEs Related to Flower Formation and Verification of Gene Expression by qRT-PCR
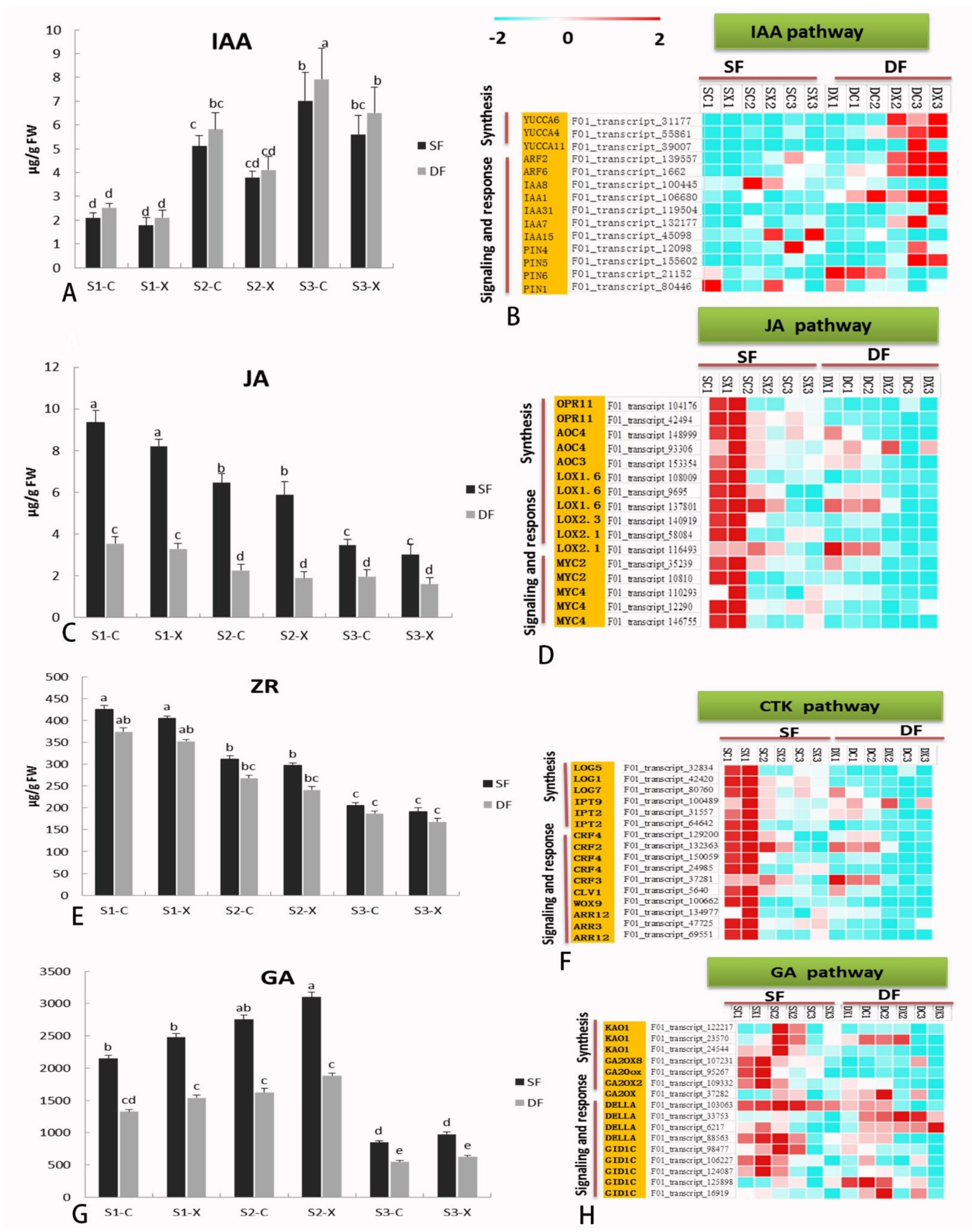
2.7. Hormone Biosynthesis and Signaling-Related Genes in Flower Development
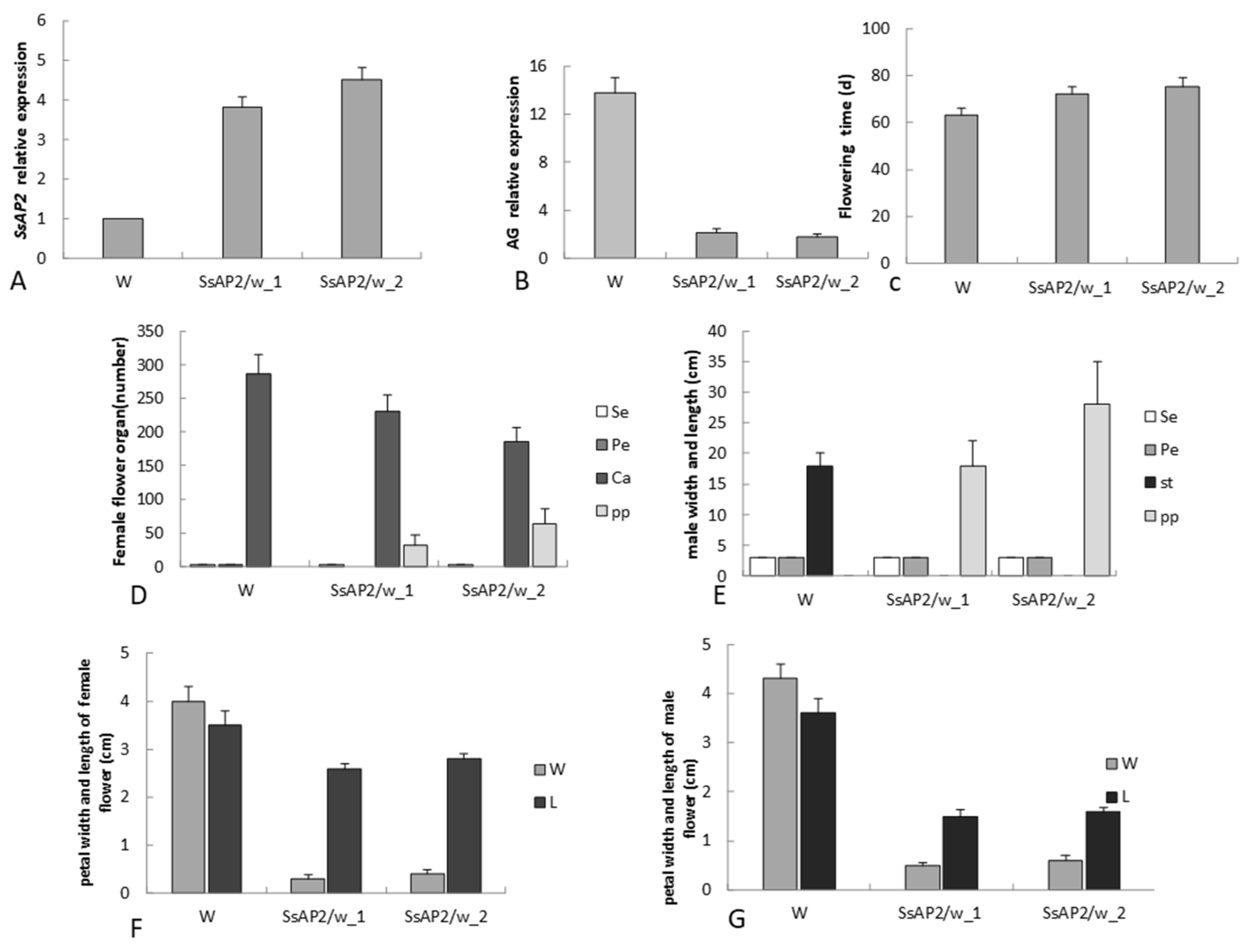
2.8. Functional Validation of SsAP2
3. Discussion
3.1. SMRT and Illumina RNA-Seq Provided Full Length Transcriptome Information for S. sagittifolia
3.2. Differentially Expressed Transcripts Reveal Genes Related to the Formation and Development of Double Flowers
3.3. Comparative Analysis the Expression of DEGs Related to Hormone Synthesis and Signal Metabolism in Single Double Flowers of S. sagittifolia
3.4. C-Function AG Gene Associated with Double Flower Formation
3.5. Function of SsAP2 in S. sagittifolia
4. Materials and Methods
4.1. Plant Materials
4.2. Preparation and Observation of Paraffin Sections
4.3. Determination of the Content of Endogenous Hormones
4.4. RNA Extraction and Library Construction
4.5. Quality Control of the Full-Length Transcriptome and UniGene Library
4.6. Annotation of Sequence Information
4.7. Other Structural Analyses
4.8. Verification of Differentially Expressed Genes (DEGs)
4.9. Construction of the Transformation Vector and Acquisition of Transgenic Plants
4.10. Floral Dip of the Inflorescence of S. sagittifolia
4.11. Identification of Transgenic S. sagittifolia
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdirashid, H.; Lenhard, M. Say it with double flowers. J. Exp. Bot. 2020, 71, 2469–2471. [Google Scholar] [CrossRef]
- Wang, Q.; Dan, N.; Zhang, X.; Lin, S.; Bao, M.; Fu, X. Identification, characterization and functional analysis of C-class genes associated with double flower trait in carnation (Dianthus caryphyllus L.). Plants 2020, 9, 87. [Google Scholar] [CrossRef]
- Meng, G.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, L.; Cao, K. Identification of loci for single/double flower trait by combining genome-wide association analysis and bulked segregant analysis in peach (Prunus persica). Plant Breed. 2019, 138, 360–367. [Google Scholar] [CrossRef]
- Espinosa-Soto, C.; Padilla-Longoria, P.; Alvarez-Buylla, E.R. A gene regulatory network model for cell-fate determination during Arabidopsis thaliana flower development that is robust and recovers experimental gene expression profiles. Plant Cell 2004, 16, 2923–2939. [Google Scholar] [CrossRef] [PubMed]
- Honma, T.; Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 2001, 409, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Mizzotti, C.; Galliani, B.M.; Masiero, S. The backstage of the ABC model: The Antirrhinum majus contribution. Plant Biosyst. 2014, 148, 176–186. [Google Scholar] [CrossRef]
- Li, X.; Li, J.; Fan, Z.; Liu, Z.; Tanaka, T.; Yin, H. Global gene expression defines faded whorl specification of double flower domestication in Camellia. Sci. Rep. 2017, 7, 3197. [Google Scholar] [CrossRef]
- Ma, J.; Shen, X.; Liu, Z.; Zhang, D.; Liu, W.; Liang, H.; Wang, Y.; He, Z.; Chen, F. DF-KjAG isolation and characterization of AGAMOUS-like genes associated with double-flower morphogenesis in Kerria japonica (Rosaceae). Front. Plant Sci. 2018, 9, 959. [Google Scholar] [CrossRef]
- Mizunoe, Y.; Kubota, S.; Kanno, A.; Ozaki, Y. Morphological variation and AGAMOUS-like gene expression in double flowers of Cyclamen persicum Mill. Hortic. J. 2015, 84, 140–147. [Google Scholar] [CrossRef]
- Nakatsuka, T.; Koishi, K. Molecular characterization of a double-flower mutation in Matthiola incana. Plant Sci. 2018, 268, 39–46. [Google Scholar] [CrossRef]
- Galimba, K.D.; Tolkin, T.R.; Sullivan, A.M.; Melzer, R.; Theissen, G.; Di Stilio, V.S. Loss of deeply conserved C-class floral homeotic gene function and C-and E-class protein interaction in a double-flowered ranunculid mutant. Proc. Natl. Acad. Sci. USA 2012, 109, E2267–E2275. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, D.; Liu, D.; Li, F.; Lu, H. Exon skipping of AGAMOUS homolog PrseAG in developing double flowers of Prunus lannesiana (Rosaceae). Plant Cell Rep. 2013, 32, 227–237. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, Z.; Li, X.; Li, J.; Yin, H. The APETALA1 and FRUITFUL homologs in Camellia japonica and their roles in double flower domestication. Mol. Breed. 2014, 33, 821–834. [Google Scholar] [CrossRef]
- Souer, E.; Rebocho, A.B.; Bliek, M.; Kusters, E.; de Bruin, R.A.M.; Koes, R. Patterning of inflorescences and flowers by the F-box protein double top and the leafy homolog aberrant leaf and flower of petunia. Plant Cell 2008, 20, 2033–2048. [Google Scholar] [CrossRef]
- Gattolin, S.; Cirilli, M.; Pacheco, I.; Ciacciulli, A.; Da Silva Linge, C.; Mauroux, J.B.; Lambert, P.; Cammarata, E.; Bassi, D.; Pascal, T.; et al. Deletion of the miR172 target site in a TOE-type gene is a strong candidate variant for dominant double-flower trait in Rosaceae. Plant J. 2018, 96, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Lee Glaettli, M.; Barrett, S.C.H. Pollinator responses to variation in floral display and flower size in dioecious Sagittaria latifolia (Alismataceae). New Phytol. 2008, 179, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Schwarz-Sommer, Z. Control of floral organ identity by homeotic MADS-box transcription factors. Results Probl. Cell Differ. 1994, 20, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hirano, H.Y. Function and diversification of MADS-box genes in rice. Sci. World J. 2006, 6, 1923–1932. [Google Scholar] [CrossRef]
- Bartlett, M.E. Changing MADS-box transcription factor protein-protein interactions as a mechanism for generating floral morphological diversity. Integr. Comp. Biol. 2017, 57, 1312–1321. [Google Scholar] [CrossRef]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The emerging importance of type I MADS-box transcription factors for plant reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef]
- May, P.; Liao, W.; Wu, Y.; Shuai, B.; Richard McCombie, W.; Zhang, M.Q.; Liu, Q.A. The effects of carbon dioxide and temperature on microRNA expression in Arabidopsis development. Nat. Commun. 2013, 4, 2145. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhang, Y.; He, Y.; Cao, Q.; Zhang, T.; Lou, L.; Cai, Q. Full-Length Transcriptome Assembly of Italian Ryegrass Root Integrated with RNA-Seq to Identify Genes in Response to Plant Cadmium Stress. Int. J. Mol. Sci. 2020, 21, 1067. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhang, L.; Chen, B.; Xiong, Z.; Chen, C.; Wang, L.; Yu, J.; Lu, C.; Wei, W. Functional identification and characterization of the Brassica napus transcription factor gene BnAP2, the ortholog of Arabidopsis thaliana APETALA2. PLoS ONE 2012, 7, e33890. [Google Scholar] [CrossRef]
- Zumajo-Cardona, C.; Pabon-Mora, N.; Ambrose, B.A. The evolution of euAPETALA2 genes in vascular plants: From plesiomorphic roles in sporangia to acquired functions in ovules and fruits. Mol. Biol. Evol. 2021, 38, 2319–2336. [Google Scholar] [CrossRef]
- Tooke, F.; Battey, N.H. A leaf-derived signal is a quantitative determinant of floral form in impatiens. Plant Cell 2000, 12, 1837–1848. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Y.; Jiang, D.; Du, S.; Deng, Z.; Wang, L.; Chen, S. Recent advances in the genomic profiling of bacterial epigenetic modifications. Biotechnol. J. 2019, 14, 1800001. [Google Scholar] [CrossRef]
- He, Z.; Su, Y.; Wang, T. Full-Length Transcriptome Analysis of Four Different Tissues of Cephalotaxusoliveri. Int. J. Mol. Sci. 2021, 22, 787. [Google Scholar] [CrossRef]
- Chen, R.; Fan, Y.; Zhou, H.; Mo, S.; Zhou, Z.; Yan, H.; Luo, T.; Huang, X.; Weng, M.; Lakshmanan, P.; et al. Global transcriptome changes of elongating internode of sugarcane in response to mepiquat chloride. BMC Genom. 2021, 22, 79. [Google Scholar] [CrossRef]
- Deng, N.; Hou, C.; Ma, F.; Liu, C.; Tian, Y. Single-molecule long-read sequencing reveals the diversity of full-length transcripts in leaves of gnetum (Gnetales). Int. J. Mol. Sci. 2019, 20, 6350. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhang, S.; Luo, C.; He, X.; Wei, S.; Jiang, W. Transcriptome analysis of starch and sucrose metabolism across bulb development in Sagittaria sagittifolia. Gene 2018, 649, 99–112. [Google Scholar] [CrossRef]
- Vandenbussche, M.; Zethof, J.; Souer, E.; Koes, R.; Tornielli, G.B.; Pezzotti, M.; Ferrario, S.; Angenent, G.C.; Gerats, T. Toward the analysis of the petunia MADS box gene family by reverse and forward transposon insertion mutagenesis approaches: B, C, and D floral organ identity functions require SEPALLATA-like MADS box genes in petunia. Plant Cell 2003, 15, 2680–2693. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shi, M.; Chen, W.; Hu, R.; Jing, D.; Wu, D.; Wang, S.; Li, Q.; Deng, H.; Guo, Q.; et al. Expression pattern and functional characterization of PISTILLATA ortholog associated with the formation of petaloid sepals in double-flower Eriobotrya japonica(Rosaceae). Front. Plant Sci. 2020, 10, 1685. [Google Scholar] [CrossRef]
- Moser, M.; Asquini, E.; Miolli, G.V.; Weigl, K.; Hanke, M.V.; Flachowsky, H.; Si-Ammour, A. The MADS-box gene MdDAM1 controls growth cessation and bud dormancy in apple. Front. Plant Sci. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Oh, J.; Shin, Y.; Ha, I.; Lee, M.; Lee, S.; Kang, B.; Kyeong, D.; Kim, D. Transcriptome Profiling of Two Ornamental and Medicinal Papaver Herbs. Int. J. Mol. Sci. 2018, 19, 3192. [Google Scholar] [CrossRef] [PubMed]
- Cronk, Q.; Mueller, N.A. Default sex and single gene sex determination in dioecious plants. Front. Plant Sci. 2020, 11, 350–361. [Google Scholar] [CrossRef]
- Mitsuda, N.; Ohme-Takagi, M. Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiol. 2009, 50, 1232–1248. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Zeisberger, E.; Vybiral, S.; Jansky, L. Endogenous antipyretics: Neuropeptides and glucocorticoids. Front. Biosci. -Landmark 2004, 9, 816–826. [Google Scholar] [CrossRef]
- Song, S.; Wang, Z.; Ren, Y.; Sun, H. Full-Length Transcriptome Analysis of the ABCB, PIN/PIN-LIKES, and AUX/LAX Families Involved in Somatic Embryogenesis of Lilium pumilum DC. Fisch. Int. J. Mol. Sci. 2020, 21, 453. [Google Scholar] [CrossRef]
- Liscum, E.; Reed, J.W. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 2002, 49, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Brioudes, F.; Joly, C.; JSzécsi Varaud, E.; Bendahmane, M. Jasmonate controls late development stages of petal growth in Arabidopsis thaliana. Plant J. 2010, 60, 1070–1080. [Google Scholar] [CrossRef]
- Kim, J. Four shades of detachment: Regulation of floral organ abscission. Plant Signal. Behav. 2013, 9, e976154. [Google Scholar] [CrossRef] [PubMed]
- Cheon, K.-S.; Nakatsuka, A.; Tasaki, K.; Kobayashi, N. Floral morphology and MADS gene expression in double-flowered Japanese evergreen azalea. Hortic. J. 2017, 86, 269–276. [Google Scholar] [CrossRef]
- Sieburth, L.E.; Running, M.P.; Meyerowitz, E.M. Genetic separation of third and fourth whorl functions of AGAMOUS. Plant Cell 1995, 7, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Sage-Ono, K.; Ozeki, Y.; Hiyama, S.; Higuchi, Y.; Kamada, H.; Mitsuda, N.; Ohme-Takagi, M.; Ono, M. Induction of double flowers in Pharbitis nil using a class-C MADS-box transcription factor with chimeric repressor gene-silencing technology. Plant Biotechnol. 2011, 28, 153–165. [Google Scholar] [CrossRef][Green Version]
- Yang, J.; Song, N.; Zhao, X.; Qi, X.; Hu, Z.; Zhang, M. Genome survey sequencing provides clues into glucosinolate biosynthesis and flowering pathway evolution in allotetrapolyploid Brassica juncea. BMC Genom. 2014, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Saito, M.; Yamada, E.; Fujita, K.; Yamagishi, N.; Yoshikawa, N.; Nishihara, M. Isolation and characterization of the C-class MADS-box gene involved in the formation of double flowers in Japanese gentian. BMC Plant Biol. 2015, 15, 182. [Google Scholar] [CrossRef]
- Tasaki, K.; Higuchi, A.; Fujita, K.; Watanabe, A.; Sasaki, N.; Fujiwara, K.; Abe, H.; Naito, Z.; Takahashi, R.; Hikage, T.; et al. Development of molecular markers for breeding of double flowers in Japanese gentian. Mol. Breed. 2017, 37, 33. [Google Scholar] [CrossRef]
- Ito, T.; Ng, K.-H.; Lim, T.-S.; Yu, H.; Meyerowitz, E.M. The homeotic protein AGAMOUS controls late stamen development by regulating a jasmonate biosynthetic gene in Arabidopsis. Plant Cell 2007, 19, 3516–3529. [Google Scholar] [CrossRef]
- Tani, E.; Polidoros, A.; Flemetakis, E.; Stedel, C.; Kalloniati, C.; Demetriou, K.; Katinakis, P.; Tsaftaris, A.S. Characterization and expression analysis of AGAMOUS-like, SEEDSTICK-like, and SEPALLATA-like MADS-box genes in peach (Prunus persica) fruit. Plant Physiol. Biochem. 2009, 47, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Salamah, A.; Rostina, I. Analysis of AGAMOUS gene expression in Hibiscus rosa-sinensis L. Single pink, crested peach, and double orange flowers. In International Conference on Mathematics and Natural Sciences; IOP Publishing: Bristol, UK, 2019; Volume 19, pp. 316–329. [Google Scholar] [CrossRef]
- Benedito, V.A.; Angenent, G.C.; Van Tuyl, J.M. Lilium longiflorum and molecular floral development: The ABCDE model. J. Exp. Bot. 2003, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, J.M.; Greenwood, J.R.; Finnegan, E.J.; Jernstedt, J.; Dubcovsky, J. APETALA 2-like genes AP2L2 and Q specify lemma identity and axillary floral meristem development in wheat. Plant J. 2020, 101, 171–187. [Google Scholar] [CrossRef]
- François, L.; Verdenaud, M.; Fu, X.; Ruleman, D.; Dubois, A.; Vandenbussche, M.; Bendahmane, A.; Raymond, O.; Just, J.; Bendahmane, M. A miR172 target-deficient AP2-like gene correlates with the double flower phenotype in roses. Sci. Rep. 2018, 8, 12912. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-H.; Lee, S.; Yun, J.; Lee, M.; Park, C.-M. The miR172 target TOE3 represses AGAMOUS expression during Arabidopsis floral patterning. Plant Sci. 2014, 215, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Kim, Y.; Dinh, T.T.; Chen, X. MiR172 regulates stem cell fate and defines the inner boundary of APETALA3 and PISTILLATA expression domain in Arabidopsis floral meristems. Plant J. 2007, 51, 840–849. [Google Scholar] [CrossRef]
- Ai, J.; Wang, Y.P.; Li, C.Y.; Guo, X.W.; Li, A.M. The changes of three endogenous hormones during flower bud differentiation of Schisandra chinensis. Chin. Mater. Med. 2006, 31, 24–26. [Google Scholar]
- Liu, L.; Wang, Z.; Su, Y.; Wang, T. Characterization and Analysis of the Full-Length Transcriptomes of Multiple Organs in Pseudotaxuschienii(W.C.Cheng). Int. J. Mol. Sci. 2020, 21, 4305. [Google Scholar] [CrossRef]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef] [PubMed]
- MISA. Available online: http://www.alz.org/what-is-dementia.asp (accessed on 25 May 2021).
- CNCI. Available online: https://github.com/www-bioinfo-org/CNCI#install-cnci (accessed on 8 June 2021).
- CPC. Available online: http://cpc.cbi.pku.edu.cn/ (accessed on 2 June 2021).
- Liu, Y.; Wang, L.; Yu, L. The principle and application of the single-molecule real-time sequencing technology. Yichuan 2015, 37, 259–268. [Google Scholar] [CrossRef]
- Zhang, C. Guilin, S. sagittifolia pollution-free cultivation technology. Yangtze River Veg. 2005, 12, 2. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, M.; Jiang, W.; Lin, Z.; Lin, Q.; Ye, Q.; Wang, W.; Xie, Q.; He, X.; Luo, C.; Chen, Q. SMRT and Illumina RNA-Seq Identifies Potential Candidate Genes Related to the Double Flower Phenotype and Unveils SsAP2 as a Key Regulator of the Double-Flower Trait in Sagittaria sagittifolia. Int. J. Mol. Sci. 2022, 23, 2240. https://doi.org/10.3390/ijms23042240
Gao M, Jiang W, Lin Z, Lin Q, Ye Q, Wang W, Xie Q, He X, Luo C, Chen Q. SMRT and Illumina RNA-Seq Identifies Potential Candidate Genes Related to the Double Flower Phenotype and Unveils SsAP2 as a Key Regulator of the Double-Flower Trait in Sagittaria sagittifolia. International Journal of Molecular Sciences. 2022; 23(4):2240. https://doi.org/10.3390/ijms23042240
Chicago/Turabian StyleGao, Meiping, Wen Jiang, Zhicheng Lin, Qian Lin, Qinghua Ye, Wei Wang, Qian Xie, Xinhua He, Cong Luo, and Qingxi Chen. 2022. "SMRT and Illumina RNA-Seq Identifies Potential Candidate Genes Related to the Double Flower Phenotype and Unveils SsAP2 as a Key Regulator of the Double-Flower Trait in Sagittaria sagittifolia" International Journal of Molecular Sciences 23, no. 4: 2240. https://doi.org/10.3390/ijms23042240
APA StyleGao, M., Jiang, W., Lin, Z., Lin, Q., Ye, Q., Wang, W., Xie, Q., He, X., Luo, C., & Chen, Q. (2022). SMRT and Illumina RNA-Seq Identifies Potential Candidate Genes Related to the Double Flower Phenotype and Unveils SsAP2 as a Key Regulator of the Double-Flower Trait in Sagittaria sagittifolia. International Journal of Molecular Sciences, 23(4), 2240. https://doi.org/10.3390/ijms23042240